Graphene
Vol.03 No.04(2014), Article ID:50633,7 pages
10.4236/graphene.2014.34008
A Comparative Study of Electronic Properties of Bulk MoS2 and Its Monolayer Using DFT Technique: Application of Mechanical Strain on MoS2 Monolayer
Sohail Ahmad1, Sugata Mukherjee2*
1Department of Physics, Faculty of Science, King Khalid University, Abha, Saudi Arabia
2S. N. Bose National Centre for Basic Sciences, Kolkata, India
Email: sohailphysics@yahoo.co.in, *sugatamukh@gmail.com, *sugata@bose.res.in
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 17 August 2014; revised 13 September 2014; accepted 12 October 2014
ABSTRACT
Electronic structure calculation of bulk and monolayer MoS2 has been performed using plane wave pseudopotential method based on density functional theory. The indirect band gap in the bulk MoS2 was found to be 0.9 eV, whereas in the monolayer-MoS2 the band gap of 1.57 eV was found to be direct one. The calculated physical parameters of monolayer MoS2 are found to be very close to the bulk MoS2 and compare well with available experimental and other theoretical results. The calculated density of states (DOS) may help explain this change in the nature of band gap in bulk and in monolayer MoS2. A further variation in band gap has been observed in MoS2 monolayer on applying biaxial strain.
Keywords:
MoS2, DFT, Electronic Properties, Mono Layer, Strain

1. Introduction
Layered transition-metal di-chalcogenides (LTMDCs) have been extensively reviewed in the recent past [1] . MoS2 is a typical example of LTMDC family of materials which attracts investigation because of its distinctive industrial applications, ranging from use as a lubricant [2] and a catalyst [3] as well as in photo-voltaics [4] and energy storage [5] . In its bulk form MoS2 is a semiconductor with an indirect band gap of about 1.23 eV while its monolayer has a direct energy gap of 1.8 eV [6] . A special attention has been paid on single layer MoS2 in the recent years. Upon thinning from the bulk [7] -[9] the electronic structure of MoS2 undergoes an interesting transition. Recently, a monolayer MoS2-based field effect transistor (FET) with HfO2 as gate insulator has been successfully implemented [10] . These ideal properties make monolayer MoS2 a very promising candidate for next generation FET and as optoelectronic devices [11] . This has raised enormous interest in exploring the extraordinary properties of mono layers of MoS2.
Theoretically, there are various possibilities of energy gap manipulation in MoS2. By reducing the layer thickness from bulk to monolayer, the indirect band gap energies in the bulk are shifted relative to the direct band gap in the monolayer limit [12] . It undergoes a transition from an indirect to direct gap exhibiting strong photoluminescence when confined in a 2D monolayer [13] . It has been suggested a way to engineer 3D semiconducting MoS2 nanoparticles with direct band gaps as well as metallic di-chalcogenides nanowires with promising catalytic and thermoelectric properties [14] . Eellis et al. carried out a study using HSE screened hybrid functional and offered improvement over semi local density functional [13] . All electron calculations including spin orbit coupling were performed [15] and confirmed indirect to direct band gap transition. Electronic structure of transition metal di-chalcogenides has been studied using ab initio theory using Troullier-Martin norm conserving, relativistic pseudopotentials in fully separable Kleinman and Bylander form [16] . They used exchange and correlation energies within LDA.
The properties of transition metal di-chalcogenides (TMDs) not only can be tuned by varying number of layers, but also can be modified by application of external field or strain engineering. Studies [17] [18] have confirmed that applying strain is one of the best possible strategies to tune the band gap, since it neither attenuates the properties nor is inefficacious for single layers. It has been predicted that straining MoS2 modifies the band gap energy and the carrier effective mass. Moreover, at strains larger than 1% the lowest lying band gap changes from direct to indirect [19] -[22] . It has been suggested that strain engineering of the band structure of MoS2 could be used to increase carrier mobility of MoS2, to create tunable photonic devices and solar cells [23] and even to control the magnetic properties of MoS2 [19] [20] . While strain perturbs the band structure of all materials, two-dimensional materials such as MoS2 can sustain strains greater than 11% [24] allowing exceptional control of material properties by strain engineering. In a recent study [25] the conduction band valley structure of a few layer MX2 by close examination of temperature dependent indirect excitation emission peaks has been explored. A study on elastic constants and electronic structures of two-dimensional monolayer MoS2 under elastic strain using first principle calculations has been made [26] . It is shown that the band gap of monolayer MoS2 undergoes a descent trend with the increase in strain. They observed a direct to indirect transition at strain of 0.01 and semiconductor to metal transition at strain of 0.10.
With the goal of understanding the electronic properties of bulk and monolayer MoS2 and strain engineering, we carried out ab initio calculations of bulk and monolayer MoS2 using gradient corrected exchange-correlation functional in DFT framework and observed a transition from indirect to direct band gap. However if this band gap can be tuned such that a semiconductor with a lower band gap or a semiconductor to metal transition can be achieved with the application of strain, then a wide range of tunable nano device can be fabricated. In the present work, therefore we study the effects of mechanical strains on the electronic properties of monolayer of MoS2. Our results suggest a way of band gap engineering in MoS2.
2. Computational Details
The calculations were performed using self consistent plane wave pseudopotential total energy method based on density functional technique as implemented in Quantum Espresso code [27] . This method has been previously used to study the electronic properties of undoped and doped graphene [28] [29] . The exchange correlation potential was approximated by generalized gradient approximation using Perdew-Wang 91 functional (GGA- PW91) [30] . The atomic positions and cell parameters were fully relaxed until an energy convergence of 10−9 eV reached. We used wave function- and charge-density cut-offs of 70 Ryd and 300 Ryd, respectively. First, we obtained lattice constants a and c by the process of total energy minimization. Optimized structure (coordinates) was used to perform self consistent calculations with a Monkhorst-Pack [31]
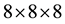 k-mesh followed by the non-self consistent calculations for band structures, density of states and partial density of states of bulk MoS2. We used
k-mesh followed by the non-self consistent calculations for band structures, density of states and partial density of states of bulk MoS2. We used
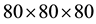 k-points mesh along the path Γ-K-M-Γ in the irreducible Brillouin zone to obtain the band structure with a very fine mesh points. However, for monolayer we used
k-points mesh along the path Γ-K-M-Γ in the irreducible Brillouin zone to obtain the band structure with a very fine mesh points. However, for monolayer we used
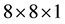 and
and
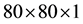 Monkhorst-Pack of k-points respectively for sampling Brillouin zone for calculations of structural properties and electronic structure. In case of monolayer MoS2, we created 15 Å vacuum along Z axis to isolate it and to prevent any interaction between the layers. The cohesive energy per atom of bulk MoS2 was calculated as Ecoh = E(MoS2) − E(Mo) − 2E(S), where E(MoS2) is the total energy of the unit cell of Molybdenum disulphide, E(Mo) is the energy of Mo atom and E(S) is the energy of S atom. The cohesive energies per atom of monolayer MoS2 was also calculated accordingly. A uniform tensile strain ranging from 0 to 10% were applied on monolayer MoS2 to study the change in behavior of its band gap.
Monkhorst-Pack of k-points respectively for sampling Brillouin zone for calculations of structural properties and electronic structure. In case of monolayer MoS2, we created 15 Å vacuum along Z axis to isolate it and to prevent any interaction between the layers. The cohesive energy per atom of bulk MoS2 was calculated as Ecoh = E(MoS2) − E(Mo) − 2E(S), where E(MoS2) is the total energy of the unit cell of Molybdenum disulphide, E(Mo) is the energy of Mo atom and E(S) is the energy of S atom. The cohesive energies per atom of monolayer MoS2 was also calculated accordingly. A uniform tensile strain ranging from 0 to 10% were applied on monolayer MoS2 to study the change in behavior of its band gap.
3. Results and Discussion
3.1. Structural Parameters
Molybdenum disulphide has a hexagonal structure consisting of S-Mo-S layers as shown in Figure 1. Bulk MoS2 has two such layers and Mo atoms of one layer are directly above the sulphur atoms of the other layer and vice versa while monolayer MoS2 has a single S-Mo-S layer. We have calculated the structural parameters of bulk and monolayer MoS2 using GGA as shown in Table 1. The results of bulk MoS2 have been compared with experimental data while the results of monolayer are compared with some other theoretical results. We find excellent agreements as can be seen in Table 1. Our calculated lattice parameters overestimate the experimental values which is an inherent feature of standard GGA functional. It is concluded that all the structural parameters calculated for monolayer MoS2 are nearly identical to the structural parameters calculated for bulk MoS2.
3.2. Electronic Band Structure and Density of States
The electronic band structure of bulk MoS2 and corresponding density of states are shown in Figure 2. The electronic band structure and density of states can be divided into three sets of bands and states respectively, separated by gaps. In the first set, bands in electronic band structure and states in density of states around −14 eV are mainly due to 3s orbital of S atom separated by large gap from second set below Fermi energy in which 3p orbital of S and 4d orbital of Mo are mainly contributing and show strong hybridization. Third set above the Fermi energy in which main contribution is due to 4d orbital of Mo is separated by band gap from second group
![]() (a) (b)
(a) (b)
![]() (c)
(c)
Figure 1. (a) Side view of bulk MoS2; (b) Top view of bulk and monolayer MoS2; and (c) Side view of mono layer MoS2. The Mo-atoms are denoted by gray and S-atoms by pale yellow balls.
![]()
Figure 2. Calculated band structure (left panel) and orbital-projected density of states (PDOS) on Mo (middle panel) and S atoms (right panel) in bulk MoS2.

Table 1. Calculated structural parameters of bulk MoS2 and mono layer MoS2 using GGA. The available results have also been given for the purpose of comparison.
below Fermi energy. A comparative band structure of MoS2 bulk and its monolayer and bilayer are shown in Figure 3. The bands on each side of the band gap are derived mainly from the 4d states of Mo and 3p states of S in bulk, bilayer and monolayer MoS2. The bands around the band gap are relatively flat, as expected from the d-character of electron states at these energies.
For bulk MoS2 the valence band maximum is at high-symmetric Γ-point and conduction band minima is in between Γ- and K-points, revealing indirect band semiconductor as can be seen in Figure 3. If we compare the band structure of bulk and monolayer MoS2, we observe that the band edge near Γ point has been shifted up by around 0.7 eV. In case of monolayer, at Λ and Σ point the band edge shifted up in such a way that the conduction band minima occurs at K-point. For monolayer the valence band maxima and conduction band minima are both at high-symmetric K-point revealing direct band gap semiconductor as can be seen in Figure 3. Thus there is a transition from indirect band gap to direct band gap as we go from bulk MoS2 to its monolayer. A PDOS comparison of bulk and mono layer MoS2 (as shown in the Figure 4) reflects that the states are essentially due to dz2 and degenerate states dxy and dx2-y2. The states dx2-y2 and dxy are degenerate in case of bulk while little bit separating in monolayer. The calculated and measured band gap for mono layer MoS2 and bulk are shown in Table 2. Our calculated band gaps are in good agreements with other theoretical values.
3.3. Tuning Electronic Properties by Biaxial Strain
To study the effect of strain on the electronic properties of monolayer MoS2, we first relaxed the atomic position and obtained the optimized geometry. We apply the uniform strain (ε) to the monolayer MoS2 in the range 0.0 - 0.10. The strained atomic structure is achieved by enlarging the hexagonal lattice a0 with an increment of εao. Similarly the atomic structure is fully optimized and the band structure is calculated. As for strained structure of MoS2, the band gap versus strain is shown in the Figure 5. The band gap is monotonic as the strain increases.
![]()
Figure 3. Calculated band structure of bulk (left), bilayer (middle) and monolayer (right) of MoS2 at high-symmetric points in the irreducible Brillouin zone. The position of valence band maxima, conduction band minima and the band gap (Eg) are indicated. For monolayer MoS2 the direct band gap occurs at K-point, unlike in other cases.
![]()
Figure 4. Calculated projected density of states (PDOS) of bulk and monolayer of MoS2 are shown for Mo(4d) and S(3p) states. The legends for the p- and d-orbitals are similar for the monolayer and bulk.
![]()
Figure 5. The variation of band gap energy with strain of monolayer MoS2.
Table 2. Energy gaps (eV) for bulk MoS2 and monolayer MoS2.
While the strain reaches 0.06, the indirect band gap vanishes. We observe a cross over in the figure. Upon ε = 0.0, the monolayer MoS2 shows a behavior of semiconductor with direct gap, at ε = 0.005 it has direct (at K) and indirect (Γ-K) band gap of almost equal amount. These results are interesting. Not only the band gap of monolayer MoS2 can be tuned by uniform strain, but also direct to indirect and semiconductor to metal transitions are controlled.
Very recently, in a paper by Das et al. [39] the strain driven direct to indirect transition in the band gap of MoS2 and ZnO has been theoretically studied. They have found such transition in MoS2 to occur at strain 0.83%, close to our calculated value of 0.5%.
4. Conclusion
In summary, we studied the structural and electronic properties of MoS2 using plane wave pseudopotential method under GGA scheme based DFT calculations. Electronic band structure and density of states calculation show many similarities between monolayer-MoS2 and bulk-MoS2 except the nature of the band gap which is found direct for monolayer-MoS2 as compared to indirect for bulk-MoS2. This observation is consistent with the theoretical prediction of indirect to direct band gap transition in going from bulk to monolayer. Such behavior, arising from d-orbital related interaction in MoS2, may also arise in other layered transition metal di-chalcoge- nides. A further variation in band gap has been observed in MoS2 monolayer on applying strain. It points out a new direction of band engineering, hence such capability can lead to engineering novel behaviors and holds promise for new applications.
Acknowledgements
Part of the calculations was performed on High-Performance cluster computer of SNBNCBS, Kolkata for which authors are thankful. SA acknowledges valuable discussion with T.P. Kaloni. This work was initiated at S.N. Bose National Centre, when one of us (SA) was a visiting scientist under the Extended Visitors Linkage Programme (EVLP), which is gratefully acknowledged.
References
- Wilson, J.A. and Yoffe, A.D. (1969) The Transition Metal Dichalcogenides Discussion and Interpretation of the Observed Optical, Electrical and Structural Properties. Advances in Physics, 18, 193. http://dx.doi.org/10.1080/00018736900101307
- Kim, Y., Huang, J.L. and Lieber, C.M. (1991) Characterization of Nanometer Scale Wear and Oxidation of Transition Metal Dichalcogenide Lubricants by Atomic Force Microscopy. Applied Physics Letters, 59, 3404. http://dx.doi.org/10.1063/1.105689
- Hu, K.H., Huand, X.G. and Sun, X.J. (2010) Morphological Effect of MoS2 Nanoparticles on Catalytic Oxidation and Vacuum Lubrication. Applied Surface Science, 256, 2517. http://dx.doi.org/10.1016/j.apsusc.2009.10.098
- Fortin, E. and Sears, W. (1982) Photovoltaics Effect and Optical Absorption in MoS2. Journal of Physics and Chemistry of Solids, 43, 881. http://dx.doi.org/10.1016/0022-3697(82)90037-3
- Reshak, A.H. and Auluck, S. (2003) Calculated Optical Properties of 2H-MoS2, Intercalated with Lithium. Physical Review B, 68, Article ID: 125101.
- Mak, K.F., Lee, C., Hone, J., Shan, J. and Heinz, T.F. (2010) Atomically Thin MoS2: A New Direct Gap Semiconductor. Physical Review Letter, 105, Article ID: 136805. http://dx.doi.org/10.1103/PhysRevLett.105.136805
- Novoselov, K., Jiang, D., Schedlin, F., Booth, T., Khotkevich, V., Morozov, S. and Geim, A. (2005) Two Dimensional Atomic Crystals. Proceedings of the National Academy of Sciences of the United States of America, 102, 10451.
- Joensen, P., Frindt, R. and Morrison, S. (1986) Single Layer MoS2. Materials Research Bulletin, 21, 457. http://dx.doi.org/10.1016/0025-5408(86)90011-5
- Coleman, J.N., Lotya, M., O’Neill, A., Bergin, S.D., King, P.J., Khan, U., Young, K., Gaucher, A., De, S., Smith, R.J., Shvets, I.V., Arora, S.K., Stanton, G., Kim, H.Y., Lee, K., Kim, G.T., Duesberg, G.S., Hallam, T., Bolland, J.J., Wang, J.J., Donegan, J.F., Grunlan, J.C., Moriarty, G., Shmeliov, A., Nicholls, R.J., Perkins, J.M., Grieveson, E.M., Theuwissen, K., McComb, D.W., Nellist, P.D. and Nicolosi, V. (2011) Two-Dimensional Nanosheets Produced by Liquid Exfoliation of Layered Materials. Science, 331, 568. http://dx.doi.org/10.1126/science.1194975
- Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V. and Kis, A. (2011) Single Layer MoS2 Transistors. Nature Nanotechnology, 6, 147. http://dx.doi.org/10.1038/nnano.2010.279
- Yoon, Y., Ganapathi, K. and Salahuddin, S. (2011) How Good Can Monolayer MoS2 Transistors Be? Nano Letters, 11, 3768-3773. http://dx.doi.org/10.1021/nl2018178
- Kumar, A. and Ahluwalia, P.K. (2012) Electronic Structure of Transition Metal Dichalcogenides Monolayers 1H-MoS2 (M = Mo, W; X = S, Se, Te) from ab Initio Theory: New Direct Band Gap Semiconductors. European Physical Journal B, 85, 186. http://dx.doi.org/10.1140/epjb/e2012-30070-x
- Eellis, J.K., Lucero, M.J. and Scuseria, G.E. (2011) The Indirect to Direct Band Gap Transition in Multilayered MoS2 as Predicted by Screened Hybrid Functional Density Functional Theory. Applied Physics Letters, 99, Article ID: 261908. http://dx.doi.org/10.1063/1.3672219
- Li, T. and Galli, G. (2007) Electronic Properties of MoS2 Nanoparticles. Journal of Physical Chemistry C, 111, 16192- 16196. http://dx.doi.org/10.1021/jp075424v
- Kadantsev, E.S. and Hawrylak, P. (2012) Electronic Structure of Single MoS2 Monolayer. Solid State Communications, 152, 909-913. http://dx.doi.org/10.1016/j.ssc.2012.02.005
- Kumar, A. and Ahluwalia, P.K. (2012) A First Principle Comparative Study of Electronic and Optical Properties of 1H-MoS2 and 2H-MoS2. Materials Chemistry and Physics, 135, 755-761. http://dx.doi.org/10.1016/j.matchemphys.2012.05.055
- Topsakal, M., Cahangirov, S. and Ciraci, S. (2010) The Response of Mechanical and Electronic Properties of Graphane to the Elastic Strain. Applied Physics Letters, 96, Article ID: 091912. http://dx.doi.org/10.1063/1.3353968
- Guinea, F., Katsnelson, M.I. and Geim, A.K. (2010) Energy Gaps and a Zero Field Quantum Hall Effect in Graphene by Strain Engineering. Nature Physics, 6, 30-33. http://dx.doi.org/10.1038/nphys1420
- Lu, P., Wu, X., Guo, W. and Zeng, X.C. (2012) Strain Dependent Electronic and Magnetic Properties of MoS2 Monolayer, Bilayer, Nanoribbons and Nanotubes. Physical Chemistry Chemical Physics, 14, 13035-13040. http://dx.doi.org/10.1039/c2cp42181j
- Pan, H. and Zhang, Y.W. (2012) Tuning the Electronic and Magnetic Properties of MoS2 Nanoribbons by Strain Engineering. Journal of Physical Chemistry C, 116, 11752-11757. http://dx.doi.org/10.1021/jp3015782
- Scalise, E., Houssa, M., Pourtois, G., Afanas’ev, V. and Stesmans, A. (2012) Strain Induced Semiconductor to Metal Transition in the Two Dimensional Honeycomb Structure of MoS2. Nano Research, 5, 43-48. http://dx.doi.org/10.1007/s12274-011-0183-0
- Shi, H., Pan, H., Zhang, Y.W. and Yakobson, B.I. (2013) Quasiparticle Band Structures and Optical Properties of Strained Monolayer MoS2 and WS2. Physical Review B, 87, Article ID: 155304. http://dx.doi.org/10.1103/PhysRevB.87.155304
- Feng, J., Qian, X., Huang, C. and Li, J. (2012) Strain Engineered Artificial Atom as a Broad Spectrum Solar Energy Funnel. Nature Photonics, 6, 866-872. http://dx.doi.org/10.1038/nphoton.2012.285
- Bertolazzi, S., Brivio, J. and Kis, A. (2011) Stretching and Breaking of Ultrathin MoS2. ACS Nano, 5, 9703-9709. http://dx.doi.org/10.1021/nn203879f
- Zhao, W.J., Ribeiro, R.M., Toh, M., Carvalho, A., Kloc, C., Neto, A.H.C. and Eda, G. (2013) Origin of Indirect Optical Transitions in Few Layer MoS2, WS2 and WSe2. Nano Letters, 13, 5627-5634. http://dx.doi.org/10.1021/nl403270k
- Yue, Q., Kang, J., Shao, Z., Zhang, X., Chang, S., Wang, G., Qin, S. and Li, J. (2012) Mechanical and Electronic Properties of Monolayer MoS2 under Elastic Strain. Physics Letters A, 376, 1166-1170. http://dx.doi.org/10.1016/j.physleta.2012.02.029
- Giannozzi, P., Baroni, S., Bonini, N., Calandra, M., Car, R., Cavazzoni, C., Ceresoli, D., Chiarotti, G.L., Cococcioni, C., Kokalj, A., Lazzeri, M., Martin-Samos, L., Marzari, N., Mauri, F., Mazarello, R., Paolini, S., Pasquarello, A., Paulatto, L., Sbraccia, C., Scandolo, S., Sclauzero, G., Seitsonen, A.P., Smogunov, A., Umari, P. and Wentzcovitch, R.M. (2009) QUANTUM ESPRESSO: A Modular and Open Source Software Project for Quantum Simulations of Materials. Journal of Physics: Condensed Matter, 21, Article ID: 395502. http://dx.doi.org/10.1088/0953-8984/21/39/395502
- Kaloni, T.P. and Mukherjee, S. (2011) Comparative Study of Graphite and Hexagonal Boron Nitride Using Pseudopotential Plane Wave Method. Modern Physics Letters B, 25, 1855-1866. http://dx.doi.org/10.1142/S0217984911027182
- Mukherjee, S. and Kaloni, T.P. (2012) Electronic Properties of Boron- and Nitrogen-Doped Graphene: A First-Prin- ciples Study. Journal of Nanoparticle Research, 14, 1059. http://dx.doi.org/10.1007/s11051-012-1059-2
- Perdew, J., Chevary, J., Vosko, S., Jackson, K., Pederson, M., Singh, D. and Fiolhais, C. (1992) Atoms, Molecules, Solids and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation. Physical Review B, 46, 6671-6687. http://dx.doi.org/10.1103/PhysRevB.46.6671
- Monkhorst, H.J. and Pack, J.D. (1976) Special Points for Brillouin Zone Integrations. Physical Review B: Solid State, 13, 5188-5192. http://dx.doi.org/10.1103/PhysRevB.13.5188
- Ataca, C., Shahin, H., Aktruk, E. and Ciraci, S. (2011) Mechanical and Electronic Properties of MoS2 Nano Ribbons and Their Defects. Journal of Physical Chemistry C, 115, 3934-3941. http://dx.doi.org/10.1021/jp1115146
- Ataca, C. and Ciraci, S. (2011) Functionalization of Single Layer MoS2 Honeycomb Structures. Journal of Physical Chemistry C, 115, 13303-13311.
- Lebegue, S. and Erikksso, O. (2009) Electronic Structure of Two-Dimensional Crystals from ab Initio Theory. Physical Review B, 79, Article ID: 115409. http://dx.doi.org/10.1103/PhysRevB.79.115409
- Boker, T., Severin, R., Muller, A., Janowitz, C., Manzke, R., Voβ, D., Kruger, P., Mazur, A. and Pollmann, J. (2001) Band Structure of MoS2, MoSe2 and α-MoTe2: Angle Resolved Photoelectron Spectroscopy and ab Initio Calculations. Physical Review B, 64, Article ID: 235305. http://dx.doi.org/10.1103/PhysRevB.64.235305
- Kobayashi, K. and Yamauchi, J. (1995) Electronic Structure and Scanning Tunneling Microscopy Image of Molybdenum Dichalcogenide Surface. Physical Review B, 51, 17085-17095. http://dx.doi.org/10.1103/PhysRevB.51.17085
- Mattheiss, L.F. (1973) Energy Bands for 2H-NbSe2 and 2H-MoS2. Physical Review Letters, 30, 784-787. http://dx.doi.org/10.1103/PhysRevLett.30.784
- Kuc, A., Zibouche, N. and Heine, T. (2011) Influence of Quantum Confinement on the Electronic Structure of Transi- tion Metal Sulfide TS2. Physical Review B, 83, Article ID: 245213. http://dx.doi.org/10.1103/PhysRevB.83.245213
- Das, R., Rakshit, B., Debnath, S. and Mahadevan, P. (2014) Microscopic Model for the Strain-Driven Direct to Indirect Band-Gap Transition in Monolayer MoS2 and ZnO. Physical Review B, 89, Article ID: 115201. http://dx.doi.org/10.1103/PhysRevB.89.115201
NOTES

*Corresponding author.