Food and Nutrition Sciences
Vol. 4 No. 9A1 (2013) , Article ID: 36092 , 10 pages DOI:10.4236/fns.2013.49A1002
Role of Free Fatty Acids in Physiological Conditions and Mitochondrial Dysfunction*
![]()
1National Center for Toxicological Research/FDA, Jefferson, USA; 2Autonomous University of San Luis Potosi, San Luis Potosi, Mexico.
Email: zbigniew.binienda@fda.hhs.gov
Copyright © 2013 Zbigniew K. Binienda et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 14th, 2013; revised June 14th, 2013; accepted June 21st, 2013
Keywords: Free Fatty Acids; Beta-Oxidation; Myristoylation; Palmitoylation; Endothelium
ABSTRACT
The role of free fatty acids (FFAs) as a source of energy and their functions in energy transport within the body are well established. Equally important is a role that FFAs play in oxidative stress following cell membrane depolarization. FFAs are physiologically active, not only as nutritional components, but also as molecules involved in cell signaling and stabilization of membranes via palmitoylation and myristoylation. Protein palmitoylation is involved in numerous cellular processes, including apoptosis, and neuronal transmission. Besides nuclear peroxisome proliferator-activated receptors that mediate the biological effects of FFAs, G protein-coupled receptors (GPCRs) that are activated by FFA, have been recently identified. Those multiple FFA receptors (FFARs), which function on the cell surface as activated FFAs, play significant roles in the regulation of energy metabolism and mediate a wide range of important metabolic processes. FFARs have been targeted in drug development for the treatment of type 2 diabetes and metabolic syndrome. FFAs upregulate transcription of uncoupling proteins, increasing their expression in brain, cardiac, and skeletal muscle that may be protective or cytotoxic, depending on the cellular energy state. Recently, FFA effects on the endothelial function and dysfunction are being recognized. FFAs play a key role in endothelium-dependent nitric oxide production. A disturbance of endothelial function, due to an imbalance in production and release of relaxing and constricting factors, has implications in the development of cardiovascular problems, such as hypertension, as well as neurotoxicity following loss of blood-brain barrier integrity. This review presents information on broad range of FFAs actions of prime importance for physiological processes. Understanding of FFA functions in the body is crucial for developing new therapeutic strategies against several metabolic disorders.
1. Introduction
In animals, many of the dietary lipids are hydrolysed to FFAs before being absorbed and utilized further for lipid synthesis. Lipids can be hydrolysed to FFAs in tissues by lipolytic enzymes, such as lipoprotein lipase, hormonesensitive lipase, and phospholipases A and C, before they are metabolised in many different pathways, including oxidation, desaturation, elongation and re-esterification [1].
FFAs play an important role as a source of energy for the body. Fat stored in adipose tissue is transported to other tissues in the form of FFAs hydrolyzed from triglycerides and combined with albumin [1]. These saturated fatty acids (SFA), such as the 18-carbon stearic acid, the 16-carbon palmitic acid, and the 14-carbon myristic acid are also important for various biological functions such as the production of hormones, cellular membrane signaling, and the stabilization processes in the body [2,3]. A biochemical process in which the body uses palmitic acid in stabilization processes by regulating G protein-coupled receptor (GPCR) signaling is called palmitoylation [4-6].
2. Physiological Role of FFAs as an Energy Substrate
FFAs Beta-Oxidation and the Carnitine Shuttle
Fatty acid beta-oxidation is a multistep metabolic pathway, by which fatty acids are utilized to produce energy. Protein transporters on the cell surface, including fatty acid translocase (FAT/CD36), tissue-specific fatty acid transport proteins (FATP), and plasma membrane bound fatty acid binding protein (FABPpm), play an important role in transporting FFAs to the cell. Inside the cell, the thioester, coenzyme A (CoA) is added to the fatty acid by fatty acyl-CoA synthase (FACS), forming long-chain acyl-CoA.
The mitochondrial membrane is impermeable to acylCoA that enters mitochondria via the “carnitine shuttle”. The first step of this biochemical process is mediated by carnitine palmitoyltransferase 1 (CPT1) that converts acyl-CoA into acylcarnitine. CPT1 binding of the long chain acyl-CoA to long chain acylcarnitine allows the fatty acid to be transported across the inner mitochondrial membrane via facilitated diffusion by carnitine translocase (CAT), which exchanges long chain acylcarnitines for carnitine. The long chain acylcarnitine is then converted back to long chain acyl-CoA by an inner mitochondrial membrane enzyme, carnitine palmitoyltransferase 2 (CPT2). The beta-oxidation pathway of fatty acid and long chain acyl-CoA yields acetyl-CoA that enters the mitochondrial tricarboxylic acid (TCA) cycle. Electron carriers, coenzymes NADH and FADH2, supplied by both fatty acid beta-oxidation and the TCA cycle, are used in the electron transport chain to produce energy stored in ATP molecules [1,7,8].
3. “Passive” Role of FFAs
3.1. Degradation of Cellular Membranes in Energy Insults
Although FFAs serve in the first instance as an important energy substrate, high concentrations of FFAs are associated with insulin resistance, fatty liver disease, atherosclerosis and myocardial dysfunction. Increased concentrations of FFAs are risk factors for the development of type 2 diabetes [9-11]. The increased metabolism of FFAs leads via an inhibition of insulin receptor substrate-1 (IRS1) and phosphoinositol 3-kinase to reduced the intracellular effects of insulin [9]. Moreover, FFAs lead directly to reduction in the uptake and phosphorylation of glucose and to a reduced glycogen synthesis. FFAs also impair the insulin signal transduction in the liver [9].
FFAs play a role in the development of fatty liver disease because 60% of the triglycerides stored in the liver originate from circulating FFAs [12]. FFAs are also independent risk factors for the development of arterial hypertension [13]. It seems likely that an alpha a1- adrenergic activation may partially be responsible for the link between FFAs and arterial hypertension [13]. In addition, FFAs raise the oxidative stress, lead to a reduced production of vasodilatory nitric oxide (NO) and reduce the insulin mediated vasodilation [13]. FFAs also stimulate the proliferation of smooth muscle cells of the vessel walls. This causes vascular hypertrophy and vessel-wall rigidity and is associated with the development of arterial hypertension [13]. A raised concentration of FFAs with subsequent increased beta-oxidation in the cardiomyocytes can be harmful for the heart in various ways. Comparatively, more oxygen is needed for producing energy through beta-oxidation than through glycolysis. This “oxygen wastage” through beta-oxidation has an extremely negative effect, particularly in diseases that are associated with increased energy requirements by the cardiomyocytes, such as heart failure or acute coronary syndromes [14,15]. In addition, FFAs contribute to the development of atherosclerosis and have a proatherosclerotic effect onendothelial cells, macrophages and smooth muscle cells of the vascular walls [16-18].
3.2. Oxidative Stress in Mitochondrial Dysfunction and FFAs
Reduction in cellular energy production can lead to a significant loss of mitochondrial membrane potential in all cells, including neurons. Energy depletion and cellular membrane depolarization are associated with mitochondrial dysfunction due to ischemia/hypoxia or exposure to toxicants lead to accumulation of intracellular Ca2+ and excitotoxicity as well as oxidative stress. Glutamate excitotoxicity contributes to oxidative stress through a disruption of reduced glutathione (GSH) synthesis. GSH acts as an endogenous scavenger of reactive oxygen species (ROS). But far more important for ROS generation is the effect of Ca2+ activation of phospholipases, liberation of FFA and their metabolism to prostaglandins or eicosanoids (Figure 1) [19-27].
Mitochondrial dysfunction due to irreversible inactivation of mitochondrial complex II by the mitochondrial inhibitor, 3-nitropropionic acid (3-NPA), results in a decreased ATP/ADP ratio and an increased lactate/pyruvate ratio [20]. The concentration of FFAs in all brain regions increases significantly as well [22]. An increase in FFAs is followed by production of ROS and activation of endogenous radical oxygen scavengers [28]. Increases in FFA levels may be correlated with uncoupling protein concentrations in heart and skeletal muscle tissue [27].
Other studies suggest that FFAs and oxidative stress lead to activation of several transcription factors including activator protein-1 (AP-1), comprised of the proteins c-Fos and c-Jun, and nuclear peroxisome proliferatoractivated receptor-alpha (PPARα) that have a bipotential role in mediating cell regeneration or death depending on conditions of cell plasticity and repair [29,30].
In vitro preliminary studies with SHSY-5Y neuroblastoma cells exposed to 10 and 30 nm ferric oxide (Fe2O3)
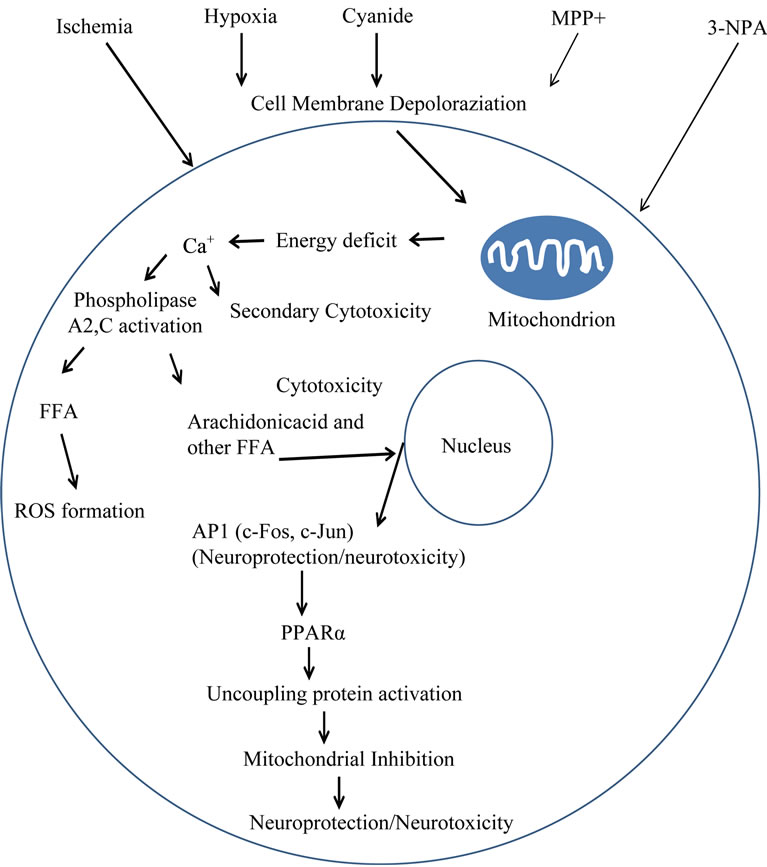
Figure 1. Schematic illustration of reactions following mitochondrial dysfunction and cellular energy deficit. AP1—activator protein-1; PPARα—peroxisome proliferator-activated receptor-alpha; ROS—reactive oxygen species.
nanoparticles (NPs) showed depletion of dopamine and conditions for oxidative stress [31]. Therefore, we examined the response of brain and liver FFAs in adult male Sprague-Dawley rats treated systemically with Fe2O3-NPs at 25, 50 and 100 mg/kg. Rats were sacrificed 72 h after injection. Concentrations of saturated FFAs (palmitic and stearic acids) in the liver and brain did not change following exposure to iron NPs. However, unsaturated brain FFAs (oleic and linoleic acids) decreased in a dose-dependent fashion in the caudate nucleus (p < 0.05). In the liver, the concentration of unsaturated FFAs increased significantly at 25 and 50 mg/kg (p < 0.05). A link between a long-term regulatory involvement of FFAs and early activation of the inducible transcription factors such as c-Fos and c-Jun proteins (AP1) in a differential response to metabolic stress in brain and liver was suggested [31].
4. “Active” (Regulatory) Role of FFAs
4.1. Biological Effects
Some FFAs are not only the essential dietary nutrients but also contribute to various physiological processes. Nuclear peroxisome proliferator-activated receptors (PPARs) act as sensors of FFAs [28]. These receptors maintain homeostasis under physiological and pathophysiological conditions by coordinating the expression of proteins that are involved in the uptake, synthesis, transport, storage, degradation and elimination of lipids. However, some of the biological effects of FFAs may be mediated by alternative mechanisms and may be mediated independently of PPARs. Recently, G protein-coupled receptors (GPCRs) activated by FFAs have been identified as FFAs receptors (FFARs) that function on the cell surface and play significant roles in nutritional regulation [32,33]. The FFARs not only act as nutrientsensing receptors, independent of PPARs, but also are known to mediate a wide range of important metabolic processes. These receptors have the ability to monitor FFA levels in the intestine, pancreas, macrophages, and adipose tissue, and are considered to be a major drug target for the treatment of type 2 diabetes due to their role in energy homeostasis [34,35].
4.2. Myristoylation and Palmitoylation
Multicellular organisms use chemical messengers to transmit signals between organelles and between cells. Relatively small hydrophobic molecules, such as FFAs, serve this signaling purpose. The two most common modifications, myristoylation and palmitoylation, differ with respect to the type and chemical nature of fatty acid attachment to the polypeptide receptor backbone.
Myristic acid, a 14-carbon saturated fatty acid, is a source of myristoyl groups utilized within the body to stabilize many different proteins, including proteins in the immune system [36]. Myristoyl group is commonly added cotransitionally to the amino terminus of glycine in receptor-associated kinases to confer the membrane localization of the enzyme. Myristoylation occurs in signal transduction via G protein when myristoyl group links the G protein to the inner surface of the plasma membrane allowing the G protein to interact with its receptor [37-40]. Myristoleic acid is biosynthesized from myristic acid by the enzyme delta-9-desaturase. This unsaturated fatty acid acylates an NH2 terminus of a protein related to signal transduction in photoreceptors in the retina [41]. Furthermore, the presence of myristoleic acid is of diagnostic value in patients with defects of longchain fatty acid oxidation [42], and it is known to be cytotoxic to tumor cells. Myristoleic acid induces apoptosis and necrosis in human LNCaP prostate cancer cells [43].
In contrast, palmitoylated proteins contain the 16- carbon saturated fatty acid palmitate, attached via a thioester linkage to one or more cysteine residues. Palmitoylation is a posttranslational reaction that appears to be mediated by a membrane-bound palmitoyl acyl transferase. Unlike myristoylation, which is generally a relatively stable lipid modification, palmitoylation is reversible through the thioester (S-acylation). Since linkage between the palmitate and protein is readily cleaved, cycles of palmitoylation and depalmitoylation occur in a regulated manner for many proteins [44].
Protein palmitoylation has been involved in numerous cellular processes, including signaling, apoptosis, and neuronal transmission [45]. Palmitoylation of proteins increases their hydrophobicity to promote protein-membrane association [46,47]. Modification of proteins through palmitoylation is involved in protein trafficking between organelles and in the segregation or clustering of proteins in membrane compartments [48-50]. Palmitoylation allows the control of protein-protein interaction [51-53], lipid raft targeting [54], and intracellular trafficking [55,56].
The palmitoylation process is involved in multiple diseases, such as degenerative Huntington’s disease, various cardiovascular and T-cell mediated immune disorders, and cancer [57].
5. Effects Induced by Fatty Acids in the Cardiovascular System
Cardiovascular disease (CVD) remains the leading cause of death worldwide, possibly as a result of population growth, increased longevity and exposure to risk factors associated with high blood pressure, obesity, hyperglycemia and hyperlipidemia [58]. The severity of cardiovascular problems has led to global concern and consequently to study of the relationship between CVD and dietary fat. Over the years, much research has been focused on the effects and imbalance between SFA, monounsaturated fatty acids, polyunsaturated fatty acids (PUFA), trans-fatty acids (TFA), and most recently, conjugated linoleic acid (CLA). It has become clear that the relationship between total fat intake and CVD is related more to the quality than to the quantity of fat consumed [59].
5.1. Endothelium
Endothelium, or intima, of the inner wall of blood vessels is a selectively permeable barrier between the blood and the vascular tissue, whose function is to mediate the integrity and metabolism of vascular wall (i.e., vascular tone and inflammatory response) through synthesis and release of vascular modulators [60]. Therefore, disturbance of endothelial function due to an imbalance in production and release of relaxing and constricting factors has implications for the development of CVD [61]. One of the most potent vasodilator agents released from the endothelium is NO, a free radical with an ultrashort half-life and highly versatile functions. NO is synthesized by endothelial nitric oxide synthase (eNOS), an endothelial Ca2+-dependent enzyme modulated by palmitoylation and myristoylation processes [62]. NO diffuses into smooth muscle cells (SMC) of the blood vessels and promotes vasodilation [63].
Some studies have reported a modulating effect of FFAs on endothelium-dependent NO production. Chronic exposure to a 2:1 (w/w) mixture of oleate/palmitate significantly reduces NO production induced by phospholipase C-activating receptor agonists, such as bradykinin and ATP-dependent vasodilators, in bovine aortic endothelial cells [64]. In rat aortic endothelial cells, palmitic acid (PA) has been reported to decrease NO bioavailability via the pAKT/PeNOS/NO pathway [65]. However, the steroidogenic acute regulatory protein inhibits PAinduced-inflammation and attenuates impairment of NO bioavilability via pAKT/PeNOS/NO pathway. It also regulates the lipid metabolism, decreasing intracellular lipid levels in endothelial cells. Those actions may offer protection against endothelial dysfunction.
Polyunsaturated fatty acids (PUFA), such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) omega-3 fatty acids, are recognized to have beneficial effects against atherosclerosis [66]. The incorporation of PUFA in the endothelial membrane phospholipids promotes synthesis of series 3 prostaglandins and series 5 leukotrienes, favoring vasodilation and anti-aggregation [67]. It has been recently shown that EPA has cardiovascular-protective effects via activation of protein kinase and up-regulation of the mitochondrial uncoupling protein-2 (UCP-2), leading to an increase in NO production [68]. The molecular mechanism underlying UCP-2 upregulation is not known. The nuclear PPAR-gamma (PPARγ) receptors could be involved, since an agonist of PPARγ promoted a significant increase in UCP-2 expression [68].
More cellular and molecular studies at the level of palmitoylation and myristoylation should be conducted as these reactions play a key role in the regulation of the eNOS activity. The identification of those signaling pathways induced by EPA in endothelial cells opens a new avenue for potential drug development in the treatment of atherosclerosis or endothelial dysfunction in human hyperlipidemia.
Endothelial dysfunction and associated inflammatory events are critical for the initiation of cardiovascular diseases such as atherosclerosis. The mechanisms by which selected fatty acids induce endothelial cell activation, oxidative stress, and inflammation are not fully understood.
5.2. Vascular Smooth Muscle and Heart
The Ca2+ channels and large-conductance calcium-activated potassium (BK) channels operate as a functional unit in the regulation of smooth muscle tone in the blood vessels [69]. A recent study has shown that DHA metabolites (16,17-EpDPE) produced in the cytochrome P450 (CYP450) pathway induce vasodilatation through activation of BK channels in vascular smooth muscle cells [70,71]. Like DHA, EPA is metabolized by CYP. However, an EPA metabolite, 17,18-epoxyeicosatetranoic acid, activates the vascular BK channels independent of intracellular Ca2+ concentration or local sarcoplasmic reticulum Ca2+ release [72].
As in the endothelium, CLA has protective effects on SMC by lowering the eicosanoid release from resting SMC; probably as a result of a reduced cellular pool of amino acids. CLA may compete with LA for the incorporation into the membrane phospholipids, interfering with the production of amino acids, and in consequence with the production of its derivatives [73,74]. In activated SMC, the reduction of cytokine-stimulated prostanoid release by cis-9, trans-11-CLA and trans-10, cis-12-CLA is mediated by the PPARγ-dependent inhibition of the NF-κB pathway, which lowers the expression of genes involved in prostanoid synthesis. This action could contribute to the anti-inflammatory and antiatherogenic effects induced by CLA [73,75].
In cardiomyocytes, eNOS is linked with the plasma membrane by the saturated fatty acids, myristic and palmitic acids, and subsequent formation of a heteromeric complex of eNOS with the caveolin-3 protein into the caveolae, which are small indentations in plasma membranes. The co-localization of caveolin-3 and eNOS may facilitate both eNOS activation by cell surface receptors and NO release at the cell surface for intercellular signaling [76]. Palmitic acid induces deregulation of cardiovascular homeostasis by a decrease in the caveolin-3 level, and consequently the translocation of eNOS from the plasma membrane to a perinuclear location in the cardiomyocytes, which is related to a decrease of the phosphorylation of eNOS at its Thr495 inhibitory site and decrease of NO production [77].
Apoptosis may play an important role in the pathogenesis of several cardiovascular diseases, such as ischemia, infarction and heart failure [78]. Autophagy has been recognized recently as an important modulator of apoptotic cell death [79]. Palmitic acid and TFA, such as vaccenic acid and elaidic acid induce apoptosis regulated by autophagy in cardiomyocytes and cardiomyoblasts, whereas EPA protects against palmitate-induced autophagy and apoptosis via the regulation of cardiolipin loss, translocation of Bcl-2-associated X protein (Bax) to the mitochondria, release of cytochrome c, and caspase-3 activation [79-82].
The mechanisms by which selected fatty acids induce endothelial cell activation, oxidative stress, and inflammation are not fully understood. Further studies will help to identify specific and selective endogenous and exogenous molecules capable of preventing the detrimental effects induced by SFA.
6. Conclusions
The “passive” role of FFAs as a source of energy, mainly in peripheral tissues but also in brain, has been well recognized. FFAs are established biomarkers of neuronal membrane degradation and have directly toxic effect in the pathogenesis of ischemic-hypoxic brain damage.
The “active” roles of unesterified fatty acids, such as the effects of polyunsaturated fatty acids on upand down-regulation of gene expression or on FFAs cell surface G coupled receptor myristoylation and palmitoylation, where they help to mediate and modify receptor signaling, are still poorly understood.
The same poor understanding pertains to the active role of FFAs in endothelial function, where they may modulate endothelium-dependent nitric oxide production or mediate inflammatory reactions in endothelial dysfunction.
Understanding mechanisms of FFA actions in the body is crucial for prevention and new drug development for multiple diseases, such as metabolic and degenerative disorders, cardiovascular and T-cell mediated immune disorders, and various forms of cancer.
REFERENCES
- W. F. Ganong, “Review of Medical Physiology,” Lange Medical Publications, Los Altos, 1985.
- C. Wilcox, J. S. Hu and E. N. Olson, “Acylation of Proteins with Myristic Acid Occurs Cotranslationally,” Science, Vol. 238, No. 4831, 1987, pp. 1275-1278. doi:10.1126/science.3685978
- I. Izawa, M. Nishizawa, Y. Hayashi and M. Inagaki, “Palmitoylation of ERBN Is Required for Its Plasma Membrane Localization,” Genes Cells, Vol. 13, No. 7, 2008, pp. 691-701. doi:10.1111/j.1365-2443.2008.01198.x
- Z. Xie, W. T. Ho and J. H. Exton, “Requirements and Effects of Palmitoylation of Rat PLD1,” Journal of Biological Chemistry, Vol. 276, No. 12, 2001, pp. 9383-9391. doi:10.1074/jbc.M009425200
- N. D. Holliday and H. M. Cox, “Control of Signaling Efficacy by Palmitoylation of the Rat Y1 Receptor,” British Journal of Pharmacology, Vol. 139, No. 3, 2003, pp. 501-512. doi:10.1038/sj.bjp.0705276
- K. Huang and A. D. El-Husseini, “Modulation of Neuronal Protein Trafficking and Function by Palmitoylation,” Current Opinion in Neurobiology, Vol. 15, No. 5, 2005, pp. 527-535. doi:10.1016/j.conb.2005.08.001
- Z. Binienda and A. Virmani, “The Mitochondriotropic Effects of L-Carnitine and Its Esters in the Central Nervous System,” Current Medical Chemistry—Central Nervous System Agents, Vol. 3, No. 4, 2003, 275-282. doi:10.2174/1568015033477659
- N. Fillmore, O. Alrob and G. D. Lopaschuk, “Animal Lipid Biochemistry. Fatty Acid β-Oxidation. Overview,” 2011. http://lipidlibrary.aocs.org/animbio/fa-oxid/index.htm
- H. Bays, L. Mandarino and R. A. DeFronzo, “Role of the Adipocyte, Free Fatty Acids, and Ectopic Fat in Pathogenesis of Type 2 Diabetes Mellitus: Peroxisomal Proliferator-Activated Receptor Agonists Provide a Rational Therapeutic Approach,” Journal of Clinical Endocrinology and Metabolism, Vol. 89, No. 2, 2004, pp. 463- 478. doi:10.1210/jc.2003-030723
- J. S. Pankow, B. B. Duncan, M. I. Schmidt, C. M. Ballantyne, D. J. Couper, R. C. Hoogeveen and S. H. Golden, “Fasting Plasma Free Fatty Acids and Risk of Type 2 Diabetes,” The Atherosclerosis Risk in Communities study. Diabetes Care, Vol. 27, No. 1, 2004, pp. 77-82. doi:10.2337/diacare.27.1.77
- A. M. Poynten, S. K. Gan, A. D. Kritekos, L. V. Campbell and D. J. Chisholm, “Circulating Fatty Acids, NonHigh Density Lipoprotein Cholesterol, and Insulin-Infused Fat Oxidation Acutely Influence Whole Body Insulin Sensitivity in Nondiabetic Men,” Journal of Clinical Endocrinology and Metabolism, Vol. 90, No. 2, 2005, pp. 1035-1040. doi:10.1210/jc.2004-0943
- K. M. Utzschneider and S. E. Kahn, “Review: The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease,” Journal of Clinical Endocrinology and Metabolism, Vol. 91, No. 12, 2006, pp. 4753-4761. doi:10.1210/jc.2006-0587
- P. A. Sarafidis and G. L. Bakris, “Non-Esterified Fatty Acids and Blood Pressure Elevation: A Mechanism for Hypertension in Subjects with Obesity/Insulin Resistance,” Journal of Human Hypertension, Vol. 21, 2007, pp. 12-19. doi:10.1038/sj.jhh.1002103
- W. C. Stanley, F. A. Recchia and G. D. Lopaschuk, “Myocardial Substrate Metabolism in the Normal and Failing Heart,” Physiological Reviews, Vol. 85, No. 3, 2005, pp. 1093-1129. doi:10.1152/physrev.00006.2004
- M. F. Oliver, “Sudden Cardiac Death: The Lost Fatty Acid Hypothesis,” QJM: An International Journal of Medicine, Vol. 99, No. 10, 2006, pp. 701-709. doi:10.1093/qjmed/hcl084
- J. F. Oram and K. E. Bornfeldt, “Direct Effects of LongChain Non-Esterified Fatty Acids on Vascular Cells and Their Relevance to Macrovascular Complications of Diabetes,” Frontiers in Bioscience: A Journal and Virtual Library, Vol. 9, 2004, pp. 1240-1253. doi:10.2741/1300
- B. Hufnagel, M. Dworak, M. Soufi, Z. Mester, Y. Zhu, J. R. Schaefer, S. Klumpp and J. Krieglstein, “Unsaturated Fatty Acids Isolated from Human Lipoproteins Activate Protein Phosphatase Type 2Cbeta and Induce Apoptosis in Endothelial Cells,” Atherosclerosis, Vol. 180, No. 2, 2005, pp. 245-254. doi:10.1016/j.atherosclerosis.2004.12.021
- E. E. Lloyd, J. W. Gaubatz, A. R. Burns and H. J. Pownall, “Sustained Elevations in NEFA Induce Cyclooxygenase-2 Activity and Potentiate THP-1 Macrophage Foam Cell Formation,” Atherosclerosis, Vol. 192, No. 1, 2007, pp. 49-55. doi:10.1016/j.atherosclerosis.2006.06.014
- E. M. Nemoto, R. W. Evans and P. M. Kochanek, “Free Fatty Acids Liberation in the Pathogenesis and Therapy of Ischemic Brain Damage,” In: N. G. Bazan, P. Braquet and M. D. Ginsburg, Eds., Neurochemical Correlates of Cerebral Ischemia, Pleanum Press, New York, 1992, pp. 183-218. doi:10.1007/978-1-4615-3312-2_10
- M. Erecińska and D. Nelson, “Effects of 3-Nitropropionic Acid on Synaptosomal Energy and Transmitter Metabolism: Relevance to Neurodegenerative Brain Diseases,” Journal of Neurochemistry, Vol. 63, No. 3, 1994, pp. 1033-1041. doi:10.1046/j.1471-4159.1994.63031033.x
- V. G. Desai, R. J. Feuers, R. W. Hart and S. F. Ali, “MPP+-Induced Neurotoxicity in Mouse Is Age-Dependent: Evidenced by the Selective Inhibition of Complexes of Electron Transport,” Brain Research, Vol. 715, No. 1-2, 1996, pp. 1-8. doi:10.1016/0006-8993(95)01255-9
- Z. Binienda and C. S. Kim, “Increase in Levels of Total Free Fatty Acids in Rat Brain Regions Following 3-Nitropropionic Acid Administration,” Neuroscience Letters, Vol. 230, No. 3, 1997, pp. 199-201. doi:10.1016/S0304-3940(97)00514-4
- T. Clausen, A. Zauner, J. E. Levasseur, A. C. Rice and R. Bullock, “Induced Mitochondrial Failure in the Feline Brain: Implications for Understanding Acute Post-Traumatic Metabolic Events,” Brain Research, Vol. 908, No. 1, 2001, pp. 35-48. doi:10.1016/S0006-8993(01)02566-5
- C. Fonck and M. Baudry, “Rapid Reduction of ATP Synthesis and Lack of Free Radical Formation by MPP+ in Rat Brain Synaptosomes and Mitochondria,” Brain Research, Vol. 975, No. 1-2, 2003, pp. 214-221. doi:10.1016/S0006-8993(03)02675-1
- Z. Binienda and A. Virmani, “The Mitochondriotropic Effects of L-Carnitine and Its Esters in the Central Nervous System,” Current Medical Chemistry—Central Nervous System Agents, Vol. 3, 2003, pp. 275-282.
- T. Kristián, “Metabolic Stages, Mitochondria and Calcium in Hypoxic/Ischemic Brain Damage,” Cell Calcium, Vol. 36, No. 3-4, 2004, pp. 221-233. doi:10.1016/j.ceca.2004.02.016
- H. Kageyama, M. Suga, M. Kashiba, J. Pka, T. Osaka, T. Kashiwa, T. Hirano, K. Nemoto, Y. Namba, D. Ricquier, J.-P. Giacobino and S. Inoue, “Increased Uncoupling Protein-2 and -3 Gene Expression in Skeletal Muscle of STZ-Induced Diabetic Rats,” FEBS Letters, Vol. 440, No. 3, 1998, pp. 450-453. doi:10.1016/S0014-5793(98)01506-3
- Z. Binienda, C. Simmons, S. Hussain, W. Slikker Jr. and S. F. Ali, “Effect of Acute Exposure to 3-Nitropropionic Acid on Activities of Endogenous Antioxidants in the Rat Brain,” Neuroscience Letters, Vol. 251, No. 3, 1998, pp. 173-176. doi:10.1016/S0304-3940(98)00539-4
- J. A. Murray, M. Panagia, D. Hauton, G. F. Gibbons and K. Clarke, “Plasma Free Fatty Acids and Peroxisome Proliferator-Activated Receptor α in the Control of Myocardial Uncoupling Protein Levels,” Diabetes, Vol. 54, No. 12, 2005, pp. 3496-3502. doi:10.2337/diabetes.54.12.3496
- L. I. Kelly, P. P. Vicario, G. M. Thompson, M. R. Candelore, T. W. Doebber, J. Ventre, M. S. Wu, R. Meurer, M. J. Forrest, M. W. Conner, M. A. Cascieri and D. E. Moller, “Peroxisome Proliferator-Activated Receptors γ and α Mediate in Vivo Regulation of Uncoupling Protein (UCP1, UCP2, UCP3) Gene Expression,” Endocrinology, Vol. 139, No. 12, 1998, pp. 4920-4927. doi:10.1210/en.139.12.4920
- Z. K. Binienda, I. A. Ross, B. Gough, S. F. Ali, S. Z. Imam and C. S. Kim, “Differential Response of Brain and Liver Free Fatty Acids Following Administration of Iron Nanoparticles in Rats,” The Toxicologist, Vol. 132, 2013, p. 505.
- A. Hirasawa, T. Hara, S. Katsuma, T. Adachi and G. Tsujimoto, “Free Fatty Acid Receptors and Drug Discovery,” Biological & Pharmaceutical Bulletin, Vol. 31, No. 10, 2008, pp. 1847-1851.doi:10.1248/bpb.31.1847
- A. Ichimura, A. Hirasawa, T. Hara and G. Tsujimoto, “Free Fatty Acid Receptors Act as Nutrient Sensors to Regulate Energy Homeostasis,” Prostaglandins & Other Lipid Mediators, Vol. 89, No. 3-4, 2009, pp. 82-88. doi:10.1016/j.prostaglandins.2009.05.003
- C. J. Nolan, M. S. Madiraju, V. Delghingaro-Augusto, M. L. Peyot and M. Prentki, “Fatty Acid Signaling in the Beta-Cell and Insulin Secretion,” Diabetes, Vol. 55, Suppl. 2, 2006, pp. 16-23. doi:10.2337/db06-S003
- K. Kotarsky, N. E. Nilsson, E. Flodgren, C. Owman and B. Olde, “A Human Cell Surface Receptor Activated by Free Fatty Acids and Thiazolidinedione Drugs,” Biochemical and Biophysical Research Communications, Vol. 301, No. 2, 2003, pp. 406-410. doi:10.1016/S0006-291X(02)03064-4
- F. Uno, J. Sasaki, M. Nishizaki, G. Carboni, K. Xu, E. N. Atkinson, M. Kondo, J. D. Minna, J. A. Roth and L. Ji, “Myristoylation of the Fus1 Protein Is Required for Tumor Suppression in Human Lung Cancer Cells,” Cancer Research, Vol. 64, No. 9, 2004, pp. 2969-2976. doi:10.1158/0008-5472.CAN-03-3702
- A. Aitken, P. Cohen, S. Santikarn, D. H. Williams, A. G. Calder, A. Smith and C. B. Klee, “Identification of the NH2-Terminal Blocking Group of Calcineurin B as Myristic Acid,” FEBS Letters, Vol. 150, No. 2, 1982, pp. 314-318.doi:10.1016/0014-5793(82)80759-X
- S. A. Carr, K. Biemann, S. Shoji, D. C. Parmalee and K. Titani, “N-Tetradecanoyl is the NH2-Terminal Blocking Group of the Catalytic Submits of Cyclic AMP-Dependent Protein Kinase from Bovine Cardiac Muscles,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 79, No. 20, 1982, pp. 6128- 6131.doi:10.1073/pnas.79.20.6128
- E. N. Olson and G. Spizz, “Fatty Acylation of Cellular Proteins. Temporal and Subcellular Differences between Palmitate and Myristate Acylation,” Journal of Biological Chemistry, Vol. 261, No. 5, 1986, pp. 2458-2466.
- E. N. Olson, “Modification of Proteins with Covalent Lipids,” Progress in Lipid Research, Vol. 27, No. 3, 1988, pp. 177-197. doi:10.1016/0163-7827(88)90012-4
- A. M. Dizhoor, L. H. Ericsson, R. S. Johnson, S. Kumar, E. Olshevskaya, S. Zozulya, T. A. Neubert, L. Stryer, J. B. Hurley and Walsh, “The NH2 Terminus of Retinal Recovering Is Acylated by a Small Family of Fatty Acids,” Journal of Biological Chemistry, Vol. 267, No. 23, 1992, pp. 16033-16036.
- W. Onkenhout, V. Venizelos, P. F. van der Poel, M. P. van den Heuvel and B. J. Poorthuis, “Identification and Quantification of Intermediates of Unsaturated Fatty Acid Metabolism in Plasma of Patients with Fatty Acid Oxidation Disorders,” Clinical Chemistry, Vol. 41, No. 10, 1995, pp. 1467-1474.
- K. Iguchi, N. Okumura, S. Usui, H. Sajiki, K. Hirota and K. Hirano, “Myristoleic acid, a Cytotoxic Component in the Extract from Serenoa Repens, Induces Apoptosis and Necrosis in Human Prostatic LNCaP cells,” Prostate, Vol. 47, No. 1, 2001, pp. 59-65.doi:10.1002/pros.1047
- M. D. Resh, “Fatty Acylation of Proteins: New Insights into Membrane Targeting of Myristoylated and Palmitoylated Proteins,” Biochimica et Biophysica Acta, Vol. 1451, No. 1, 1999, pp. 1-16. doi:10.1016/S0167-4889(99)00075-0
- S. I. Patterson, “Posttranslational Protein S-Palmitoylation and the Compartmentalization of Signaling Molecules in Neurons,” Biological Research, Vol. 35, 2002, pp. 139-150.doi:10.4067/S0716-97602002000200005
- M. Bijlmakers and M. Marsh, “The On-Off Story of Protein Palmitoylation,” Trends in Cell Biology, Vol. 13, No. 1, 2003, pp. 32-42. doi:10.1016/S0962-8924(02)00008-9
- K. Huang and A. D. El-Husseini, “Modulation of Neuronal Protein Trafficking and Function by Palmitoylation,” Current Opinion in Neurobiology, Vol. 15, 2005, pp. 527-535. doi:10.1016/j.conb.2005.08.001
- S. J. Plowman and J. F. Hancock, “Ras Signaling from Plasma Membrane and Endomembrane Microdomains,” Biochimica at Biophysica Acta, Vol. 1746, 2005, pp. 274-283. doi:10.1016/j.bbamcr.2005.06.004
- A. Mor and M. R. Philips, “Compartmentalized Ras/ MAPK Signaling,” Annual Review of Immunology, Vol. 24, 2006, pp. 771-800. doi:10.1146/annurev.immunol.24.021605.090723
- L. P. Wright and M. R. Philips, “Thematic Review Series Lipid Posttranslational Modifications. CAAX Modification and Membrane Targeting of Ras,” Journal of Lipid Research, Vol. 47, 2006, pp. 883-891. doi:10.1194/jlr.R600004-JLR200
- X. Yang, O. V. Kovalenko, W. Tang, C. Claas, C. S. Stipp and M. E. Hemler, “Palmitoylation Supports Assembly and Function of Integrin-Tetraspanin Complexes,” Journal of Cell Biology, Vol. 167, No. 6, 2004, pp. 1231-1240. doi:10.1083/jcb.200404100
- B. Zhou, L. Liu, M. Reddivari and X. A. Zhang, “The Palmitoylation of Metastasis Suppressor KAII/CD82 Is Important for Its Motility and Invasiveness-Inhibitory Activity,” Cancer Research, Vol. 64, No. 20, 2004, pp. 7455-7463.doi:10.1158/0008-5472.CAN-04-1574
- K. L. Clark, A. Oelke, M. E. Johnson, K. D. Eilert, P. C. Simpson and S. C. Todd, “CD81 Associates with I4-3-3 in a Redox-Regulated Palmitoylation-Dependent Manner,” Journal of Biological Chemistry, Vol. 279, No. 19, 2004, pp. 19401-19406.doi:10.1074/jbc.M312626200
- S. W. Wong, M. J. Kwon, A. M. Choi, H. P. Kim, K. Nakahira and D. H. Hwang, “Fatty Acids Modulate TollLike Receptor 4 Activation Through Regulation of Receptor Dimerization and Recruitment into Lipid Rafts in a Reactive Oxygen Species-Dependent Manner,” Journal of Biological Chemistry, Vol. 284, No. 40, 2009, pp. 27384- 27392. doi:10.1074/jbc.M109.044065
- E. V. Kalinina and L. D. Fricker, “Palmitoylation of Carboxypeptidase D. Implications for Intracellular Trafficking,” Journal of Biological Chemistry, Vol. 278, No. 11, 2003, pp. 9244-9249. doi:10.1074/jbc.M209379200
- I. Navarro-Lerida, M. M. Corvi, A. A. Barrientos, F. Gavilanes, L. G. Berthiaume and I. Rodriguez-Crespo, “Palmitoylation of Inducible Nitric Oxide Synthase at Cys-3 Is Required for Proper Intracellular Traffic and Nitric Oxide Synthesis,” Journal of Biological Chemistry, Vol. 279, No. 53, 2004, pp. 5682-55689. doi:10.1074/jbc.M406621200
- J. M. Draper and C. D. Smith, “Palmitoyl Acyltransferase Assays and Inhibitors,” Molecular Membrane Biology, Vol. 26, No. 1, 2009, pp. 5-13. doi:10.1080/09687680802683839
- World Health Organization, “World Health Statistics 2012,” World Health Organization, Geneva, 2012.
- M. U. Jakobsen, E. J. O’Reilly, B. L. Heitmann, M. A. Pereira, K. Bälter, G. E. Fraser, U. Goldbourt, G. Hallmans, P. Knekt, S. Li, P. Pietinen, D. Spiegelman, J. stevens, J. Virtamo, W. C. Willett and A. Ascherio, “Major Types of Dietary Fat and Risk of Coronary Heart Disease: A Pooled Analysis of 11 Cohort Studies,” The American Journal of Clinical Nutrition, Vol. 89, No. 5, 2009, pp. 1425-1432.doi:10.3945/ajcn.2008.27124
- [61] D. B. Cines, E. S. Pollak, C. A. Buck, J. Loscalzo, G. A. Zimmerman, R. P. McEver, J. S. Pober, T. M. Wick, B. A. Konkle, B. S. Schwartz, E. S. Barnathan, K. R. McCrae, B. A. Hug, A.-M. Schmidt and D. M. Stern, “Endothelial Cells in Physiology and in the Pathophysiology of Vascular Disorders,” Blood, Vol. 91, No. 10, 1998, pp. 3527- 3561.
- [62] J. D. Pearson, “Normal Endothelial Cell Function,” Lupus, Vol. 9, No 3, 2000, pp. 183-188. doi:10.1191/096120300678828299
- [63] J. Liu, G. Garcia-Cardena and W. C. Sessa, “Biosynthesis and Palmitoylation of Endothelial Nitric Oxide Synthase: Mutagenesis of Palmitoylation Sites, Cysteines-15 and/or -26, Argues Against Depalmitoylation-Induced Translocation of the Enzyme,” Biochemistry, Vol. 34, No. 38, 1995, pp. 12333-12340.doi:10.1021/bi00038a029
- [64] C. Clapp, J. Aranda, C. González, M. C. Jeziorski and G. M. de la Escalera, “Vasoinhibins: Endogenous Regulators of Angiogenesis and Vascular Function,” Trends in Endocrinology & Metabolism, Vol. 17, No. 8, 2006, pp. 301- 307.doi:10.1016/j.tem.2006.08.002
- [65] Y. Tang and G. Li, “Chronic Exposure to High Fatty Acids Impedes Receptor Agonist-Induced Nitric Oxide Production and Increments of Cytosolic Ca2+ Levels in Endothelial Cells,” Journal of Molecular Endocrinology, Vol. 47, No. 3, 2011, pp. 315-326. doi:10.1530/JME-11-0082
- [66] D. Tian, Y. Qiu, Y. Zhan, X. Li, X. Zhi, X. Wang, L. Yin and Y. Ning, “Overexpression of Steroidogenic Acute Regulatory Protein in Rat Aortic Endothelial Cells Attenuates Palmitic Acid-Induced Inflammation and Reduction in Nitric Oxide Bioavailability,” Cardiovascular Diabetology, Vol. 11, No. 144, 2012, pp. 1-12.
- [67] J. Dyerberg, H. O. Bang, E. Stoffersen, S. Moncada and J. R. Vane, “Eicosapentaenoic Acid and Prevention of Thrombosis and Atherosclerosis?” The Lancet, Vol. 312, No. 8081, 1978, pp. 117-119. doi:10.1016/S0140-6736(78)91505-2
- [68] M. Hirafuji, T. Machida, N. Hamaue and M. Minami, “Cardiovascular Protective Effects of n-3 Polyunsaturated Fatty Acids with Special Emphasis on Docosahexaenoic Acid,” Journal of Pharmacological Sciences, Vol. 92, No. 4, 2003, pp. 308-316.doi:10.1254/jphs.92.308
- [69] Y. Wu, C. Zhang, Y. Dong, S. Wang, P. Song, B. Viollet and M.-H. Zou, “Activation of the AMP-Activated Protein Kinase by Eicosapentaenoic Acid (EPA, 20:5n-3) Improves Endothelial Function in Vivo,” PLoS ONE, Vol. 7, No. 4, 2012, Article ID: e35508. doi:10.1371/journal.pone.0035508
- [70] M. T. Nelson, H. Cheng, M. Rubart, L. F. Santana, A. D. Bonev, H. J. Knot and W. J. Lederer, “Relaxation of Arterial Smooth Muscle by Calcium Sparks,” Science, Vol. 270, No. 5236, 1995, pp. 633-637. doi:10.1126/science.270.5236.633
- [71] L.-H. Lai, R.-X. Wang, W.-P. Jiang, X.-J. Yang, J.-P. Song, X.-R. Li and G. Tao, “Effects of Docosahexaenoic Acid on Large-Conductance Ca2+-activated K+ Channels and Voltage-Dependent K+ Channels in Rat Coronary Artery Smooth Muscle Cells,” Acta Pharmacologica Sinica, Vol. 30, No. 3, 2009, pp. 314-320. doi:10.1038/aps.2009.7
- [72] R.-X. Wang, Q. Chai, T. Lu and H.-C. Lee, “Activation of Vascular BK Channels by Docosahexaenoic Acid Is Dependent on Cytochrome P450 Epoxygenase Activity,” Cardiovascular Research, Vol. 90, No. 2, 2011, pp. 344- 352. doi:10.1093/cvr/cvq411
- [73] H. C. Hercule, B. Salanova, K. Essin, H. Honeck, J. R. Falck, M. Sausbier, P. Ruth, W.-H. Schnuck, F. C. Luft and M. Gollasch, “Vasodilator 17,18-Epoxyeicosatetraenoic Acid Targets the Pore-Forming BK α Channel Subunit in Rodents,” Experimental Physiology, Vol. 92, No. 6, 2007, pp. 1067-1076. doi:10.1113/expphysiol.2007.038166
- [74] R. Ringseis, A. Müller, C. Herter, S. Gahler, H. Steinhart and K. Eder, “CLA Isomers Inhibit TNFα-Induced Eicosanoid Release from Human Vascular Smooth Muscle Cells via a PPARγ Ligand-Like Action,” Biochimica et Biophysica Acta—General Subjects, Vol. 1760, No. 2, 2006, pp. 290-300.
- [75] R. Ringseis, S. Gahler, C. Herter and K. Eder, “Conjugated Linoleic Acids Exert Similar Actions on Prostanoid Release from Aortic and Coronary Artery Smooth Muscle Cells,” International Journal for Vitamin and Nutrition Research, Vol. 76, No. 5, 2006, pp. 281-289. doi:10.1024/0300-9831.76.5.281
- [76] U. Ikeda, M. Shimpo, Y. Murakami and K. Shimada, “Peroxisome Proliferator-Activated Receptor-γ Ligands Inhibit Nitric Oxide Synthesis in Vascular Smooth Muscle Cells,” Hypertension, Vol. 35, No. 6, 2000, pp. 1232- 1236. doi:10.1161/01.HYP.35.6.1232
- [77] O. Feron and J.-L. Balligand, “Caveolins and the Regulation of Endothelial Nitric Oxide Synthase in the Heart,” Cardiovascular Research, Vol. 69, No. 4, 2006, pp. 788-797.doi:10.1016/j.cardiores.2005.12.014
- [78] C. J. Knowles, M. Dionne, M. Cebova and I. M. Pinz, “Palmitate-Induced Translocation of Caveolin-3 and Endothelial Nitric Oxide Synthase in Cardiomyocytes,” OnLine Journal of Biological Sciences, Vol. 11, No. 2, 2011, pp. 27-36.doi:10.3844/ojbsci.2011.27.36
- [79] V. P. M. van Empel, A. T. A. Bertrand, L. Hofstra, H. J. Crijns, P. A. Doevendans and L. J. De Windt, “Myocyte Apoptosis in Heart Failure,” Cardiovascular Research, Vol. 67, No. 1, 2005, pp. 21-29. doi:10.1016/j.cardiores.2005.04.012
- [80] A. Eisenberg-Lerner, S. Bialik, H.-U. Simon and A. Kimchi, “Life and Death Partners: Apoptosis, Autophagy and the Cross-Talk between Them,” Cell Death & Differentiation, Vol. 16, No. 7, 2009, pp. 966-975. doi:10.1038/cdd.2009.33
- [81] C. Leroy, S. Tricot, B. Lacour and A. Grynberg, “Protective Effect of Eicosapentaenoic Acid on Palmitate-Induced Apoptosis in Neonatal Cardiomyocytes,” Biochimica et Biophysica Acta, Vol. 1781, No. 11-12, 2008, pp. 685-693.doi:10.1016/j.bbalip.2008.07.009
- [82] S. Ghavami, R. H. Cunnington, B. Yeganeh, J. J. L. Davies, S. G. Rattan, K. Bathe, M. Kavosh, M. J. Los, D. H. Freed, T. Klonisch, G. N. Pierce, A. J. Halayko and I. M. C. Dixon, “Autophagy Regulates Trans Fatty Acid-Mediated Apoptosis in Primary Cardiac Myofibroblasts,” Biochimica et Biophysica Acta (BBA)—Molecular Cell Research, Vol. 1823, No. 12, 2012, pp. 2274-2286.
- [83] S. Cetrullo, B. Tantini, F. Flamigni, C. Pazzini, A. Facchini, C. Stefanelli, C. M. Caldarera and C. Pignatti, “Antiapoptotic and Antiautophagic Effects of Eicosapentaenoic Acid in Cardiac Myoblasts Exposed to Palmitic Acid,” Nutrients, Vol. 4, No. 2, 2012, pp. 78-90. doi:10.3390/nu4020078
Abbreviations
FFAs free fatty acids
SFA saturated fatty acids
FFARs multiple FFA receptors
GPCR G protein-coupled receptor
FAT/CD36 fatty acid translocase
FATP fatty acid transport proteins
FABPpm plasma membrane bound fatty acid binding protein
CoA coenzyme A
FACS fatty acyl-CoA synthase
CPT1 carnitine palmitoyltransferase 1
CAT carnitine translocase
CPT2 carnitine palmitoyltransferase 2
TCA tricarboxylic acid cycle
IRS1 insulin receptor substrate-1
NO nitric oxide
GSH reduced glutathione
ROS reactive oxygen species
3-NPA 3-nitropropionic acid
AP1 activator protein-1
PPARα peroxisome proliferator-activated receptor-alpha
NPs nanoparticles
CVD cardiovascular disease
SMC smooth muscle cells
PUFA polyunsaturated fatty acids
TFA trans-fatty acids
CLA conjugated linoleic acid
LA linoleic acid
eNOS endothelial nitric oxide synthase
EPA eicosapentaenoic acid
DHA docosahexaenoic acid
UCP-2 uncoupling protein-2
CYP450 cytochrome P450
BK calcium-activated potassium channels
Bax Bcl-2-associated X protein
NOTES
*Disclaimer: The views presented in this article are those of the authors and do not necessarily represent the views of the US Food and Drug Administration.