Graphene
Vol.2 No.3(2013), Article ID:34766,7 pages DOI:10.4236/graphene.2013.23015
Band Gap Opening of Graphene by Noncovalent π-π Interaction with Porphyrins
1Physics of Nanodevices, Zernike Institute for Advanced Materials, University of Groningen, Groningen, The Netherlands
2Kavli Institute of Nanoscience, Delft University of Technology, Delft, The Netherlands
Email: *arramel0780@yahoo.com
Copyright © 2013 Arramel et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 12, 2013; revised June 18, 2013; accepted July 5, 2013
Keywords: Band Gap; Graphene; Scanning Tunneling Spectroscopy; Porphyrins; π-π Stacking
ABSTRACT
Graphene has been recognized as a promising 2D material with many new properties. However, pristine graphene is gapless which hinders its direct application towards graphene-based semiconducting devices. Recently, various ways have been proposed to overcome this problem. In this study, we report a robust method to open a gap in graphene via noncovalent functionalization with porphyrin molecules. Two type of porphyrins, namely, iron protoporphyrin (FePP) and zinc protoporphryin (ZnPP) were independently physisorbed on graphene grown on nickel by chemical vapour deposition (CVD) resulting in a bandgap opening in graphene. Using a statistical analysis of scanning tunneling spectroscopy (STS) measurements, we demonstrated that the magnitude of the band gap depends on the type of deposited porphyrin molecule.The π-π stacking of FePP on graphene yielded a considerably larger band gap value (0.45 eV) than physisorbed ZnPP (0.23 eV). We proposed that the origin of different band gap value is governed due to the metallic character of the respective porphyrin.
1. Introduction
The development of novel techniques to produce highly crystalline monolayer graphene on the surface of transition metals is bringing graphene-based applications a step closer [1-6]. However, the absence of band gap around the Fermi level in graphene is hampering its application in nanoelectronic and photonic devices [2], since an energy gap is necessary to fabricate field effect transistors with a large current on-off ratio [7,8]. This fact has motivated the study of different methods to open a band gap in graphene. The band structure of graphene is mainly governed by its special electronic properties where π and π* bands are crossing at the so called Dirac neutrality point. The opening of a gap in graphene can be realized by breaking its symmetry in various approaches: defect formation [9], chemical dopants [10-12], electric fields [13-15], and interaction with gases [16].
The noncovalent stacking of aromatic organic molecules on graphene through π-π interaction is emerging as a promising route to tailor the electronic properties of graphene [17], motivated by the study of the interaction between large aromatic molecules and graphene [18-20]. Among all possible aromatic molecules, porphyrins are of primary interest in molecular electronics due to their rich electronic/photonic properties (including charge transport, energy transfer, light absorption or emission) [21]. Individual porphyrin molecules physorbed on different substrates have shown that their structure can be wellresolved using scanning tunneling microscopy (STM) [22-24]. In particular, metalloporphyrins (with a metal ion at the central part of the porphyrin) have attracted much interest due to their peculiar electronic properties [21]. However, a microscopic knowledge of the topographic and the local electronic structure of physisorbed porphyrin molecules on monolayer graphene is still lacking.
In this work the π-π stacking of porphryins on graphene and its effect on the electronic properties of graphene is investigated. Two different types of porphyrin molecules, namely, iron protoporphyrin and zinc porphryins were physisorbed on CVD graphene grown on nickel. By means of a systematic statistical analysis of scanning tunnelling spectroscopy measurements, we probe the opening of an energy bandgap in graphene in the vicinity of physisorbed porphyrins. Although optical experiments (photo electron spectroscopy or UV-VIS experiments) can be useful to probe the electronic properties of a fully functionalized graphene surface, scanning tunneling spectroscopy allows one the chance to study even single molecules and to investigate of the electronic properties of the sample with high accuracy and spatial resolution.
2. Experiments
STM measurements were carried out using a PicoLE STM (Agilent) equipped with low tunneling current STM scanner head (type: N9501-A) and digital instrument from Agilent Technologies model N960A. STM Tips were obtained by mechanically cutting 0.25 mm Pt0.8Ir0.2 wire (Goodfellow). The STM images were acquired in constant current operating mode, in air at room temperature. Typical scanning parameters to obtain STM images are in the range of 10 to 15 pA with the tip bias voltage ranging from −0.8 to −1.2 V. The STM images were treated by standard flattening procedure without further image processing treatment. The porphyrins were used without any further purification from Aldrich. A solution 1.63 mM of FePP in uvasol grade chloroform was prepared. Further, a droplet of 10 µL of the porphyrin solution was dropcasted onto CVD-graphene surface and the solvent was left to evaporate for 3h in open air condition. The same procedure was also applied for the deposition of ZnPP.
3. Results and Discussion
We have studied and used commercially available graphene/multi-layered graphene grown by chemical vapour deposition on polycrystalline nickel [25]. The as received samples have been first characterized by scanning tunnelling microscopy and spectroscopy. Figure 1 shows the STM topography, measured in constant current mode, of the surface of the CVD graphene on nickel, showing a characteristic rippled structure (a Moiré pattern) due to mismatching between the graphene and nickel lattices. The topographic line profile, measured along the black line in Figure 1(c), shows the graphene atomic corrugation superimposed to the Moiré pattern of graphene (Figure 1(d)). Iron porphyrins (FePP) are then deposited onto the CVD graphene surface by drop casting.
Figure 2(b) shows the STM topography of the CVD graphene surface after the deposition of FePP molecules. There are two main features associated with the presence of FePP molecules on the surface: small and chain-like protuberances (see Figure 2(c)). While chain-like structures present typical lengths ranging from 4.5 nm to 7 nm and 0.6 nm in height, higher resolution images reveals that the small protuberances have a star-like shape with
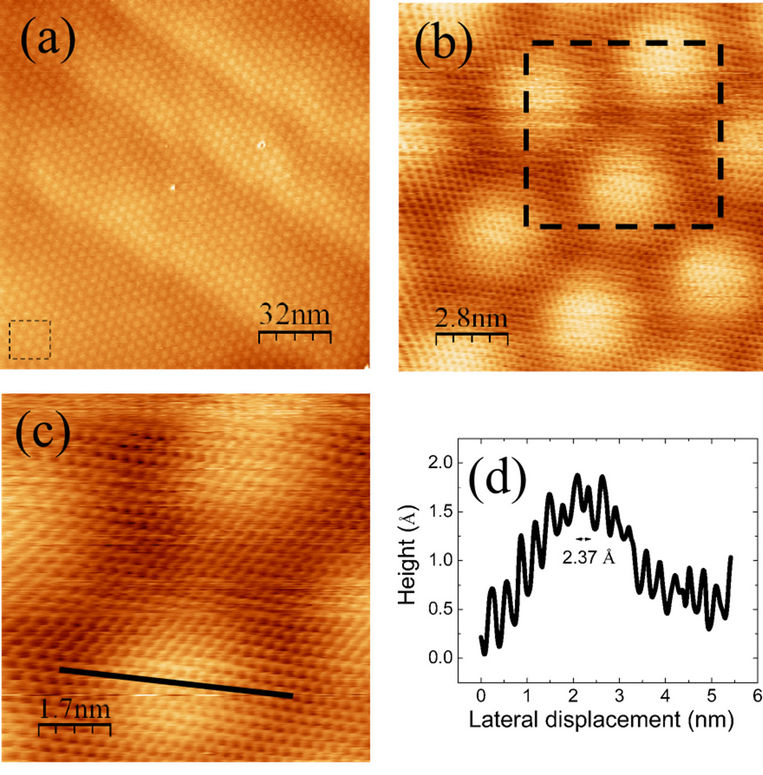
Figure 1. A representative STM image of pristine CVDgraphene probed in ambient condition; (a) 160 × 160 nm2 STM image covered with single layer of graphene (It = 0.1 nA; V = 0.5 V; Z-scale = 2.05 nm); (b) Super mesh of single layer graphene decorated with a hexagonal moiré feature throughout the entire surface area (It = 0.1 nA; V = 0.3 V; Z-scale = 5.01 Å); (c) Carbon atoms arranged in the honeycomb lattice is markedly the evidence of single layer graphene on nickel substrate (It = 0.1 nA; V = 0.3 V; Z-scale = 5.01 Å); (d) A corresponding cross section profile drawn from a solid black line in Figure 1(c).
0.26 nm in height and approximate dimension of 13 Å × 19 Å (Figure 2(d)). These lateral dimensions are in agreement with the dimensions of a single porphyrin molecule obtained by first principle calculations (16 Å × 16 Å) [26]. The resolved six-star shape, however, do not perfectly match to the expected geometrical structure of porphyrin molecules (four-star shape).
The additional features in the observed star shape can be due to the splitting of the local density of the state (LDOS) of one phenyl ring. Another interesting finding is that FePP molecules are preferable physisorbed at the valleys of the Moiré structure of graphene (Figure 2(c)). The origin of the chain-like structures is possibly driven by a noncovalent interaction created between the next neighboring FePP extended up to few nanometers. To evaluate further the effect of the molecule-substrate (MS) interaction, we present another example of self-assembled porphyrin on graphene using Zincporphyrin (ZnPP) molecules with a weaker interaction with the graphene surface [27]. The molecular structure is illustrated in Figure 2(e). We deposited the ZnPP in a similar way as the FePP. The ZnPP molecule is mainly constructed of a zinc metal at the central propyrins backbone which is a rather weak transition metal in comparison to Fe. Inter-
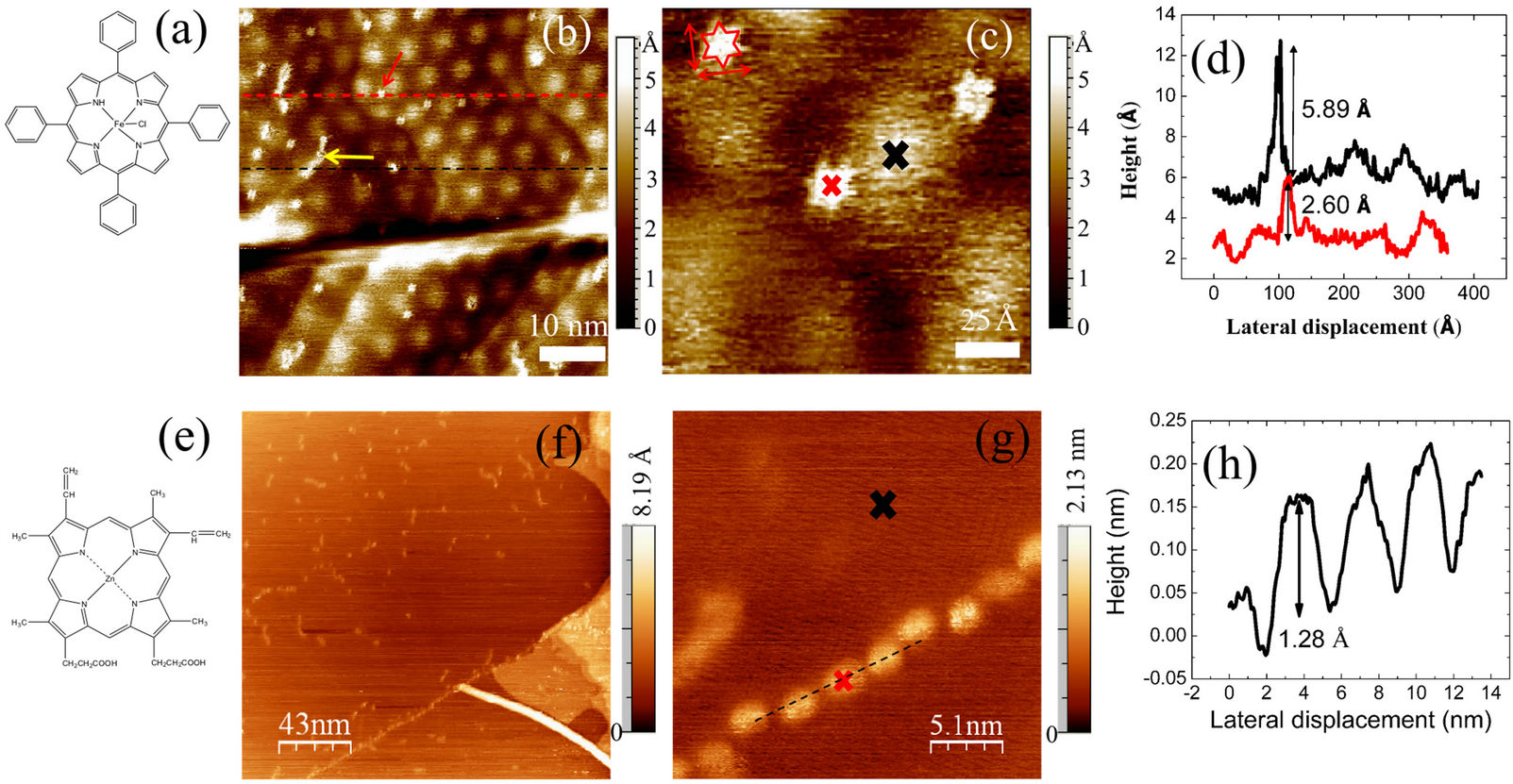
Figure 2. Single molecule detection of porphyrins on CVD-grown graphene; (a) 5,10,15,20-Tetraphenyl-21H,23H-porphine iron(III) chloride (abbreviated as FePP); (b) Porphyrin molecules decorating the terraces of single layer graphene (It = 0.1 nA; V = 0.5 V). FePP can be found at the valleys of the Moiré pattern of graphene; (c) Single porphyrin molecules with star-like shape were observed at the vicinity of hills of the rippled structure of graphene on Ni; (d) Topographic line profile alongthe black and red lines highlighted in Fig. (b); (e) 7,12-Diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropionic acid, zinc complex (abbreviated as ZnPP); (f) STM image of ZnPP molecules deposited on the surface of CVD graphene; (g) In high resolution, a chain-like structure of ZnPP is clearly observed; (h) Lateral profile is extracted from the black dashed line in Figure 2(g).
estingly, a chain-like structure was observed in the STM image (Figure 2(f)). The chain structures of ZnPP are slightly lower in height compare to the FePP and they can be much longer (Figure 2(h)). Petukhov and coworkers observed a similar shape in the manganese gridlike assembly molecules [28]. In this particular system, the molecular chain is governed by the coordination bond of the ligands which form a grid-like structure. Note that we could not resolve the Moiré pattern in graphene at the vicinity of the ZnPP as it is previously shown in FePP, indicating that the modification of the electronic properties induced by the physisorption of ZnPP is more extensive than that of FePP.
We expect that the two different structural arrangements of the molecules are related to the strength of the metallic centre of porphyrins. While FePP strongly interacts with graphene, resulting in the physisorption of localized individual molecules on the surface, the ZnPPgraphene interaction is rather weak and thus individual ZnPP molecules preferably interact among themselves and form large chain-like aggregates on graphene surface (Figure 2(f)). In addition to that, the absence of phenyl rings at the corners of ZnPP did not give additional molecular rigidity of porphryins backbone.
In order to study the effect of the deposition of porphyrin molecules on the electronic properties of graphene, we used a statistical analysis of tunneling spectroscopic measurements [29,30]. The STS measurements were carried out as follows: A total of 1000 tunneling current vs voltage traces (I-V traces) were measured at several spots on the sample by interrupting the feedback control loop during the measurements and sweeping the tip bias voltage between −1.5 V and 1.5 V (0.01 second per trace). The differential conductance vs. voltage (dI/dV vs V) was obtained by numerically differentiated the I-V traces using a moving average of 10 - 20 datapoints to smooth the resulting numerical derivative. Then the whole set of 1000 (I-V and dI/dV) vs V is plotted in a 2D histogram shape which shows the distribution of the dI/dV vs V of the sample. To construct these 2D histograms, both the bias voltage and the dI/dV axes are divided into a number of bins forming a N by N matrix (200 × 200 in our case). Each datapoint whose dI/dV and V values are within the interval of one bin, adds one count to it. The number of counts in each bin was then represented with a color scale.
After the deposition of FePP onto the graphene surface, we measured the I-V traces directly on top of six porphyrin molecules (one of them is indicated by the red cross in Figure 2(c). The tunneling differential conductance at low bias shows a strong reduction (Figure 3(c)) in comparison to pristine graphene (Figure 3(a)). In fact, the tunneling differential conductance traces present zero conductance for a wide range in bias, a characteristic of semiconducting behaviour while for pristine graphene the low bias differential conductance value is significantly larger than zero. Surprisingly, when this measurement is repeated on graphene several angstroms away from a single porphyrin molecule (at the topmost point of Moiré as indicated by the black cross in Figure 2(c) the characteristic I-V traces (Figure 3(d)) are different than that of pristine graphene (Figure 3(a)). The tunneling differential conductance at low bias is strongly reduced. Indeed, we also observed a significant number of traces with zero conductance, indicating the opening of a band gap in graphene mediated by the interaction with the vicinal porphryin molecule. This modification of the electronic properties of graphene seems to be due to the adsorption of porphyrins as regions far from the porphyrins show a conducting behavior similar to pristine graphene (Figure 3(b)).
Further, we analysed the tunneling differential conductance in the histogram bin close to zero conductance value (between 0 pS and 2.5 pS) to gain a deeper insight of the reduction of the differential tunneling conductance by the physisorption of porphyrins on graphene. Therefore, we extracted 1D histograms at zero differential conductance from the 2D histograms as presented in Figure 4.
The 1D histogram of modified graphene (close to a physisorbed molecule) shows a reasonable number of counts that are concentrated close to the zero bias. Moreover, by fitting the 1D histogram to a Gaussian curve we estimate a bandgap value of 0.45 eV. Interestingly, although the π-π interaction with graphene is rather weak this value is quite comparable to the band gap opened in CVD graphene by the chemisorption due to hydrogen plasma treatment [29]. The semiconducting behaviour observed directly on top of the porphyrin molecules by STS is more marked and the estimated energy gap is larger (0.70 eV) (Figure 4(d)). The 1D histograms obtained for pristine CVD graphene and for graphene far from the porphyrin molecules are similar and do not show a significant number of counts which demonstrates a fairly conducting behaviour. According to these measurements, the semiconducting behaviour observed on top of the FePP molecules physisorbed on graphene is extended to the adjacent graphene (up to 10 - 20 nm in distance) while graphene far away from the molecules remains fairly conducting (as the initial CVD graphene).
We also investigate the local electronic properties of the chain-like features formed by ZnPP molecules by means of the same statistical STS approach. Figures 5(a)

Figure 3. A comparison of the I-V and dI-dV graphene before and after FePP deposition; The data is presented as 2D histrograms, each one built from 1000 individual traces; (a) I-V of pristine CVD graphene measured on top of Moiré (we also observed a similar I-V shape of pristine graphene in the the valleys of Moiré); (b) The I-V of CVD graphene after FePP deposition acquired on top of hill of Moiré far from the physisorbed molecules; (c) The collected I-V traces acquired on top of the porphyrin molecule marked in Figure 2(c). (d) I-V of CVD graphene after FePP deposition taken on different hills of the Moiré within 10 - 20 nm in radius of a porphyrin molecule; (e)-(h) The corresponding differential conductances (obtained by numerical differentiation) are presented next to each corresponding measurements.
and (b) present the collected I-V traces on pristine graphene and graphene far from the physisorbed ZnPP,
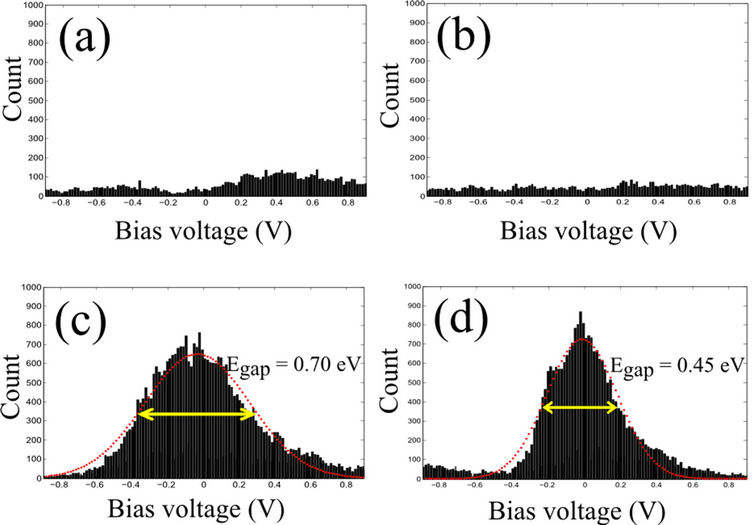
Figure 4. One dimensional histogram extracted from the respective black dashed lines in Figures 3(e)-(h); We considered the datapoints that fall in the bin with differential conductance; (a) and (b) 1D histograms for pristine CVD graphene and graphene after FePP deposition (far from the molecules) are featureless, showing a negligible amount of datapoints with zero differential conductance. The observed counts are due to the noise of the dI/dV traces; (c) and (d) 1D histograms for a single FePP molecule and for the graphene close to a FePP molecule which show a clear accumulation of datapoints with zero differential conductance at low bias. A Gaussian fit is used to estimate the band gap.
sharing an ohmic behaviour for low biases. In fact, the tunnelling differential conductance in Figures 5(e) and (f) shows that the minimum differential conductance value is significantly larger than zero. On the other hand, the I-V and the dI/dV spectra measured on top of the ZnPP chain structure (Figure 5(c)) and on a graphene region 10 - 20 nm close to the ZnPP molecules (Figure 5(d)) show a strong reduction on the differential conductance with a non-negligible presence of datapoints with zero differential conductance. This resembles again the semiconducting behaviour observed on top of the FePP molecules and on graphene close to FePP molecules.
In order to estimate the band gap opening of graphene, 1D histograms were extracted from the bin with zero dI/dV value in the 2D histograms as shown in Figure 6. Figure 6(d) shows that close to the chain of ZnPP molecules there is an accumulation of datapoints with zero differential conductance for low bias voltage, in agreement with an opening of a band gap with a value of 0.23 eV. The resulted band gap value after ZnPP deposition is almost a factor of two lower than that of FePP. This is in agreement with the fact that the semiconducting behavior measured directly on top of the chains of ZnPP molecules was less marked than that measured on top of the FePP molecules (Figure 6(c)). In fact, the 1D histograms extracted from STS measurements on top of the ZnPP molecules show an accumulation of datapoints with zero differential conductance for a range of around 0.37 mV of bias voltage. For pristine graphene (Figure 6(a)) and
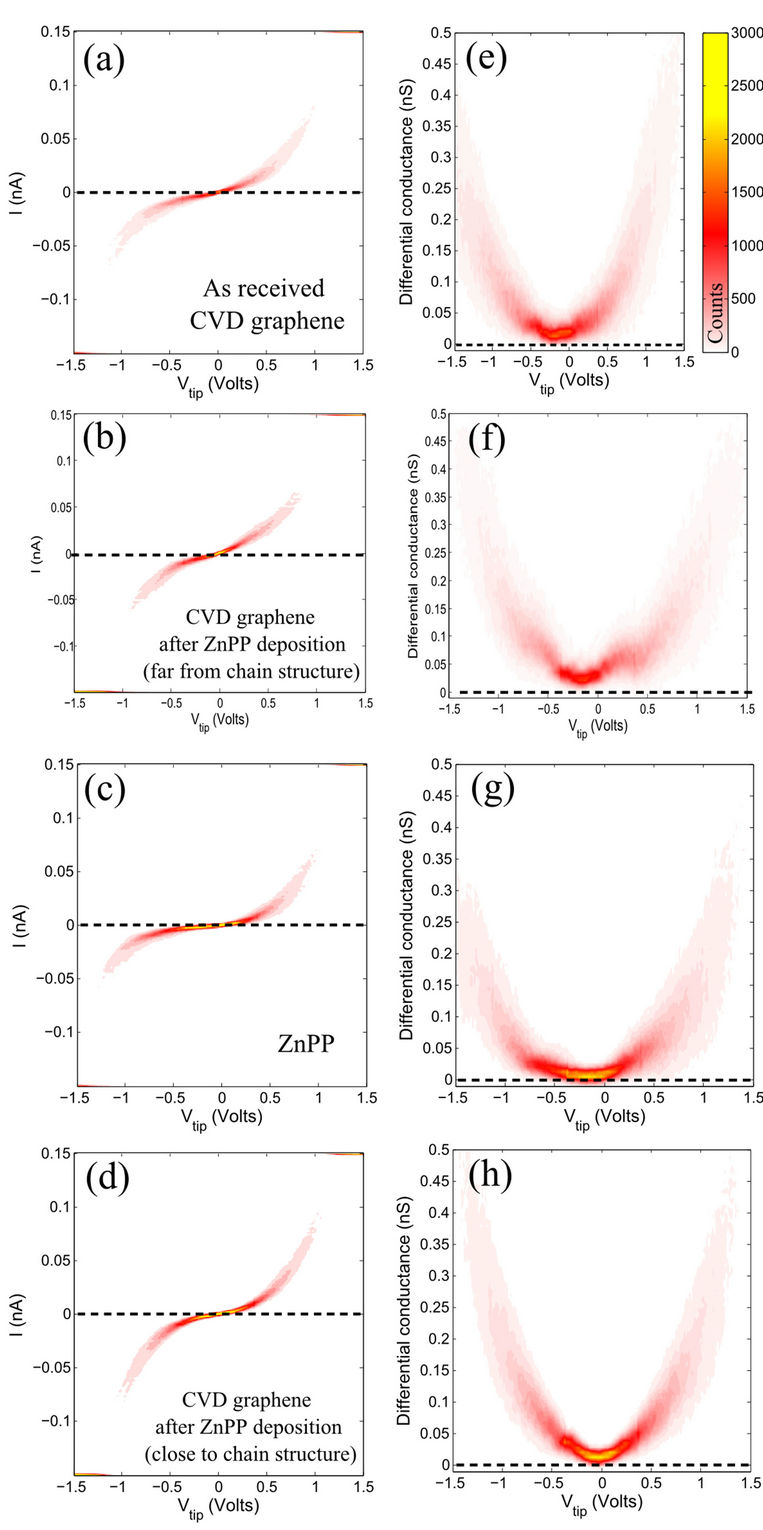
Figure 5. A comparison of the I-V and dI-dV graphene before and after ZnPP deposition. The data is presented as 2D histrograms, each one built from 1000 individual traces; (a) I-V of pristine CVD graphene; (b) The I-V of CVD graphene after ZnPP deposition acquired on top of hill of Moiré far from the physisorbed molecules; (c) The collected I-V traces acquired on top of the porphyrin chain-like structure marked in Figure 2(g); (d) I-V of CVD graphene after ZnPP deposition taken on different hills of the Moiré within 10 - 20 nm in radius of a porphyrin molecule; (e)-(h) The corresponding differential conductance (obtained by numerical differentiation) is presented next to each corresponding measurements.
graphene far from the ZnPP molecules (Figure 6(b)) the 1D histograms do not show any clear accumulation of datapoints with zero differential conductance, compatible
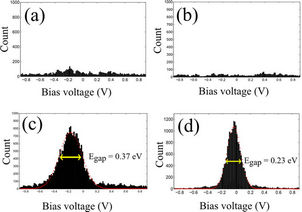
Figure 6. One dimensional histogram extracted from the respective black dashed lines in Figures 5(e)-(h); (a) and (b) 1D histograms for pristine CVD graphene and graphene after ZnPP deposition (far from the molecules) are featureless, showing a negligible amount of datapoints with zero differential conductance. The observed counts are due to the noise of the dI/dV traces; (c) and (d) 1D histograms on top of the chain-like ZnPP structure and for the graphene close to the chain of molecules which show a clear accumulation of datapoints with zero differential conductance at low bias. A Gaussian fit is used to estimate the band gap.
with a metallic behaviour. In addition, it is unlikely that the Coulomb repulsion plays role in these types of molecules based on the theoretical calculation carried out by Panchmatia and coworkers [31].
In general, the STS spectra show that the tunneling probability is very small in certain bias regimes for tunneling into the porphyrin molecule. The origin of this observation might be due to the existence of very localized states of Fe and/or Zn in this bias regime which prevents a tunneling event due to a Coulomb repulsion. However, this is not unlikely for these type of molecules based on electronic structure calculations by Panchmatia et al. [31]. Indeed our finding is in agreement with this calculation, if the molecule would act as a Coulomb island, the change in the electronic properties would be extremely localized in the surface and it is clearly not the case (fairly away from the molecule the dI/dVs show still a gap).
These results indicate that porphyrin molecules physisorbed on top of CVD graphene show a marked semiconducting behaviour which is extended to the vicinal graphene while the graphene far from the porphyrins remains fairly conducting. Moreover, the magnitude of the opened band gap depends on the strength of the molecule-graphene interaction which can be engineered by a proper selection of the porphyrin metal core [32-34]. The effect which is presented in our work is very local. Therefore, the transport measurements would require a fully covered graphene to observe any bandgap opening. This type of approach requires a different kind of sample compare to our findings. We believe that this is a first step towards this other proposed experiment.
4. Conclusions
In conclusion, we investigated the opening of a band gap in graphene by physisorption of porphyrin molecules. We have performed a statistical analysis of the tunneling spectra that allow to monitoring the opening a gap of graphene via adsorption of different porphyrin molecules (FePP and ZnPP).
We find that the presence of porphyrin molecules physisorbed on the graphene substrate induces a semiconducting behaviour in the surrounding graphene layer up to 10 - 20 nm far from the molecules. Interestingly, the magnitude of the bandgap opened in graphene can be controlled via the selection of the metal core of the porphyrin molecules that determines the graphene-molecule interaction. We believe that this work will open more opportunities to build other hybrid systems based on the noncovalent π-π stacking of aromatic molecules on graphene.
5. Acknowledgements
We thank to T. C. Pijper for DFT calculations. Technical assistance from S. Bakker, J. G. Holstein, and B. H. J. Wolfs is thankfully acknowledged. Financial support from Fundamental Research on Matter (FOM) program G-08 is also acknowledged and supported by the Zernike Institute for Advanced Materials.
REFERENCES
- J. Wintterlin and M. L. Bocquet, “Graphene on Metal Surfaces,” Surface Science, Vol. 603, No. 10-12, 2009, pp. 1841-1852. doi:10.1016/j.susc.2008.08.037
- X. S. Li, et al., “Large-Area Synthesis of High-Quality and Uniform Graphene Films on Copper Foils,” Science, Vol. 324, No. 5932, 2009, pp. 1312-1314. doi:10.1126/science.1171245
- C. Berger, et al., “Ultrathin Epitaxial Graphite: 2D Electron Gas Properties and a Route toward Graphene-Based Nanoelectronics,” The Journal of Physical Chemistry B, Vol. 108, No. 52, 2004, pp. 19912-19916. doi:10.1021/jp040650f
- C. H. Lui, L. Liu, K. F. Mak, G. W. Flynn and T. F. Heinz, “Ultraflat Graphene,” Nature, Vol. 462, No. 7271, 2009, pp. 339-341. doi:10.1038/nature08569
- D. Martoccia, et al., “Graphene on Ru(0001): A 25 × 25 Supercell,” Physical Review Letters, Vol. 101, No. 12, 2008, p. 126102. doi:10.1103/PhysRevLett.101.126102
- J. Coraux, A. T. N’Diaye, C. Busse and T. Michely, “Structural Coherency of Graphene on Ir(111),” Nano Letters, Vol. 8, No. 2, 2008, pp. 565-570. doi:10.1021/nl0728874
- O. C. Compton and S. T. Nguyen, “Graphene Oxide, Highly Reduced Graphene Oxide, and Graphene: Versatile Building Blocks for Carbon-Based Materials,” Small, Vol. 6, No. 6, 2010, pp. 711-723. doi:10.1002/smll.200901934
- J. Lin, et al., “Gating of Single-Layer Graphene with Single-Stranded Deoxyribonucleic Acids,” Small, Vol. 6, No. 10, 2010, pp. 1150-1155. doi:10.1002/smll.200902379
- X. C. Dong, et al., “Symmetry Breaking of Graphene Monolayers by Molecular Decoration,” Physical Review Letters, Vol. 102, No. 13, 2009, p. 135501. doi:10.1103/PhysRevLett.102.135501
- D. Elias, et al., “Control of Graphene’s Properties by Reversible Hydrogenation: Evidence for Graphane,” Science, Vol. 323, No. 5914, 2009, pp. 610-613. doi:10.1126/science.1167130
- R. Balog, et al., “Bandgap Opening in Graphene Induced by Patterned Hydrogen Adsorption,” Nature Material, Vol. 9, 2010, pp. 315-319. doi:10.1038/nmat2710
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, “Controlling the Electronic Structure of Bilayer Graphene,” Science, Vol. 313, No. 5789, 2006, pp. 951- 954. doi:10.1126/science.1130681
- Y.W. Son, M. L. Cohen and S. G. Louie, “Half-Metallic Graphene Nanoribbons,” Nature, Vol. 444, No. 7117, 2006, pp. 347-349. doi:10.1038/nature05180
- E. Rudberg, P. Salek and Y. Luo, “Nonlocal Exchange Interaction Removes Half-Metallicity in Graphene Nanoribbons,” Nano Letters, Vol. 7, No. 8, 2007, pp. 2211- 2213. doi:10.1021/nl070593c
- Y. B. Zhang, et al., “Direct Observation of a Widely Tunable Bandgap in Bilayer Graphene,” Nature, Vol. 459, No. 7248, 2009, pp. 820-823. doi:10.1038/nature08105
- S. Y. Zhou, D. A. Siegel, A. V. Fedorov and A. Lanzara, “Metal to Insulator Transition in Epitaxial Graphene Induced by Molecular Doping,” Physical Review Letters, Vol. 101, No. 8, 2008, p. 086402. doi:10.1103/PhysRevLett.101.086402
- Z. Zhang, H. Huang, X. Yang and L. Zang, “Tailoring Electronic Properties of Graphene by π-π Stacking with Aromatic Molecules,” The Journal of Physical Chemistry Letters, Vol. 2, No. 22, 2011, pp. 2897-2905. doi:10.1021/jz201273r
- X. R. Wang, et al., “N-Doping of Graphene through Electrothermal Reactions with Ammonia,” Science, Vol. 324, No. 5928, 2009, pp. 768-771. doi:10.1126/science.1170335
- X. C. Dong, D. L. Fu, W. J. Fang, Y. M. Shi, P. Chen and L. J. Li, “Doping Single-Layer Graphene with Aromatic Molecules,” Small, Vol. 5, No. 12 , 2009, pp. 1422-1426. doi:10.1126/science.1170335
- Y. H. Zhang, K. G. Zhou, K. F. Xie, J. Zeng, H. L. Zhang and Y. Peng, “Tuning the Electronic Structure and Transport Properties of Graphene by Noncovalent Functionalization: Effects of Organic Donor, Acceptor And Metal Atoms,” Nanotechnology, Vol. 21, No. 6, 2010, p. 065201. doi:10.1088/0957-4484/21/6/065201
- J. Otsuki, “STM Studies on Porphyrins,” Coordination Chemistry Reviews, Vol. 254, No. 19-20, 2010, pp. 2311- 2341. doi:10.1016/j.ccr.2009.12.038
- N. J. Tao, G. Cardenas, F. Cunha and Z. Shi, “In Situ STM and AFM Study of Protoporphyrin and Iron(III) and Zinc(II) Protoporphyrins Adsorbed on Graphite in Aqueous Solutions,” Langmuir, Vol. 11, No. 11, 1995, pp. 4445-4448. doi:10.1021/la00011a043
- T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, “Selective Assembly on a Surface of Supramolecular Aggregates with Controlled Size and Shape,” Nature, Vol. 413, No. 6856, 2001, pp. 619-621. doi:10.1038/35098059
- N. Wintjes, et al., “A Supramolecular Multiposition Rotary Device,” Angewandte Chemie International Edition, Vol 46, No. 22, 2007, pp. 4089-4092. doi:10.1002/anie.200700285
- www.graphene-supermarket.com
- Hyperchem Version 6.0.
- P. Gargiani, M. Angelucci, C. Mariani and M. G. Betti, “Metal-Phthalocyanine Chains on the Au(110) Surface: Interaction States versus d-Metal States Occupancy,” Physical Review B, Vol. 81, No. 8, 2010, p. 085412. doi:10.1103/PhysRevB.81.085412
- K. Petukhov, et al., “STM Spectroscopy of Magnetic Molecules,” Coordination Chemistry Review, Vol. 253, No. 19-20, 2009, pp. 2387-2398. doi:10.1016/j.ccr.2009.01.024
- M. Wojtaszek, N. Tombros, A. Caretta, P. H. M. van Loosdrecht and B. J. van Wees, “A Road to Hydrogenating Graphene by a Reactive Ion Etching Plasma,” Journal Applied Physics, Vol. 110, No. 6, 2011, p. 063715. doi:10.1063/1.3638696
- A. Castellanos-Gomez, M. Wojtaszek, Arramel, N. Tombros and B. J. van Wees, “Reversible Hydrogenation and Bandgap Opening of Graphene and Graphite Surfaces Probed by Scanning Tunneling Spectroscopy,” Small, Vol. 8, No. 10, 2012, pp. 1607-1613. doi:10.1002/smll.201101908
- P. M. Panchmatia, B. Sanyal and P. M. Oppeneer, “GGA + U Modeling of Structural, Electronic, and Magnetic Properties of Iron Porphyrin-Type Molecules,” Chemical Physics, Vol. 343, No. 1, 2008, pp. 47-60. doi:10.1016/j.chemphys.2007.10.030
- W. Dou, S. Huang, R. Q. Zhang and C. S. Lee, “Molecule-Substrate Interaction Channels of Metal-Phthalocyanines on Graphene on Ni(111) Surface,” Journal of Chemical Physics, Vol. 134, No. 9, 2011, p. 094705. doi:10.1063/1.3561398
- H. Vázquez, W. Gao, F. Flores and A. Kahn, “Energy Level Alignment at Organic Heterojunctions: Role of the Charge Neutrality Level,” Physical Review B, Vol. 71, No. 4, 2005, p. 041306(R). doi:10.1103/PhysRevB.71.041306
- F. Evangelista, et al., “Electronic States of CuPc Chains on the Au(110) Surface,” Journal Chemical Physics, Vol. 131, No. 17, 2009, p. 174710. doi:10.1063/1.3257606
NOTES
*Corresponding author.