Case Reports in Clinical Medicine
Vol.03 No.12(2014), Article ID:52166,2 pages
10.4236/crcm.2014.312135
Successful Tacrolimus therapy extended by everolimus in retroperitoneal angiomyolipoma after lung transplantation: a case report
Tamás Erdélyi1, Anikó Bohács1, Krisztina Vincze1, Zsuzsanna Kováts1, Veronika Sárosi2, András Sinkovicz2, György Balázs3, Veronika Müller1
1Department of Pulmonology, Semmelweis University, Budapest, Hungary
21st Department of Internal Medicine, Medical School, University of Pécs, Pécs, Hungary
3Department of Radiology, The Heart and Vascular Center, Semmelweis University, Budapest, Hungary
Email: muller.veronika@med.semmelweis-univ.hu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 21 October 2014; revised 20 November 2014; accepted 8 December 2014
ABSTRACT
Lymphangioleiomyomatosis (LAM) is a rare disease that affects women, especially in child-bear- ing age. Clinical manifestations include angiomyolipoma, pneumothorax, chylothorax, cystic chan- ges of lungs and progressive pulmonary failure. In this article, we report a case of lung transplantation (LuTX) for end stage pulmonary LAM and the treatment of angiomyolipoma showing growth after LuTX resulting in complete remission with combination therapy of everolimus and tacrolimus.
Keywords:
Lymphangioleiomyomatosis, Everolimus, Angiomyolipoma, Lung transplant

1. Introduction
Lymphangioleiomyiomatosis (LAM) is a multisystem disease occurring in lung, abdomen and axial lymphatics [1] . This disease is rare―its prevalence is 1 in 1,000,000 in the whole population―and it usually affects young women. LAM has two types: sporadic LAM and cases associated with the genetic disease tuberous sclerosis [2] . Tuberous sclerosis is caused by the mutations of genes TSC1 and TSC2 which regulate the proteins hamartin and tuberin. These proteins regulate their signal by means of mammalian target of rapamycin (mTOR) which controls cell size and growth [3] . LAM has many similarities to cancer including inappropriate cell growth and survival, infiltration of surrounding tissues, metastasis formation via blood and lymphatic circulation. LAM can be considered as an elegant, monogenic model of neoplasia which be categorized as benign or malignant [4] . Pulmonary manifestations of LAM include pneumothorax―at least two thirds of all patients―chylothorax and cystic changes of the lung which results in progressive decline in pulmonary function. In the advanced phases of the disease the last therapeutic option is lung transplantation (LuTX) [5] . Angiomyolipomas belong to the extrapulmonary manifestations of LAM. In most cases, angiomyolipomas involve the axial lymphatics in the upper retroperitoneum (RP).
2. Case report
A 42-year-old woman developed progressive exercise induced dyspnea in 2008. Spirometry confirmed partially reversible obstructive ventilatory disorder and initial treatment with inhaled corticosteroid plus long acting β2 receptor agonist was introduced. Despite treatment dyspnea progressed and lung function declined. In 2010 chest CT confirmed cystic pulmonary changes with fibrosis and emphysema, abdominal and inguinal lymph node enlargement. Subsequently surgical lung biopsy was performed and histology confirmed changes typical for LAM. Deteriorating progression of pulmonary function and recurring pneumothorax led to intensive care unit treatment in March 2011 requiring mechanical ventilation. Due to end stage lung disease she was presented at the LuTX committee and was listed for LuTX (lung functional values before transplantation: FVC 2.30 L (68% predicted), FEV1: 0.93 L (32% predicted), FEV1/FVC: 40%).
Bilateral LuTX was performed in Vienna in January 2012. Postoperatively she received triple immunosuppressive therapy including tacrolimus, mycophenolate mofetil and prednisolon. After two weeks surveillance bronchoscopy confirmed A2B1-2 rejection, following steroid shot treatment control transbronchial lung biopsy was negative for rejection (A0B0). Lung function has been gradually improving. The six-month chest-abdo- minal CT scan showed an enlargement in the angiomyolipoma located in the upper RP (Figure 1(a)) compared to the preoperative state. In October 2012, 9 months after TX everolimus (plasma level: 2.31 ng/ml) was added to tacrolimus (plasma level: 7.7 ng/ml) therapy, while combination with mycophenolate mofetil and prednisolon maintained. Six month after the introduction of combination therapy with mTOR inhibitor the angiomyolipoma was not detectable on the one year chest-abdominal CT (Figure 1(b)). The patient is since on everolimus, tacrolimus, mycophenolate mofetil and prednisolon therapy with the same plasma level as the combined therapy started, showing excellent graft function (lung functional values October 2014: FVC 3.63 L (110% predicted), FEV1: 2.59 L (91% predicted), FEV1/FVC: 71%), and no signs of abdominal disease on the abdominal CT scan performed 2 years after LuTX.
3. Discussion
LAM is a rare cause of respiratory failure requiring LuTX in young women. Angiomyolipoma is commonly observed and specific treatment of the disease is still unavailable. Angiomyolipomas with a diameter < 4 cm are at
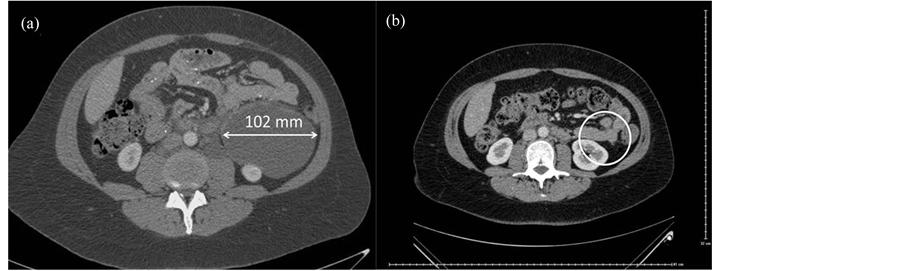
Figure 1. (a) Abdominal angiomyolipoma in LAM patient 6 months after LuTX; (b) Complete remission of angiomyolipoma in LAM patient after 6 month of treatment with everolimus- tacrolimus combination therapy.
a low risk of progression and ultrasound follow-ups are sufficient every 1 or 2 years. In our case the angiomyolipoma was asymptomatic, however due to its extent (102 mm) surgical removal might be suggested due the possibility of life-threatening haemorrhage [2] . This benign tumor could not be removed surgically as LAM is a multisystem disease and intervention would not have stopped the development of angiomyolipomas at subsequent sites. Although the tumors have oestrogen and progesterone receptors there is no firm evidence of efficacy of hormone treatment in this disease [2] . A comprehensive study examined the effects of the mTOR inhibitor sirolimus on angiomyolipomas. A 12 month treatment with sirolimus led to significant reduction (53.2% ± 26.6%, p < 0.001) within a group of 25 patients in a phase II study [6] . The efficacy of sirolimus is the result of the known pathomechanism of LAM: tuber in and hamartin proteins are responsible for inhibiting mTOR complex which regulates cell growth. Without this inhibiting signal cell proliferation pathway is uncontrolled [7] . Based on these results and experiences with everolimus in lung transplantation we decided to start treatment with a combination of tacrolimus and everolimus. CT performed 6 months after the combination therapy resulted in complete remission of RP angiomyolipoma. In a previous case report the treatment of angiomyolipoma in LAM patient receiving LuTX included combination therapy of everolimus with cyclosporine, mycophenolatemofetil and steroids. This treatment resulted in a reduction of angiomyolipoma by ~30% after 6 months of treatment [7] . The difference between the effects of cyclosporin and tacrolimus might have a significant impact of everolimus on angiomyolipoma cells.
4. Conclusion
In our LAM patient requiring lung transplantation, the combined use of everolimus plus tacrolimus resulted in complete remission of the angiomyolipoma 6 months after treatment without influencing perfect graft function. According to our results using everolimus and tacrolimus combination in LAM, it is considered to be an adequate and safe way of significantly reducing or even reaching full remission of angiomyolipomas after LuTX in these patients.
References
- Glasgow, C.G., EL-Chemaly, S. and Moss, J. (2012) Lymphatics in Lymphangioleiomyomatosis and Idiopathic Pulmonary Fibrosis. European Respiratory Review: An Official Journal of the European Respiratory Society, 21, 196-206. http://dx.doi.org/10.1183/09059180.00009311
- Johnson, S.R. (2006) Lymphangioleiomyomatosis. The European Respiratory Journal, 27, 1056-1065.
- Casanova, A. and Ancochea, J. (2011) Lymphangioleiomyomatosis: New Therapeutic Approaches. Archivos de Bronconeumologia, 47, 579-580. http://dx.doi.org/10.1016/j.arbres.2011.06.008
- Henske, E.P. and McCormack, F.X. (2012) Lymphangioleiomyomatosis―A Wolfinsheep’s Clothing. The Journal of Clinical Investigation, 122, 3807-3816. http://dx.doi.org/10.1172/JCI58709
- Kpodonu, J., Massad, M.G., Chaer, R.A., et al. (2005) The US Experience with Lung Transplantation for Pulmonary Lymphangioleyomiomatosis. The Journal of Heart and Lung Transplantation: The Official Publication of the International Society for Heart Transplantation, 24, 1247-1253. http://dx.doi.org/10.1016/j.healun.2004.09.013
- Bissler, J.J., McCormack, F.X., Young, L.R., et al. (2008) Sirolimus for Angiomyolipoma in Tuberous Sclerosis Complex or Lymphangioleiomyomatosis. The New England Journal of Medicine, 358, 140-151. http://dx.doi.org/10.1056/NEJMoa063564
- Bujalance-Cabrera, C., Vaquero-Barrios, J.M. and Redel-Montero, J. (2012) Reduction in Size of Renal Angiomyolipoma after Treatment with Everolimus in Lung Transplantation Due to Lymphangioleiomyomatosis. Archivos de Bronconeumologia, 48, 479-481. http://dx.doi.org/10.1016/j.arbres.2012.02.002