Journal of Behavioral and Brain Science
Vol.3 No.5(2013), Article ID:36389,10 pages DOI:10.4236/jbbs.2013.35040
Biomarkers for Alzheimer’s Disease: Imagination or Reality-View and Review!
1Rabin Medical Center, Campus Hasharon, Petah Tikva, Israel
2EMMS Nazareth, The Nazareth Hospital, Faculty of Medicine Bar-Ilan University, Nazareth, Israel
Email: zaherarmaly@nazhosp.com, bishara@nazhosp.com, *bowirrat@netvision.net.il
Copyright © 2013 Mustafa Yassin et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 30, 2013; revised June 5, 2013; accepted June 20, 2013
Keywords: Biomarkers; Alzheimer’s Disease; Amyloid-β; Tau; Cerebrospinal Fluid; Blood
ABSTRACT
The Alzheimer’s disease (AD) is the most widespread chronic, insidious neurodegenerative disease causing dementia in elderly and leading to a massive burden on AD individuals, their families, and on social and health care systems. Its diagnosis is subjective, definite AD can only be diagnosed after pathological brain specimens are examined by either biopsy or autopsy, and it covers 50% - 70% of all dementia cases. It is estimated that, by 2050, the number of people aged 80 years or older will approach 370 million worldwide and that 50 percent of those aged 85 years or older will be afflicted with AD. Causes of the disease are multifactorial; where genetics and environmental risk factors work in harmony to cause the disease. Neuropathological features of AD depend on finding of extracellular deposits of β-amyloid peptides (Aβ) that lead to senile plaque formation and intracellular neurofibrillary tangles of hyperphosphory lated tau. However, increasing evidence has suggested that inflammation may play a critical role in AD pathogenesis as well. In the era of genome and sophisticated technology, AD early diagnosis still indecisive and valid biomarkers for AD to be used in routine clinical practice have met with dissatisfaction. Indeed, the relatively unchanged levels of plasma β-amyloid in AD, and a lack of analytical sensitivity for assays for the axonal damage marker have increased the effort to find an alternative ultra-sensitive assay for pathological markers in peripheral blood. We believe that early presymptomalogical practical inexpensive strategies, for characterizing a potential surrogate marker in blood and CSF for AD is warranted, are of interest because they: 1) confirm diagnosis; 2) enable epidemiological screening; 3) identify distinct groups of patients (predictive testing); 4) monitor progression and response to treatment and aid in design/implementation of optimal therapeutic regimens; 5) further the study of the brain-behaviour relationship underlying neurodegeneration.
1. Introduction
Alzheimer’s disease (AD), essentially, is multifactorial disease and a genetically heterogeneous disorder characterized by insidious chronic and progressive loss of neurons in discrete areas of the brain, causing debilitating symptoms and globally decreasing cognitive function such as dementia, loss of memory, loss of sensory or motor capability, decreased overall quality of life and well-being, and eventually, patient’s premature death. Not to mention the long sufferance and the heavy economical and emotional load placed on families and caregivers. No satisfactory cure is available yet; at best, existing treatments are symptomatic in nature and do not completely prevent or significantly impede the progression of the disease [1,2].
AD covers 50% - 70% of all dementia cases that affects millions of people globally. It is estimated that, by 2050, the number of people aged 80 years or older will approach 370 million worldwide and that 50 percent of those aged 85 years or older will be afflicted with AD [3,4].
However, rapid progress towards understanding the molecular underpinnings of AD is revolutionizing drug discovery for this condition. Furthermore, the development of models for this disorder is accelerating efforts to translate insights related to neurodegenerative mechanisms into disease-modifying therapies. Neither of Myriad approaches that had been undertaken to identify biomarkers such as neurophysiological, imaging and cognitive testing in addition to newer sophisticated technologies for instance biochemical, proteomic, metabanomic and gene array profiling of tissue and biofluids from patients, and despite generous international effort, neither has proved satisfactory [5,6].
Hence, clear vision and an understanding of pathological progression of the illness can help to identify points of intervention and lead to promising therapeutic approaches. A fundamental approach for reducing the burden of AD is thus to slow or halt progression, and ultimately, to prevent the onset of the disease process. Strategies for neurorescue, neurorepair, or neuroprotection are being actively pursued by the basic, translational, and clinical research communities [7].
2. Overview of Biomarkers Properties
Access to molecular and biochemical markers of AD would complement clinical approaches, and further the goals of early and accurate diagnosis. Hence, the importance of the biological biomarkers studies which are quantitative measurements that provide information about; intrinsic biological processes, a disease circumstances and risk of developing an illness (antecedent biomarkers), assist in diagnosing disease (diagnostic biomarkers), response to treatment (prognostic biomarkers), providing much-needed insight into preclinical and clinical data, all of these are still valid procedures for early detection of AD [8].
Detection the subject’s susceptibility to the disease prior to appearance of prodromal signs, and detection of neural dysfunction before irreversible cellular damage, will be tremendously valuable for developing; prevention and intervention strategies and early treatments. From here stems the truthfulness and reliability of biomarker to distinguishbetween normal and interested disease [9].
In fact, biomarkers are set of factors used to measure anatomic, physiologic, biochemical, pharmacological, or molecular parameters associated with the presence and severity of particular disease states or processes in humans and animals [10,11].
These characteristics are objectively measured and carefully evaluated as indicators of normal biologic or pathogenic processes or pharmacological responses to a therapeutic intervention. Actually, define biomarker panels comprehensively to quantify risk, assess prognosis, and determine response to therapy [12]. Years ago, biomarkers were primarily physiological indicators such as blood pressure or heart rate. More recently, biomarker is becoming an important tool in different disciplines such as in field of oncology, immunology, cardiovascular diseases and metabolic diseases [13]. Synonym for molecular biomarker, such as elevated prostate specific antigen (PSA) as a molecular biomarker for prostate cancer, or using enzyme assays as liver function tests [14].
Biomarkers also cover the use of molecular indicators of environmental exposure in epidemiologic studies such as human papilloma virus (HPV) or certain markers of tobacco exposure such as 4-(methylnitrosamino)-1-(3- pyridyl)-1-butanone (NNK) which is a nitrosamine present in tobacco that is a potent procarcinogen. It is activated by CYP2A6, and plays a role as a biomarker of exposure to cigarette smoke, produced upon the curing of tobacco [15,16]. Also, Genomic biomarkers have principal role in investigation diseases; such as Apolipoproteine Epsilon-4 allele (APOE-ε4) for AD, and HLA for looking for narcolepsy, CD19, Sialophorin, CD11 integrin cluster, and IL-4 receptor for Crohn’s disease. Other methods and assays are used like; Serum or Spinal fluid substance, Neuroimaging and Physiologic parameters [17-20].
2.1. Biomarkers and Alzheimer’s Disease (AD)
2.1.1. The Cerebrospinal Fluid (CSF) Biomarkers
Plethora of biomarkers has been evaluated for AD. According to the literature more than 25 potential biomarkers for AD had previously been identified and new ones are still under investigation [21].
The CSF is the direct target because of its straight contact with the extracellular space of the brain where many biochemical processes in the brain take place and are reflected in the CSF. Since AD pathology is restricted to the brain, CSF is an obvious source and justified biomarkers for AD. Indeed, early biomarker discovery efforts for AD is based on immunoassays to detect and measure pathophysiological molecules of AD, such as Cerebrospinal fluid-derived β-amyloid protein 1-42, total tau protein, and phosphorylated tau (181,199,231) protein [22].
Several of these immunoassays demonstrated effectiveness in improving AD diagnosis, and these molecules continue to be used as potential diagnostic biomarkers. Two categories of AD biomarker studies are available today in the literature: the first, aims to prove potential biomarkers that already exist, and the second, looks for clues novel AD biomarkers with an emphasis on research using Neuroimaging. This article will address the different aspects of AD biomarkers with an emphasis on the most significant and prevalent ones [23].
2.1.2. Amyloid Beta (Aβ)
Aβ is composed of a family of peptides produced by proteolytic cleavage of the type I transmembrane straddling glucoprotein amyloid precursor protein (APP). In fact, one of the extracellular pathological hallmarks lesions of AD is the accumulation of aggregated Aß42 peptide in amyloid plaques. Aß42 makes up less than 10% of total Aß and it is the initial and major component of amyloid plaque deposits in AD [24]. Increased Aß42 production throughout life and failure of Aß clearance mechanisms lead to accumulation of oligomerization of Aß42 in limbic and association cortices [25] (Figure 1).
The detection that Aβ42 peptide forms the essential component of AD plaques and that is secreted by cells led to examinations of Aβ42 in the CSF. Previous studies showed a decrease in CSF-Aβ42 to about 40% - 50% of control levels has been found in AD in several papers [26].
It is not clear why Aβ42 is reduced in AD patients, but it is thought that its decrease reflects trapping of Aß42 in the amyloid plaques in the brain. Indeed, studies suggest that decreased CSF Aß42 correlates well with the levels of amyloid plaques in the AD brain as determined by amyloid imaging [27]. Decreased CSF Aß42 appears to be an early marker for AD and might predict conversion to AD when combined with CSF tau measures. Considering the diagnostic sensitivity and specificity levels of Aβ42 in CSF-AD patients that ranged between 80% and 90%, may enhance it’s used as potential test in combinations with other tests [28-30].
2.1.3. Total Tau Protein
While tau has long been implicated in neurodegenerative conditions, its functions in the adult brain and the precise
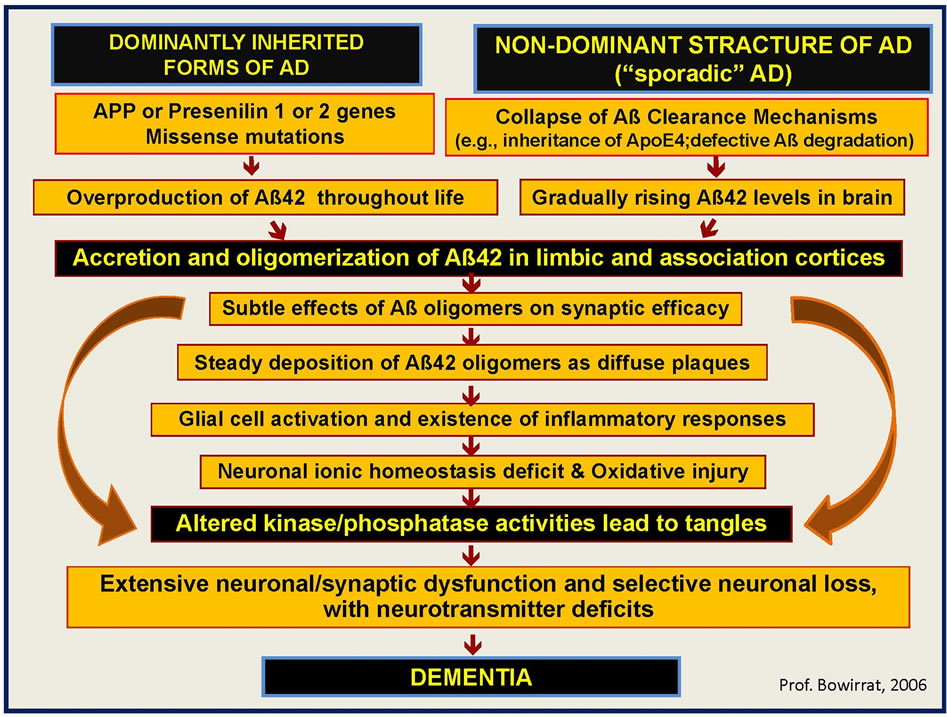
Figure 1. The figure describes the pathways leading and underlying the mechanism of dementia, where dominantly inherited form (APP, PRESINILINS 1 & 2 GENES) and non-dominant inherited forms (Sporadic form of Alzheimer’s diseases—AD, inheritance of ApoE4 and defective Aß degradation) works in harmony to cause the diseases. Indeed the overproduction of Aß42 throughout life as result of missense mutations in the dominantly inherited form and the collapse of Aß Clearance Mechanisms in the sporadic form can lead to the accumulation of the Aß42 oligomers responsible for the cell damage in different region of the brain with extensive neuronal/synaptic dysfunction and selective neuronal loss, with neurotransmitter deficits. The final outcome will be dementia.
mechanisms by which it contributes to neuronal dysfunction and degeneration in these disorders remain to be elucidated.
Physiologically, tau proteins are an intracelluar microtubule-associated protein acting as stabilizers microtubules in the cell cytoskeleton, and pathologically, tau proteins characterize the main component relating to intraneuronal changes in AD patients.
A flurry of recent studies has challenged major dogmas in this field, including the vision that filamentous tau aggregates are the most pernicious forms of tau, that failure of tau function plays a major role in the pathogenesis of tauopathies. Provocative discoveries suggest that tau regulates neuronal excitability and that it is required for Aβ and other neurotoxins to cause neuronal deficits, aberrant network activity and cognitive decline [31].
Microtubule-associated protein tau becomes abnormally hyperphosphorylated in AD and accumulates as tangles of paired helical filaments in neurons undergoing degeneration. Because of alternative splicing of TmRNA, there are six isoforms ranging in size from 352 to 441 amino acids, with molecular weights of approximately 50 - 65 kDa [32].
In normal situations, tau is coordinated by phosphorylation. In abnormal conditions, tau becomes hyperphosphorylated (phospho-tau) and accumulates as paired helical filaments that aggregate into masses inside the neuronsas neurofibrillary tangles (NFT), which represent one of the hallmarks of AD [33].
The logic for considering tau as biomarker is the presence of abnormal intraneuronal aggregates of phosphotau observed in many tauopathies, including AD, Tau aggregates can be examined in the Brain and peripheral fluids. Biochemical and Immunohistochemical properties of Tau cumulative in brain permit postmortem categorization and differential diagnosis of tauopathies [34]. The first report on CSF T-T as a biomarker for AD was published in 1993. In that paper, an enzyme-linked immunosorbent assay (ELISA) with a polyclonal reporter antibody was used [35].
Total tau concentrations especially phosphory lated tau (181,199,231) proteins can be measured in the CSF as Aß, and show a good correlation with the diagnosis of AD [36].
Previous studies have established 300% increase in the concentration of total-Tau in CSF Alzheimer’s patients 70 years and older versus control subjects younger than 50 years [(>600 pg/mL vs <200 pg/mL), respectively] [21]. A strong correlation between age and t-tau in healthy individuals has been determined with a cut off value of >500 pg/mL (>70 years) versus 450 pg/mL (<70 years) [37]. CSF t-tau levels in AD patients have a sensitivity of 90% and specificity of 81% compared to healthy Control [38]. Compared to other dementias, the sensitivity and specificity drops to 50% - 60% [39].
Notwithstanding, the relative high sensitivity and specificity that CSF T-tau level plays as discriminator between AD patients and control, but its presence in other neurological diseases for instance, vascular dementia, progressive supranuclear palsy, corticobasal degeneration, cerebral autosomal dominant arteriopathy with subcortical infarcts, leuko encephalopathy (CADASIL), and its notable high concentration in Spongiform Encephalopathies (3000 pg/mL), deceases its validity as AD specific biomarker [40,41]. This notion boost the idea that some proteins such as p-tau may simply be generally associated with neurode generation and cellular failure, as they have been linked to other neurological disorders and finally demilitarize it legitimacy as a specific AD pathology biomarker, despite its statistical significance.
2.1.4. Phosphorylated Tau
Tau proteins belongs to a group of proteins referred to as microtubule-associated phosphoproteins that are abundant in neurons in the central nervous system and are less common elsewhere. It has been almost 38 years since tau was discovered as a heat resistant and limited affected by acid treatment without loss their function [42].
Remarkably soluble neuronal microtubule-associated protein that normally functions to support the assembly and stabilization of the microtubule cytoskeleton [31].
There is significant evidence that a deviations from normal phosphorylation process (Hyperphosphorylation) results in tau dysfunction and modification of the conformation of tau and decreasing its affinity to micro-tubules [32].
Discussions with regulatory authorities gain momentum defining the role of tau biomarkers for trial designs and how they may be further qualified for surrogate marker status [43].
Numerous studies have evaluated the diagnostic value of CSF markers for AD cases, finding high CSF-T and P-T, and low CSF-Aβ42, with sensitivity figures around 85% - 90%, but with lower specificity against other dementia disorders.
2.2. Blood Biomarkers & Neurotransmitter Deficits as Potential Biomarkers for AD
Because of the simplicity and relatively the omnipresent venepuncture procedure to collect blood from AD patients everywhere, knowing its cheapness, noninvasive routine procedure, possible repeatability measurements, and time-saving method comparing to CSF lumbar puncture, many scientists comment hope to find out a valid peripheral blood biomarkers. Despite, the hesitation on the ability of the peripheral blood to supply information and complete image about pathological mechanisms taken place on brain like transport abnormal molecules who may denote evident about what happened in the AD brain given its indirect connectivity as result of the delimitation by the blood-brain barrier (BBB)-confines its usefulness as keystone biomarker and decrease its sensitivity and specificity as biomarker. However, challenges continuous to hunt for a specific clue biomarker of interest who involve in various mechanisms such as inflamemation, senescence, apoptosis, and cerebrovascular dysfunctions.
Neurotransmitter Deficits as Potential Biomarkers for AD
Alzheimer’s disease (AD) per se has complex pathophysiology, involving multiple biochemical pathways. The loss of neuronal cells leads to deficits in various neurotransmitters. Changes in the cholinergic, serotonergic, glutamatergic noradrenergic, dopaminergic, GABAergic and somatostatinergic neurons and the potential involvement of inflammatory, oxidative and hormonal pathways were investigated to determine their roles in AD [44].
Consequently, these pathways were carefully planned as potential targets for AD early detection, treatment and prevention strategies. In this manuscript we will spotlight on some important neurotransmitters involved in pathology of this devastating disease. Indeed, in the brain the cholinergic pathway represented by acetylcholine (Ach) which is a critical neurotransmitter responsible for information processing, memory; learning and attention. Decreasing levels of ACh synthesis are significantly correlated with increasing severity of dementia in patients with AD [45]. Cortical deficiencies Ach contribute to a loss of cholinergic neurons in the basal forebrain, reduced acetylcholine (Ach) levels, and adecrease in the synthesis of choline acetyltransferase, the enzyme that catalyzes the synthesis of ACh from choline and acetyl coenzyme-A by 35% to 50% in AD comparing to healthy subjects. Treatments such as, Acetylcholinesterase inhibitors (Donepezil Hydrochloride-Aricept), Tacrine which is a centrally acting anticholinesterase and indirectly acting as cholinergic agonist (parasympathomimetic), the reversible cholinesterase inhibitor (rivastigmine tartrate-Exelon) and Ach receptor agonists, including nicotine, have been used to treat AD. The slight success of these medications suggests that, in addition to Ach deficiency, other deficit neurotransmitters may play a role and may involved together with Ach reduction to cause the profound deteriorations in the cognitive impairment.
The glutamatergic pathway involvement in AD comes from its responsibility on different input and output pathways responsible for thinking and remembering, and is driven from the connection between the hippocampus and neocortix [46].
All such pathways activity depends on signaling mediated by the neurotransmitter glutamate. In intact brain glutamatergic cycles start in the hippocampus mitochondria neural cells, where the conversion of glutamine to glutamate takes place by the enzyme glutaminase and end on the packaging of the glutamate molecule into vesicles, glutamate released from vesicles into the synaptic cleft leading to increase free glutamate [47].
The interaction between glutamatergic receptor and postsynaptic neurons provokes transmission of neural signals, followed by quick synaptic glutamate levels restoration to normal concentrations, however, through the rapid uptake of unbound glutamate molecules by nearby glial cells, which subsequently convert these glutamate molecules to glutamine [48]. In AD patients’ interruption of gulamatergic cycle at the point of glial cell reuptake of free glutamine from the synapse is noticeable, and was confirmed by many studies who demonstrate decease levels of glutamate reuptake in the cortex in addition to the reduction uptake by vesicular glutamate transported [49]. As a result of the diminution in vesicular glutamate uptake, fewer glutamate will be stored in each vesicle, then neurons are left with low neurotransmitter molecules to release into the synaptic cleft in times of neuronal activity, consequently, the neural impulse will attenuated, leading to inadequate neurotransmission [50].
According to this hypothesis, it seems that low clearance of free glutamate from the synapse leads to high accumulation and high concentrations under resting conditions leading to abnormalities on neuronal signaling and neural death. In fact, the glutamatergic hypothesis links cognitive decline in patients with Alzheimer’s to neuronal damage resulting from over activation of Nmethyl-d-aspartate (NMDA) receptors by glutamate [51].
The sustained low-level activation of NMDA receptors, which are pivotal in learning and memory, may result from deficiencies in glutamate reuptake by astroglial cells in the synaptic cleft [51].
In 2003, the FDA approved a glutamate antagonist memantine (Namenda) for the treatment of moderate to severe AD [52].
The medication helps block the activity of the neurotransmitter glutamate by binding to N-methyl-D-aspartate (NMDA) receptors on the surface of brain cells. If glutamate levels are too low, cognitive problems develop. If levels are too high, glutamate over stimulates nerve cells, leading to cell death [53].
Serotonin (5-hydroxytryptamine or 5-HT) is synthesized from the amino acid tryptophan and considered a key neurotransmitter in the central and peripheral nervous systems [54].
Its pathways spread extensively throughout the brainstem, the cerebral cortex and the spinal cord, forming the principal serotonin centers in the caudal and rostral raphe nuclei [55].
It was observed that transmission of serotonin descending from the rostral raphe nuclei to the temporal lobe is decreased in AD patients, and there is an extensive degeneration of serotonergic neurons, with corresponding losses of the serotonin (5HT) transporter (5HTT), which is responsible for the reuptake of 5HT from the synaptic cleft. Studies indicates that allelic variation of the 5HTT gene promoter (5HTT gene-linked polymorphic region, 5HTTLPR) determines high or low 5HT uptake in normal human brain [56].
Because of the extensive serotonergic denervation that has been observed in the AD brain and the important role played by serotonin (5-HT) in executive functions control, this neurotransmitter system has become a focus of concerted research efforts to identify new treatments for AD [56,57].
3. Summary
Ones of the major novelties in the field of medical science are represented by biomarkers. Biomarkers are objectively and impartially measured and evaluated as an indicator of physiological processes, pathological processes or pharmacological responses to the applied therapy [58,59].
The scientific rationale and regulatory acceptance for biomarkers in medicine and in drug development has become more fully recognized, and biomarkers in medicine provide the platform for all players in this increasingly crucial area to converse and join forces to find confident biomarkers. Because of the complexity and pathological ramification of the disease, and after careful consideration to almost all medical literature who investigated AD biomarkers, we are convinced that joint action and cross-institutional standards must be employed in order to validate a novel biomarker.
Planned synergistic collaborations between academic institutions, pharmaceutical companies and supporting organizations for the establishment of standards and networks for the identification and qualification of biological marker candidates are urgently warranted.
Indeed, Interruption of the cascade of events that underlying AD pathology by novel therapeutic strategies that guide to even small delays, or even reject the onset and progression of the AD would significantly decrease the global burden of the disease, and a blessing advancement in deceasing sufferance of the patients.
Presently, Aβ1-42, t-tau and p-tau in CSF are considered the most consistent, sensitive and specific biomarkers for AD; and recently these biomarkers were found to be useful in differential dementia diagnosis. Patients with progressive supranuclear palsy had normal CSF biomarker values in 90%. Patients with CreutzfeldtJakob disease demonstrated an extremely high CSF t-tau at a relatively normal CSF p-tau. CSF AD biomarker profile was seen in 47% of patients with dementia with Lewy bodies (DLB), 38% in corticobasal degeneration (CBD), and almost 30% in frontotemporal lobar degeneration (FTLD) and vascular dementia (VaD). Concordance between clinical and neuropathologic diagnosis was 85% and CSF markers reflected neuropathology in 94% [60]. However, CSF collection, despite its importance, is an invasive procedure and many researchers steer clear of it, and sensitive assays to detect pathological molecules in CSF are still unavailable [61-64].
Therefore, more attention was focus toward bloodbased biomarkers; namely many biomarkers in blood depending on pathophysiological processes have been identified, such as amyloid plaque formation (Aβ), Aβ autoantibodies, platelet amyloid precursor protein (APP) isoforms, inflammation (cytokines), oxidative stress (vitamin E, isoprostanes), lipid metabolism (Apolipoprotein Epsilon-4 allele (ApoE-ε4), 24S-hydroxycholesterol), 3- nitrotyrosin, α-1-antichymotrypsin, C-reactive protein, C1q complementary systems, and vascular disease (homocysteine, lipoprotein) [65-68].
Previous studies found that plasma concentrations of Aβ do not correlate with those in CSF, and no consistent change in plasma Aβ over time in AD patients, and no cross-sectional Aβ differences between AD patients and controls was found that would allow plasma Aβ concentrations to be used as a diagnostic measure [68]. James D. Doecke et al., (2012) identify panel of plasma biomarkers that distinguish individuals with AD from cognitively healthy control with high sensitivity (85%) and specificity (93%) cross-validated accuracy measures, the revealed biomarkers panel that included markers significantly increased (cortisol, pancreatic polypeptide, insulin like growth factor binding protein 2, β2 microglobulin, vascular cell adhesion molecule 1, carcinoembryonic antigen, matrix metalloprotein 2, CD40, macrophage inflammatory protein 1α, superoxide dismutase, and homocysteine) and decreased (apolipoprotein E, epidermal growth factor receptor, hemoglobin, calcium, zinc, interleukin 17, and albumin) in AD [69]. Indeed [69], Plasma biomarker results confirm cerebrospinal fluid studies reporting increased levels of pancreatic polypeptide and N-terminal protein B-type brain natriuretic peptide in patients with AD and mild cognitive impairment. Incorporation of plasma biomarkers yielded high sensitivity with improved specificity, supporting their usefulness as a screening tool. The ApoE genotype was associated with a unique biochemical profile irrespective of diagnosis, highlighting the importance of genotype on blood protein profiles [70].
J. B. Toledo et al. (2013) mentioned that different inflammatory and metabolic pathways have been associated with AD. The authors recruited 818 subjects and studied multi-analyte panels using a large number of molecules that point to the affected pathways and also studied the relationship between a panel of plasma biomarkers and presence of AD-like brain atrophy patterns using MRI. Their results indicate that stress and insulin responses and cytokines associated with recruitment of inflammatory cells in Mild Cognitive Impairment-AD are associated with its characteristic AD-like brain atrophy pattern and correlate with clinical changes or CSF biomarkers [71].
Thus, a neuropathological diagnosis which leads to synchronization and correlation between CSF biomarkers and blood biomarkers levels may relatively reflect the pathological processes in the brain and increase the sensitivity, specificity and validity for putative biomarkers [72,73].
The most promising approach to diagnose AD is the combination of numerous biomarkers, and isolated tests should not be used alone. Unifying multiplex methological techniques in one assay; using standardized tests for measuring peripheral proteins to avoid contradictory results; and combining panels of existing biomarkers pattern who reflect the whole spectrum of abnormal proteins deposited in the brain or surveying the range of proteins in plasma (proteomics) will show promise for discovering biomarker profiles that are characteristic of AD, yet distinct from non-demented patients or patients with other forms of dementia. Furthermore, these proteins might serve as interesting biomarker candidates for multiple biomarkers strategies [74].
We believe that we should consider the centenarian’s hypothesis, where 90% of all of the centenarians were still independently functioning at the average age of 93 years by searching for their protective genes and genetic markers. Recent studies found 281 genetic markers that are 61% accurate in predicting who 100 years old is. These markers point to at least 130 genes, many of which have been shown to play roles in chronic diseases including the basic biological mechanisms of aging. Therefore, centenarian’s tremendous survival advantage may in great part due to the existence of longevity associated genetic variants that counteract or restrain the onset and the progression or even escape age-related diseases like AD. Our driving hypothesis is that we can examine these genes and signature markers among AD patients to verify if they possess the protective genes or markers, and to use this model as a diagnostic test [75-77].
Finally, we believe that prior to search an accurate biomarker, we must be confident of the actual diagnostic guidelines for AD. Reevaluation of current criteria, changes in the mentality and the way that many clinicians diagnose AD are mandatory, because diagnosis of AD is still largely based on exclusion criteria of secondary causes and other forms of dementia.
Despite the frustrations, enormous efforts have been made and others energetic efforts are ongoing to find adequate biomarkers characterized by high sensitivity and specificity for this complex neurodegenerative disorder. The future is full of surprises and simple answer to complex disease will be sooner.
REFERENCES
- B. V. Zlokovic, R. Deane, J. Sallstrom N. Chow and J. M. Miano, “Neurovascular Pathways and Alzheimer Amyloid Beta-Peptide,” Brain Pathology, Vol. 15, No. 1, 2005, pp. 78-83. doi:10.1111/j.1750-3639.2005.tb00103.x
- J. L. Cummings, H. V. Vinters, G. M. Cole and S. Khachaturian, “Alzheimer’s Disease: Etiologies, Pathophysiology, Cognitive Reserve, and Treatment Opportunities,” Neurology, Vol. 5, No. 1, 1998, pp. S2-S17.
- Y. H. Suh, F. Cacabelos, R. Fernández-Novoa and L. Corzo, “Phenotypic Profiles and Functional Genomics in Dementia with a Vascular Component,” Neurology Research, Vol. 26, No. 5, 2004, pp. 459-480. doi:10.1179/016164104225017677
- C. L. Lendon, F. Ashall and A. M. Goate, “Exploring the Etiology of Alzheimer Disease Using Molecular Genetics,” JAMA, Vol. 277, No. 10, 1997, pp. 825-831. doi:10.1001/jama.1997.03540340059034
- S. Henley, G. P. Bates and S. Tabrizi, “Biomarkers for Neurodegenerative Diseases,” Current Opinion in Neurology, Vol. 18, No. 6, 2005, pp. 698-705. doi:10.1097/01.wco.0000186842.51129.cb
- K. Furiea and S. Gisele, “Biomarkers in Neurology,” Clinical Trials in the Neurosciences, Vol. 25, 2009, pp. 55-61. doi:10.1159/000209475
- A. Bowirrat, M. Yassin, M. Abir, B. Bisharat and Z. Armaly, “Traditional and Modern Medicine Harmonizing the Two Approaches in the Treatment of Neurodegeneration (Alzheimer’s Disease—AD),” In: S. Marcelo and R. de Medeiros, Eds., Complementary Therapies for the Contemporary Healthcare, InTech Prepress, Novi Sad, 2012, pp. 181-212.
- N. G. Frangogiannis, “Biomarkers: Hopes and Challenges in the Path from Discovery to Clinical Practice,” Translational Research, Vol. 159, No. 4, 2012, pp. 197-204. doi:10.1016/j.trsl.2012.01.023
- M. Shaw, M. Korecka, C. M. Clark, V. M. Y. Lee and J. Q. Trojanowski, “Biomarkers of Neurodegeneration for Diagnosis and Monitoring Therapeutics,” Nature Reviews Drug Discovery, Vol. 6, No. 4, 2007, pp. 295-303. doi:10.1038/nrd2176
- G. Downing, “Biomarkers Definitions Working Group. Biomarkers and Surrogate Endpoints,” Clinical Pharmacology & Therapeutics, Vol. 69, 2001, pp. 89-95.
- J. Lee, V. Devanarayan, U. Barrett, R. Weiner, J. Allinson, S. Fountain, S. Keller, I. Weinryb, M. Green and L. Duan, “Fit-for-Purpose Method Development and Validation for Successful Biomarker Measurement,” Pharmaceutical Research, Vol. 23, No. 2, 2006, pp. 312-328. doi:10.1007/s11095-005-9045-3
- R. Frank and R. Hargreaves, “Clinical Biomarkers in Drug Discovery and Development,” Nature Reviews Drug Discovery, Vol. 2, No. 7, 2003, pp. 566-580. doi:10.1038/nrd1130
- H. Harald, L. Simone and Z. S. Khachaturian, “Development of Biomarkers to Chart All Alzheimer’s Disease Stages: The Royal Road to Cutting the Therapeutic Gordian Knot,” Alzheimer’s & Dementia, Vol. 8, No. 4, 2012, pp. 312-336. doi:10.1016/j.jalz.2012.05.2116
- S. M. Dhanasekaran, “Delineation of Prognostic Biomarkers in Prostate Cancer,” Nature, Vol. 412, No. 6849, 2001, pp. 822-826. doi:10.1038/35090585
- P. D. Wagner, P. Maruvada and S. Srivastava, “Molecular Diagnostics: A New Frontier in Cancer Prevention,” Expert Review of Molecular Diagnostics, Vol. 4, No. 4, 2004, pp. 503-511. doi:10.1586/14737159.4.4.503
- P. Gerde, B. A. Muggenburg, T. Stephens, J. L. Lewis, K. H. Pyon and A. R. Dahl, “A Relevant Dose of 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone Is Extensively Metabolized and Rapidly Absorbed in the Canine Tracheal Mucosa,” Cancer Research, Vol. 58, No. 7, 1998, pp. 1417-1422.
- V. Prashanthi, “Effect of Apolipoprotein E on Biomarkers of Amyloid Load and Neuronal Pathology in Alzheimer Disease,” Annals of Neurology, Vol. 67, No. 3, 2010, pp. 308-316.
- J. A. Johnston and M. Hill, “113 The Risk and Gamble We Take with Our Patients on Dopamine Agonists,” Journal of Neurology, Neurosurgery & Psychiatry, Vol. 83, No. 3, 2012, pp. e1-e1. doi:10.1136/jnnp-2011-301993.155
- C. H. Golias, “Adhesion Molecules in Cancer Invasion and Metastasis,” Hippokratia, Vol. 9, No. 1, 2005, pp. 106-114.
- H. A. Kader, V. T. Tchernev, E. Satyaraj, S. Lejnine, G. Kotler, S. F. Kingsmore and D. Patel, “Protein Microarray Analysis of Disease Activity in Pediatric Inflammatory Bowel Disease Demonstrates Elevated Serum PLGF, IL-7, TGF-β1, and IL-12p40 Levels in Crohn’s Disease and Ulcerative Colitis Patients in Remission versus Active Disease,” The American Journal of Gastroenterology, Vol. 100, No. 2, 2005, pp. 414-423. doi:10.1111/j.1572-0241.2005.40819.x
- O. Carrette, I. Demalte, A. Scherl, O. Yalkinoglu, O. Corthals, G. Burkhard and J. C. Sanchez, “A Panel of Cerebrospinal Fluid Potential Biomarkers for the Diagnosis of Alzheimer’s Disease,” Proteomics, Vol. 3, No. 8, 2003, pp. 1486-1494. doi:10.1002/pmic.200300470
- B. Ibach, H. Binder, M. Dragon, S. Poljansky, E. Haen, E. Schmitz and G. Hajak, “Cerebrospinal Fluid tau and β- Amyloid in Alzheimer Patients, Disease Controls and an Age-Matched Random Sample,” Neurobiology of Aging, Vol. 27, No. 9, 2006, pp. 1202-1211. doi:10.1016/j.neurobiolaging.2005.06.005
- M. Davis, S. Hanson and B. Altevogt, “Neuroscience Biomarkers and Biosignatures: Converging Technologies, Emerging Partnerships: Workshop Summary,” National Academies Press, Washington DC, 2008.
- N. R. Graff-Radford, J. E. Crook, J. Lucas, B. F. Boeve, D. S. Knopman, R. J. Ivnik and S. G. Younkin, “Association of Low Plasma Abeta42/Abeta40 Ratios with Increased Imminent Risk for Mild Cognitive Impairment and Alzheimer Disease,” Archives of Neurology, Vol. 64, No, 3, 2007, p. 354. doi:10.1001/archneur.64.3.354
- D. Butler and B. A. Bahr, “Oxidative Stress and Lysosomes: CNS-Related Consequences and Implications for Lysosomal Enhancement Strategies and Induction of Autophagy,” Antioxidants & Redox Signaling, Vol. 8, No. 1-2, 2006, pp. 185-196.
- K. Blennow, E. Vanmechelen and H. Hampel, “CSF Total tau, Ab42 and Phosphorylated tau Protein as Biomarkers for Alzheimer’s Disease” Molecular Neurobiology, Vol. 24, No. 1-3, 2001, pp. 87-97. doi:10.1385/MN:24:1-3:087
- A. M. Fagan, M. A. Mintun, R. H. Mach, S. Y. Lee, C. S. Dence, A. R. Shah, G. N. LaRossa, M. L. Spinner, W. E. Klunk, C. A. Mathis, S. T. DeKosky, J. C. Morris and D. M. Holtzman, “Inverse Relation between in Vivo Amyloid Imaging Load and Cerebrospinal Fluid Abeta42 in Humans,” Annals of Neurology, Vol. 59, No. 3, 2006, pp. 512-519. doi:10.1002/ana.20730
- A. M. Fagan, C. M. Roe, C. Xiong, M. A. Mintun, J. C. Morris and D. M. Holtzman, “Cerebrospinal Fluid tau/ beta-Amyloid(42) Ratio as a Prediction of Cognitive Decline in Nondemented Older Adults,” Archives of Neurology, Vol. 64, No. 3, 2007, pp. 343-349. doi:10.1001/archneur.64.3.noc60123
- M. Sjögren, P. Davidsson and A. Aallin, “Decreased CSF β-Amyloid42 in Alzheimer’s Disease and Amyotrophic Lateral Sclerosis May Reflect Mismetabolism of β- Amyloid Induced by separate mechanisms,” Dementia and Geriatric Cognitive Disorders, Vol. 13, 2002, pp. 112-118. doi:10.1159/000048642
- D. Strozyk, K. Blennow, L. R. White and L. J. Launer, “CSF Aβ42 Levels Correlate with Amyloid-Neuropathology in a Population-Based Autopsy Study,” Neurology, Vol. 60, No. 4, 2003, pp. 652-656. doi:10.1212/01.WNL.0000046581.81650.D0
- M. Morris, S. Maeda, K. Vossel and L. Mucke, “The Many Faces of Tau,” Neuron, Vol. 70, No. 3, 2011, pp. 410-426. doi:10.1016/j.neuron.2011.04.009
- L. Buée, T. Bussiere, V. Buee-Scherrer, A. Delacourte and P. R. Hof, “Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders,” Brain Research Reviews, Vol. 33, No. 1, 2000, pp. 95-130. doi:10.1016/S0165-0173(00)00019-9
- C. Feijoo, D. G. Campbell, R. Jakes, M. Goedert and A. Cuenda, “Evidence That Phosphorylation of the Microtubule-Associated Protein Tau by SAPK4/p38δ at Thr50 Promotes Microtubule Assembly,” Journal of Cell Science, Vol. 118, No. 2, 2005, pp. 397-408. doi:10.1242/jcs.01655
- K. Boekhoorn, D. Terwel, B. Biemans, P. Borghgraef, O. Wiegert, G. J. Ramakers and P. L. Lucassen, “Improved Long-Term Potentiation and Memory in Young tauP301L Transgenic Mice before Onset of Hyperphosphorylation and Tauopathy,” The Journal of Neuroscience, Vol. 26 No. 13, 006, pp. 3514-3523.
- M. Vandermeeren, M. Mercken and E. Vanmechelen, “Detection of τ Proteins in Normal and Alzheimer’s Disease Cerebrospinal Fluid with a Sensitive Sandwich Enzyme-Linked Immunosorbent Assay,” Journal of Neurochemistry, Vol. 61, 1993, pp. 1828-1834. doi:10.1111/j.1471-4159.1993.tb09823.x
- O. Hansson, H. Zetterberg, P. Buchhave, E. Londos, K. Blennow and L. Minthon, “Association between CSF Biomarkers and Incipient Alzheimer’s Disease in Patients with Mild Cognitive Impairment: A Follow-Up Study,” The Lancet Neurology, Vol. 5, No. 3, 2006, pp. 228-234. doi:10.1016/S1474-4422(06)70355-6
- A. Lanari and L. Parnetti, “Cerebrospinal Fluid Biomarkers and Prediction of Conversion in Patients with Mild Cognitive Impairment: 4-Year Follow-Up in a Routine Clinical Setting,” The Scientific World Journal, Vol. 9, 2009, pp. 961-966. doi:10.1100/tsw.2009.106
- N. Mattsson, H. Zetterberg, O. Hansson, N. Andreasen, L. Parnetti, M. Jonsson and K. Blennow, “CSF Biomarkers and Incipient Alzheimer Disease in Patients with Mild Cognitive Impairment,” The Journal of the American Medical Association, Vol. 302, No. 4, 2009, pp. 385-393. doi:10.1001/jama.2009.1064
- G. Zanusso, M. Fiorini, P. G. Righetti and S. Monaco, “Specific and Surrogate Cerebrospinal Fluid Markers in Creutzfeldt—Jakob Disease,” Advances in Neurobiology, Vol. 2, 2011, pp. 455-467.
- P. Formichi, L. Parnetti, E. Radi, G. Cevenini, M. T. Dotti and A. Federico, “CSF Levels of β-Amyloid 1-42, Tau and Phosphorylated Tau Protein in CADASIL,” European Journal of Neurology, Vol. 15, No. 11, 2008, pp. 1252-1255. doi:10.1111/j.1468-1331.2008.02277.x
- P. M. Stanford, G. M. Halliday, W. S. Brooks, J. B. Kwok, C. E. Storey, H. Creasey and P. R. Schofield, “Progressive Supranuclear Palsy Pathology Caused by a Novel Silent Mutation in Exon 10 of the Tau Gene Expansion of the Disease Phenotype Caused by Tau Gene Mutations,” Brain, Vol. 123, No. 5, 2011, pp. 880-893. doi:10.1093/brain/123.5.880
- D. W. Cleveland, S. Y. Hwo and M. W. Kirschner, “Physical and Chemical Properties of Purified Tau Factor and the Role of Tau in Microtubule Assembly,” Journal of Molecular Biology, Vol. 116, No. 2, pp. 1977, pp. 227- 247.
- H. Hampel, K. Blennow, L. M. Shaw, Y. C. Hoessler, H. Zetterberg and J. Q. Trojanowski, “Total and Phosphorylated Tau Protein as Biological Markers of Alzheimer’s Disease,” Experimental Gerontology, Vol. 45, No. 1, 2009, pp. 30-40. doi:10.1016/j.exger.2009.10.010
- K. J. Reinikainen, H. Soininen and P. J. Riekkinen, “Neurotransmitter Changes in Alzheimer’s Disease: Implications to Diagnostics and Therapy,” Journal of Neuroscience Research, Vol. 27, No. 4, 1990, pp. 576-586. doi:10.1002/jnr.490270419
- S. T. DeKosky and S. W. Scheff, “Synapse Loss in Frontal Cortex Biopsies in Alzheimer’s Disease: Correlation with Cognitive Severity,” Annals of Neurology, Vol. 27, No. 5, 1990, pp. 457-464. doi:10.1002/ana.410270502
- M. G. Giovannini, A. Rakovska, R. S. Benton, M. Pazzagli, L. Bianchi and G. Pepeu, “Effects of Novelty and Habituation on Acetylcholine, GABA, and Glutamate Release from the Frontal Cortex and Hippocampus of Freely Moving Rats,” Neuroscience, Vol. 106, No. 1, 2001, pp. 43-53. doi:10.1016/S0306-4522(01)00266-4
- A. Schousboe, “Transport and Metabolism of Glutamate and GABA in Neurons are Glial Cells,” International Review of Neurobiology, Vol. 22, No. 1, 1981, pp. 1-45.
- L. Hertz and H. R. Zielke, “Astrocytic Control of Glutamatergic Activity: Astrocytes as Stars of the Show,” Trends in Neurosciences, Vol. 27, No. 12, 2004, pp. 735- 743. doi:10.1016/j.tins.2004.10.008
- J. T. Greenamyre, “The Role of Glutamate in Neurotransmission and in Neurologic Disease,” Archives of neurology, Vol. 43, No. 10, 1986, pp. 1058-1063. doi:10.1001/archneur.1986.00520100062016
- P. G. Haydon, “GLIA: Listening and Talking to the Synapse,” Nature Reviews Neuroscience, Vol. 2, No. 3, 2001, pp. 185-193.
- P. T. Francis, “The Interplay of Neurotransmitters in Alzheimer’s Disease,” CNS Spectrums, Vol. 10, Suppl. 18, 2005, pp. 6-20.
- M. R. Farlow, “NMDA Receptor Antagonists,” Geriatrics, Vol. 59, No. 6, 2004, pp. 22-27.
- J. D. Rothstein, L. J. Martin and R. W. Kuncl, “Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis,” New England Journal of Medicine, Vol. 326, No. 22, 1992, pp. 1464-1468. doi:10.1056/NEJM199205283262204
- J. Masson, C. Sagne, M. Hamon and S. El Mestikawy, “Neurotransmitter Transporters in the Central Nervous System,” Pharmacological Reviews, Vol. 51, No. 3, 1999, pp. 439-464.
- J. P. Hornung, “The Human Raphe Nuclei and the Serotonergic System,” Journal of Chemical Neuroanatomy, Vol. 26, No. 4, 2003, pp. 331-343. doi:10.1016/j.jchemneu.2003.10.002
- R. Mössner, A. Schmitt, Y. Syagailo, M. Gerlach, P. Riederer and K. P. Lesch, “The Serotonin Transporter in Alzheimer’s and Parkinson’s Disease,” Advances in Research on Neurodegeneration, Vol. 60, 2000, pp 345-350.
- W. J. Geldenhuys and C. J. Van der Schyf, “Serotonin 5-HT6 Receptor Antagonists for the Treatment of Alzheimer’s Disease,” Current Topics in Medicinal Chemistry, Vol. 8, No. 12, 2008, pp. 1035-1048. doi:10.2174/156802608785161420
- G. Downing (Biomarkers Definitions Working Group), “Biomarkers and Surrogate Endpoints,” Clinical Pharmacology and Therapeutics, Vol. 69, No. 3, 2001, pp. 89- 95.
- J. Lee, V. Devanarayan, U. Barrett, R. Weiner, J. Allinson, S. Fountain, S. Keller, I. Weinryb, M. Green and L. Duan, “Fit-for-Purpose Method Development and Validation for Successful Biomarker Measurement,” Pharmaceutical Research, Vol. 23, No. 2, 2006, pp. 312-328. doi:10.1007/s11095-005-9045-3
- [61] N. S. M. Schoonenboom, F. E. Reesink, N. A. Verwey, M. I. Kester, C. E. Teunissen, P. M. van de Ven, Y. A. L. Pijnenburg, M. A. Blankenstein, A. J. Rozemuller, P. Scheltens and W. M. van de Flier, “Cerebrospinal Fluid Markers for Differential Dementia Diagnosis in a Large Memory Clinic Cohort,” Neurology, Vol. 78, No. 1, 2012, pp. 47-54. doi:10.1212/WNL.0b013e31823ed0f0
- [62] C. Humpel, “Identifying and Validating Biomarkers for Alzheimer’s Disease,” Trends in Biotechnology, Vol. 29, No. 1, 2011, pp. 26-32. doi:10.1016/j.tibtech.2010.09.007
- [63] K. Buerger, E. M. Pirttila, R. Zinkowski, I. Alafuzoff, S. J. Teipel, J. DeBernardis, D. Kerkman, C. McCulloch, H. Soininen and H. Hampel, “CSF Phosphorylated Tau Protein Correlates with Neocortical Neurofibrillary Pathology in Alzheimer’s Disease,” Brain, Vol. 129, No. 11, 2006, pp. 3035-3041. doi:10.1093/brain/awl269
- [64] S. Engelborghs, K. Sleegers, P. Cras, N. Brouwers, S. Serneels, E. De Leenheir, J. J. Martin, E. Vanmechelen, C. Van Broeckhoven and P. P. De Deyn, “No Association of CSF Biomarkers with APOE e4, Plaque and Tangle Burden in Definite Alzheimer’s Disease,” Brain, Vol. 130, No. 9, 2007, pp. 2320-2326. doi:10.1093/brain/awm136
- [65] K. Buerger, I. Alafuzoff, M. Ewers, T. Pirttila, R. Zinkowski and H. Hampel, “No Correlation between CSF Tau Protein Phosphorylated at Threonine 181 with Neocortical Neurofibrillary Pathology in Alzheimer’s Disease,” Brain, Vol. 130, No. 10, 2007, p. e82. doi:10.1093/brain/awm140
- [66] M. C. Irizarry, “Biomarkers of Alzheimer Disease in Plasma,” NeuroRX, Vol. 1, No. 2, 2004, pp. 226-234. doi:10.1602/neurorx.1.2.226
- [67] N. Rösier, I. Wichart and K. A. Jellinger, “Current Clinical Neurochemical Diagnosis of Alzheimer’s Disease,” Laboratoriums Medizin, Vol. 26, No. 3-4, 2002, pp. 139- 148. doi:10.1046/j.1439-0477.2002.02031.x
- [68] R. Mayeux, L. S. Honig and M. X. Tang, “Plasma A[beta]40 and A[beta]42 and Alzheimer’s Disease: Relation to Age, Mortality, and Risk,” Neurology, Vol. 61, No. 9, 2003, pp. 1185-1190. doi:10.1212/01.WNL.0000091890.32140.8F
- [69] P. M. Mehta, T. Pirttila and B. A. Patrick, “Amyloid Beta Protein 1-40 and 1-42 Levels in Matched Cerebrospinal Fluid and Plasma from Patients with Alzheimer Disease,” Neuroscience Letters, Vol. 304, No. 1-2, 2001, pp. 102- 106. doi:10.1016/S0304-3940(01)01754-2
- [70] J. D. Doecke, S. M. Laws, N. G. Faux, W. Wilson, S. C. Burnham, C. P. Lam, A. Mondal, J. Bedo, et al., “BloodBased Protein Biomarkers for Diagnosis of Alzheimer Disease,” Archives of Neurology, Vol. 69, No. 10, 2012, pp. 1318-1325. doi:10.1001/archneurol.2012.1282
- [71] H. D. Soares, W. Z. Potter, E. Pickering, M. Kuhn, F. W. Immermann, D. M. Shera, M. Ferm, R. A. Dean, A. J. Simon, F. Swenson, J. A. Siuciak, J. Kaplow, M. Thambisetty, et al., “Biomarkers Consortium Alzheimer’s Disease Plasma Proteomics Project. Plasma Biomarkers Associated with the Apolipoprotein E Genotype and Alzheimer Disease,” Archives of Neurology, Vol. 69, No. 10, 2012, pp. 1310-1317.
- [72] J. B. Toledo, X. Da, P. Bhatt, D. A. Wolk, S. E. Arnold, L. M. Shaw, J. Q. Trojanowski and C. Davatzikos, “Relationship between Plasma Analytes and SPARE-AD Defined Brain Atrophy Patterns in ADNI,” PLoS One, Vol. 8, No. 2, 2013, Article ID: e55531. doi:10.1371/journal.pone.0055531
- [73] R. Mayeux, A. M. Saunders, S. Shea, S. Mirra, D. Evans, A. D. Roses, B. T. Hyman, B. Crain, M. X. Tang and C. H. Phelps, “Utility of the Apolipoprotein E Genotype in the Diagnosis of Alzheimer’s Disease,” New England Journal of Medicine, Vol. 338, 1998, pp. 506-511. doi:10.1056/NEJM199802193380804
- [74] R. S Britschgi, C. Herbert, Y. Takeda-Uchimura, A. Boxer, K. Blennow, L. F. Friedman, D. R. Galasko, M. Jutel, A. Karydas, A. Kaye and J. Leszek, “Classification and Prediction of Clinical Alzheimer’s Diagnosis Based on Plasma Signaling Proteins,” Nature Medicine, Vol. 13, 2007, pp. 1359-1362.
- [75] P. Sebastiani, N. Solovieff, A. T. DeWan, K. M. Walsh, A. P. Stephen, H. Efthymia, M. S. Andersen, D. A. Dworkis, J. B. Wilk, R. H. Myers and M. H. Steinberg, “Perls Genetic Signatures of Exceptional Longevity in Humans,” PloS One, Vol. 7, No. 1, 2012, p. e29848. doi:10.1371/journal.pone.0029848
- [76] S. L. Andersen, P. Sebastiani, L. Feldman, “Health Span Approximates Life Span Amongmany Supercentenarians: Compression of Morbidity at the Approximate Limit of Life Span,” The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, Vol. 67, No. 4, 2012, pp. 395-405.
- [77] A. B. Newman, N. W. Glynn, C. A. Taylor, P. Sebastiani, T. T. Perls, R. Mayeux, K. Christensen, J. M. Zmuda, S. Barral, J. H. Lee and E. M. Simonsick, “Health and Function of Participants in the Long Life Family Study: A Comparison with Other Cohorts,” Aging (Albany NY), Vol. 3, No. 1, 2011, pp. 63-76.
- [78] T. Perls and N. Barzilai, “100 Semi-Supercentenarians and Older as a Proposed Sample Set for the Archon Genomics X PRIZE Validation Protocol,” Nature Precedings, 2011, in press. doi:10.1038/npre.2011.5756.1
NOTES
*Corresponding author.