Modern Research in Inflammation
Vol.3 No.1(2014), Article ID:42751,12 pages DOI:10.4236/mri.2014.31002
Autoinflammatory diseases in childhood
![]()
1Department of Pediatric Rheumatology, Medical Faculty, Ege University, Izmır, Turkey; *Corresponding Author: betulsozeri@yahoo.com
2Department of Pediatric Rheumatology, Cerrahpasa Medical Faculty, Istanbul University, Istanbul, Turkey
Copyright © 2014 Betul Sozeri1, Ozgur Kasapcopur. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Betul Sozeri1, Ozgur Kasapcopur. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 20 November 2013; revised 20 December 2013; accepted 28 December 2013
KEYWORDS
Autoinflammatory Diseases; Familial Mediterranean Fever; Diagnosis; Pathogenesis; Treatment
ABSTRACT
Autoinflammatory diseases are defined as recurrent attacks of systemic inflammation that are often unprovoked (or triggered by a minor event) related to a lack of adequate regulation of the innate immune system. Within the past decade, the list of autoinflammatory diseases has included cryopyrin-associated periodic syndromes, familial Mediterranean fever, mevalonate kinase deficiency, tumor necrosis factor receptor-associated periodic syndrome, hereditary pyogenic disorders, pediatric granulomatous autoinflammatory diseases, idiopathic febrile syndromes (systemic-onset juvenile idiopathic arthritis, PFAPA syndrome), complement dysregulation syndromes and Behçet’s disease. The hereditary autoinflammatory diseases are a group of Mendelian disorders characterized by seemingly unprovoked fever and localized inflammation. Autoinflammatory diseases can activate NOD-like receptors and inflammasome products including especially interleukin 1β. In this review, it focuses on how recent advances have impacted hereditary autoinflammatory diseases.
1. INTRODUCTION
The term autoinflammatory diseases (AID) describes a group of inherited disorders that are characterized by exaggerated innate immune responses leading to recurrent episodes of fever and inflammation that affects multiple organs including the skin, serosal membranes, joints, gastrointestinal tube, central nervous system, etc.
Most AIDs typically manifest in childhood, although a limited number of patients may experience disease onset during adulthood. The autoinflammatory diseases has included cryopyrin-associated periodic syndromes, familial Mediterranean fever, mevalonate kinase deficiency, tumor necrosis factor receptor-associated periodic syndrome, hereditary pyogenic disorders, pediatric granulomatous autoinflammatory diseases, idiopathic febrile syndromes (systemic-onset juvenile idiopathic arthritis, PFAPA syndrome), complement dysregulation syndromes and Behçet’s disease.
The aim of this review is to focus on how recent advances have impacted diagnosis, pathogenesis, and treatment of hereditary autoinflammatory diseases
2. PATHOGENESIS
These conditions can be distinguished from autoimmune disorders by the absence of autoantibodies or antigen specific T cells. The disease mechanisms in AIDs involve innate immune regulation of cytokines and neutrophilic inflammation [1]. IL-1β is a potent pro-inflammatory cytokine, which is synthesized early in response to infection and tissue injury by cells of the innate immune system [2]. (Figure 1).
The cells of the innate immune system, primarily epithelial, dendritic, polymorphonuclear, and macrophage cells, act not only as an immediate barrier, but also as effectors in the evolution of the inflammatory response. The innate immune system recognizes pathogen-associated molecular patterns and damage associated molecular patterns by several complex mechanisms, involving cell associated pattern recognition receptors and soluble recognition molecules. Nucleotide oligomerization domain—like receptors (NLRs) within the cell and other
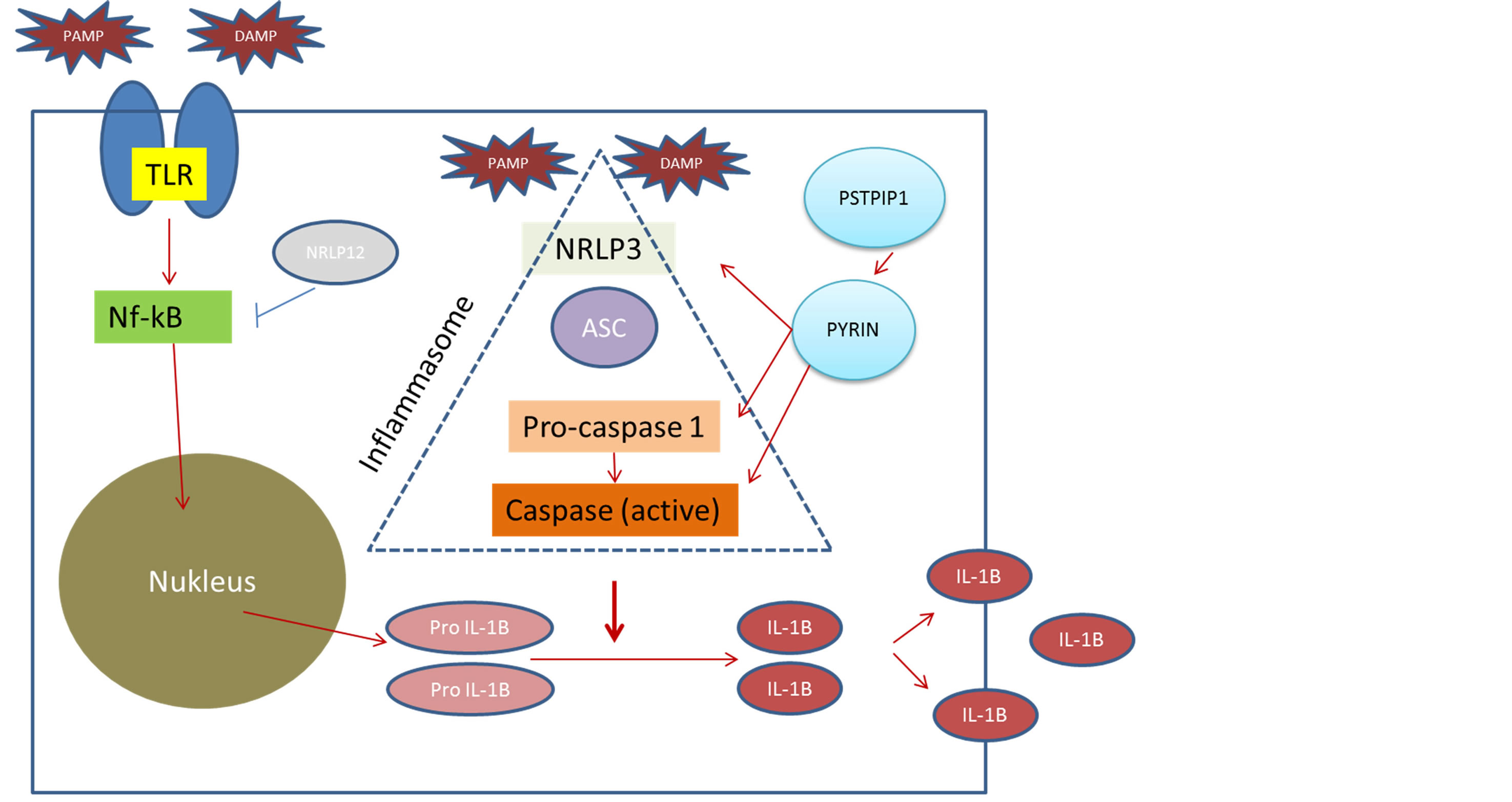
Figure 1. Inflammatory pathways related to the autoinflammatory diseases. DAMP, damage-associated molecular patterns; IL, interleukin; NF-kB, nuclear factor-kB; NLR, nucleotide oligomerization domain-like receptors; NLRPs, NLRs with pyrin-domaincontaining proteins; PAMP, pathogen-associated molecular patterns; PSTPIP, praline-serine-hreonine phosphatase-interacting protein; TLR, toll-like receptor.
receptors further mediate intracellular innate immune system processes and development of the inflammatory response. NLRPs (NLRs with pyrindo-main—containing proteins) are a subfamily of the NLRs. NLRP3 assembles other proteins to form the inflammasome complex in response to cytoplasmic pathogen-associated molecular patterns and damage-associated molecular patterns, which also triggers the expression of proinflammatory genes by transcription factors (eg, nuclear factor-kB). The inflammasome complex involving NLRP3 recruits and activates caspase 1, a protease that cleaves pro-interleukin (IL)-1β and IL-18 to their active forms. The common pathogenic pathway of the autoinflammatory syndromes involves the excessive production and activity of these proinflammatory cytokines and molecules, not as a result of external stimuli but as a result of mutations in different proteins that regulate these pathways [1,3].
2.1. Familial Mediterranean Fever (FMF)
Familial Mediterranean fever is an autosomal recessive disease characterized by recurring self-limited short episodes of fever and serositis, resulting in pain in the abdomen, chest, joints and muscles. It is the most common of the periodic hereditary fevers. This mainly affects Middle Eastern populations and other ethnic groups living around the Mediterranean basin, such as Jews, Armenians, Turks, Arabs, with high prevalence (1/200- 1/1000); also, it is not considered rare in Italy, Spain and Greece [4-6]. Almost 60% of patients have a disease onset before 5 years and almost all patients before the second decade of life [6,7].
Clinical presentation:
The disease typically presents with recurrent episodes of fever, associated with acute abdominal pain and large joint arthritis that last 1 to 3 days [8. Generally, a typical attack lasts between 12 and 72 hours, and raises a peak within 12 hours of onset. The interval between attacks is variable from few weeks to months or years. The attack may be triggered by common factors such as cold exposure, emotional or physical stress, infections or menstruation [9].
Recurrent fever is characterized by temperature from 38˚C to 40˚C, it can partially respond to antipyretics or steroids administration, while antibiotics do not have any effect. In course of fever, laboratory exams can typically show a neutrophilic leukocytosis, together with the increase of inflammation indexes, such as erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), serum amyloid-A (SAA) and fibrinogen, which disappear in inter-current well-being periods, except in patients with persistent subclinical inflammation [10].
Abdominal pain: These are experienced by 90% of affected individuals and start with the sudden onset of fever and pain affecting the entire abdomen [1]. Physical examination reveals board-like rigidity of the abdominal muscles, rebound tenderness, abdominal distension, and loss of peristaltic sounds. Radiographs reveal multiple small air-fluid levels in the small bowel. Joint involvement: Large joints may be affected by arthritis or arthralgia in more than 50% of patients (chiefly knees, hips and ankles). These occur suddenly, and may be precipitated by minor trauma or effort, such as prolonged walking. Gradual resolution of the signs and symptoms are after peaking in 24 - 48 hours. The attacks are commonly in the hip or knee but may occur in other joints such as the ankle, shoulder, temporomandibular joint, or sternoclavicular joint. The joint remains swollen and painful, as in chronic monoarthritis [11,12].
Chest attacks: Pleuritis is experienced by approximately 45% of patients with FMF and are the sudden onset of an acute, one-sided febrile pleuritis, which resolves within 48 hours. Pericarditis is a rare occurrence. It is characterized by retrosternal pain [13]. Other clinical manifestations of FMF include protracted febrile myalgia, which generally responds to steroid therapy [14], and also erysipelas-like erythema, aseptic meningitis (also known as Mollaret syndrome) and vasculitides (HenochSchönlein purpura and polyarteritis nodosa) entities that are generally present in ≤5% of patients [15].
Tel Hashomer criteria (Table 1) are often used to make the diagnosis [16]. Yalçinkaya and colleagues recently validated the sensitivity and specificity of Tel Hashomer criteria in 170 FMF children [17]. The results were compared with sensitivity and specificity of a proposed new set of 5 criteria for the FMF diagnosis in childhood, including fever (axillary temperature ≥ 38˚C), abdominal pain, chest pain, arthritis (for all the conditions the number of the attacks has to be ≥3, with a 6 - 72 hours of duration), and family history of FMF. The presence of 2 of these 5 criteria resulted to have a higher specificity compared to that of Tel Hashomer criteria (93.6% versus 54.6%, respectively) (Table 2) [17]. The validation of the Yalçinkaya criteria in a French population of children using an appropriate control group did not show a better contribution to FMF than the Tel Hashomer criteria [18] while a recent evaluation of sensitivity of Yalçinkaya and Tel Hashomer criteria in 110 Turkish children affected found values of 93% and 100% respectively, indicating a also high sensitivity of the new set of criteria in patients with a mutation at a single allele [19].
Genetic diagnosis:
The Mediterranean fever gene (MEFV) which is composed of 10 exons, located on 16p13 a 781-amino-acid protein termed pyrin [20,21]. The major role of pyrin is the regulation of caspase-1 activation. The inflammatory phenotypes of FMF are induced by IL-1β and NF-Kβ, which are abnormally activated by FMFassociated mutations of pyrin [22]. Mutations in MEFV may result in less active pyrin [3]. Nowadays, 218 MEFV mutations have been identified for the phenotypic variance seen in the disease [23]. M694V, V726A, M680I, M694I (con-

Table 1. Tel-Hashomer criteria for the diagnosis of familial Mediterranean fever [19].
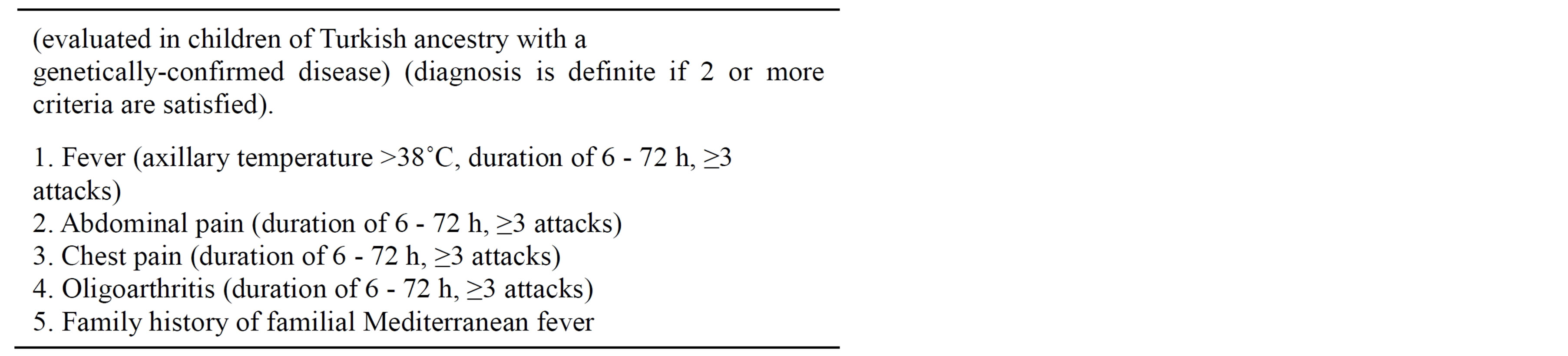
Table 2. Yalcinkaya set of criteria for the diagnosis of familial Mediterranean fever in childhood [20].
servative mutations clustered in exon 10) and E148Q (clustered in exon 2) are considered as common mutations related to FMF [24-26]. Mattit et al. [27] tested for these mutations in 83 unrelated patients who fulfilled the international FMF criteria and 242 unrelated apparently healthy controls. Among the 83 patients, 30.1% were homozygotes, 39.8% compound heterozygotes, 19.3% heterozygotes, and 10.8% had no identifiable mutation.
M694V is more commonly seen among Sephardic Jews, Turks, and Armenians; E148Q among European and Turks patients; M694I is more frequent among Arabs; M680I is detected particularly among Armenians [27-31].
The carrier rate for FMF has been calculated to be as high as 1:3 - 1:7 in North African Jews, Iraqi Jews, Armenians, and Turks [29-32].
Studies have cast considerable doubt on whether FMF is, in fact, a traditional autosomal recessive disease. Booty et al. [33] performed an extensive search for a second MEFV mutation in 46 patients diagnosed clinically as having FMF and carrying only one high-penetrance FMF mutation. They did not identify a second MEFV mutation in any of the patients screened, and haplotype analysis did not identify a common haplotype that might be associated with the transmission of a second FMF allele. They also found no significant difference in pyrin levels between patients with a single mutation and those with a double mutation. The authors concluded that there exists a significant subset of patients with FMF who have only one MEFV mutation and that in such patients detection of a single mutation seems to be sufficient in the presence of clinical symptoms for the diagnosis of FMF and the initiation of a trial of colchicine.
In the literature, vary authors discussed possible explanations as to how a person carrying only one MEFV mutation can present with the clinical manifestations [33-35]: one of these explanation is the presence of less common mutations missed by routine testing, second one is digenic inheritance (the interaction of two genes resulting in the expression of a phenotype) is known to occur with autoinflammatory diseases. The third possibility is the epigenetics which changes in gene expression that do not involve changes in the underlying DNA sequence.
Complications:
The most important long-term complication is progressive systemic type AA amyloidosis [36,37]. AA amyloidosis is caused by the extra-cellular deposition of amyloid fibrils, which culminates in multi-organ dysfunction, particularly of the kidneys.
Treatment:
Colchicine which is a tricyclic alkaloid extracted from two plants of the lily family is the principal therapy in FMF [38,39]. Colchicine reduces attack frequency, decreases severity and shortens duration of the acute attacks in most FMF patients [38,39]. Colchicine inhibits microtubule polymerization by binding to tubulin, marked effects are exerted on leukocytes and, as recently demonstrated, on NACHT-LRRPYDcontaining protein 3 (NALP3; cryopyrin) activity in macrophages [40,41]. Also, colchicine modulates the expression of pyrin and interacts in the cytosol. Its most effective results have been obtained in the prophylaxis of FMF, while the administration during the attacks is ineffective; during the attack a non-steroidal anti-inflammatory drug can be administered [42]. The dose of colchicine in adults is 1 mg/day. In children, dosage should be arranged according to age: The oral starting doses should be: ≤0.5 mg/day (for children <5 years of age); 1.0 mg/day (for children 5 - 10 years of age); above the age of 10 years, patients can take more than 1.0 mg colchicine daily [39,43]. Colchicine dosage should be increased in a stepwise fashion (e.g. 0.25 mg/step) up to a maximum of 2.0 mg/day in order to control the disease in patients who do not clinically respond to the standard dosage (therapy).
Colchicine often leads to gastrointestinal side effects. Padeh et al. [44] reported the most common side effect is diarrhea and appear the initiation of treatment. Also, colchicine can enhance B12 malabsorption and in rare cases can cause alopecia and bone marrow suppression. Macrolides, diltiazem, grapefruit and cyclosporine should not be taken with colchicine as fatal toxicity can occur [45].
Colchicine prevents febrile attacks in more than 60% of patients and significantly reduces the number of attacks in another 20% - 30%. Five to ten percent of patients do not respond to therapy, but most of them are non-compliant [4,39].
The patients who had either incomplete disease control on colchicine alone, or were unable to use colchicine due to side effects, or had FMF in association with vasculitis who can be treated with several agents: Corticosteroids, Interferon alpha, TNF blocking agents, IL-1 blocking agents (Anakinra-canakinumab) [39,46]
2.1.1. Mevalonate Kinase Deficiency
Periodic fever associated with mevalonate kinase deficiency (MKD) was originally identified in 1984 in six patients of Dutch ancestry with a long history of recurrent attacks of fever of unknown cause and a high serum Ig D level by Jos van der Meer in 1985 [47]. This is a metabolic disorder resulting from insufficient activity of the enzyme mevalonate kinase, coded by the MVK gene involved in cholesterol and isoprene biosynthesis. Mevalonate kinase activity is usually reduced to 5% - 10% of normal with excessive accumulation of mevalonic acid [48]. Increased mevalonic acid or isoprenoid end products cause IL-1 overproduction through the activation of the inflammasome [49,50].
The disease usually starts in infancy or early childhood: typical flares are irregular, last 3 - 7 days, have an abrupt onset and can be induced by vaccinations, infections and menses, being staggered by asymptomatic periods of several weeks [51]. Severe abdominal pain often accompanied by vomiting and/or diarrhea is the most frequent manifestation associated with fever attacks. Irritability and cervical lymphadenopathy are common features too and splenomegaly may be found in about half of patients during acute episodes. Marked lymph node enlargement and splenomegaly help to distinguish clinically MKD from FMF [52]. Mucocutaneous manifestations are frequent and include erythematosus macules. Neutrophilia and elevated acute phase reactants are present during fever attacks. Increased plasma levels of IgD (>100 UI/ml) during fever episodes and in basal conditions have been considered in the past as a hallmark of the disease. However, the specificity of this finding is low [53,54]. The polyclonal elevation of serum IgD is found mostly in patients older than 3 years, but this is not exclusive for MKD, while in 20% of patients there is no increase of serum IgD [10]. A clarifying clue to the diagnosis is the increased urinary excretion of mevalonic acid during febrile flares [54]. Amyloidosis is rare seen in MKD (only in 2.9% of cases) [55].
MKD is an autosomal recessive disease. So far more than 130 substitutions or deletions of the MVK gene have been reported [56] (http://fmf.igh.cnrs.fr/infevers/). The most common mutation in MVK gene is the V377I variant, which is exclusively associated with the mild phenotype of MKD with some residual MVK activity [57]. Some variants (i.e., V310M, A334T) are closely associated with a severe MA phenotype and severely impaired cellular MVK activity [58].
Fever attacks usually respond dramatically to the administration of steroids (prednisone: 1 mg/kg/day in a single dose or with a short course of 3 to 5 days). Different reports have shown the potential benefit of anti-tumor necrosis factor therapy with etanercept (0.8 mg/kg/week by subcutaneous injection) and anti-IL-1 therapy with anakinra (subcutaneously administered at the daily dosage of 1 mg/kg/day) [59,60]. However, due to the high frequency of the fever episodes, some patients may need almost continuous treatment.
2.1.2. Tumor Necrosis Factor Receptor-Associated Periodic Fever Syndrome
Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) exhibits autosomal dominant inheritance pattern and long-lasting fever episodes. TRAPS is caused by missense mutations in the p55 TNF receptor (or TNFR1A), encoded by the TNF super family receptor 1A gene [61] (TNFRSF1A). A total of 114 sequence variants of the TNFRSF1A have been recorded so far; of which 75 are associated with a TRAPS phenotype [56] (http://fmf.igh.cnrs.fr/infevers/). There are an increasing number of studies evaluating the role that specific TNFRSF1A mutations might have in the establishment of a constitutive inflammation through the secretion of proinflammatory cytokines as IL-1, interleukin-12 and interleukin-8 [62]. The status of two sequences, R92Q and P46L, has not been fully determined [63-67] These mutations are frequently seen in normal controls (up to 9% of the population), do not alter the protein structure, and may represent milder mutations of low penetrance [3,67].
The age of onset can be variable (from infancy to over 50 years) and the clinical picture is characterized by febrile episodes lasting 3 - 4 weeks, even though attacks shorter than 5 days have been reported, recurring at least 2 - 6 times each year, combined with abdominal pain, diarrhea, arthralgia, localized myalgia and variable migratory skin manifestations [10,66]. Typical are centrifugal muscle edema with chronic fasciitis and the characteristic periorbital edema with painful conjunctivitis [68].
In the most severe forms of TRAPS, clinical signs of inflammation are almost permanent and require daily use of corticosteroids, leading to dependence and requiring the use of other anti-inflammatory drugs. Colchicine does not seem to prevent recurrences of TRAPS attacks. Etanercept and other TNF inhibitors have provided various degrees of clinical improvement and allowed savings of steroids in many cases [69-71]. The IL-1 receptor antagonist anakinra has also shown substantial clinical benefit in TRAPS patients and prevented disease relapses at the dose of 1.5 mg/kg/day by subcutaneous injection for a period of 15 days after the attack onset [72].
2.1.3. Cryopyrin-Associated Periodic Syndromes
Three autosomal-dominant syndromes constitute cryopyrin-associated periodic syndromes (CAPS): (1) familial cold autoinflammatory syndrome (FCAS), (2) MuckleWells syndrome (MWS), and (3) neonatal-onset multisystem inflammatory disease (NOMID). They are caused by single base mutations on the NLRP3 gene (NOD-like receptor 3, also known as cold-induced autoinflammatory syndrome 1, CIAS1) located on the long arm of chromosome 1 encoding the protein cryopyrin [73,74]. Mutations of the NLRP3 gene are found in almost 70% of patients with a CAPS phenotype. No NLRP3 mutations are found in about 50% of patients with NOMID and 25% to 33% of patients with MWS; these patients have a similar phenotype and response to treatment as mutation-positive patients [75]. Mutations lead to cryopyrins with gain-of-function, which cause a constitutive activation of the inflammasome and IL-1 overproduction [76]. FCAS and MWS are familiar, while cases of NOMID are sporadic.
FCAS is characterized by urticarial rash and fever spikes of short duration (usually <24 h) induced by cold exposure. Arthralgia and conjunctivitis are also common. Other symptoms observed following cold exposure include profuse sweating, drowsiness, headache, extreme thirst and nausea [77]. Muckle-Wells syndrome is characterized by recurrent episodes of urticarial and fever that may develop in early infancy. Acute phase reactants are elevated during fever episodes, and may also persist slightly increased during free intervals. During the course of the disease, neurosensorial deafness (60%) and polyarthritis may develop [78]. Amyloid A (AA) amyloidosis is a complication of the late stage of the disease [79-81]. NOMID is the most severe expression of NLRP3 mutations: patients present a chronic rash at birth and develop a characteristic hypertrophic arthropathy involving both knees with premature ossification of patella, chronic aseptic meningitis and papilledema [82]. Many affected individuals present a dysmorphic faces characterized by frontal bossing, saddle back nose and midface hypoplasia, causing a sibling-like resemblance [68]. Mental retardation and seizures have also been reported. Patients exhibit persistent elevation of acute phase reactants, leukocytosis and chronic anemia [83-86]. Renal amyloidosis has been observed in 25% of MWS patients and 20% of NOMID ones [87]. Up to the present some 100 variants have been associated with any of the 3 phenotypic forms (http://fmf.igh.cnrs.fr/infevers/) [56]. Almost all the observed mutations are found in exon 3 of the NLRP3 gene, coding for the NACHT domain of cryopyrin that plays a crucial role in oligomerisation of the protein.
The pivotal role of cryopyrin in driving caspase-1 activation and the massive secretion of mature IL-1β observed in cryopyrin-mutated individuals suggested that anti-IL-1 treatment could represent an effective therapy. Anakinra has been the first biologic designed for the selective blockade of IL-1: however, its short plasma half-life requires a daily subcutaneous administration at the dose of 1 - 3 mg/kg/day by subcutaneous injection (only some patients with MWS might tolerate anakinra administrations at least every 2 days, remaining in ongoing remission); the dosage required for NOMID is variable and might need escalations varying from 1 to 10 mg/kg/day [88]. Two more recent IL-1 antagonists have been licensed for FCAS and MWS, rilonacept (a dimeric fusion protein, designed for subcutaneous administration at weekly intervals, FDA-approved in 2008) and canakinumab (a fully human monoclonal anti-IL-1 antibody, designed for subcutaneous administration once every 4 - 8 weeks, FDA-approved in 2009), both with a highly favorable safety profile [89,90].
The most frequent features of autoinflammatory diseases are compared in Table 3. Diagnostic algorithm was shown in Figure 2.
2.2. NLRP12-Associated Autoinflammatory Disorder
This disorder also known as familial cold autoinflammatory syndrome 2 (FCAS 2) which is a rare genetic disease caused by NLRP12 mutations transmitted with autosomal dominant inheritance. NLRP12 has been shown to play a role in the regulation of the pro-inflammatory NF-κβ pathway, accelerated secretion of IL-1 secondary to a deregulated redox state has been suggested as an alternative pathogenic mechanism [91,92]. These patients suffer from recurrent bouts of fever lasting 5 - 10 days accompanied by headache, joint symptoms and skin rash triggered by cold exposure.
2.2.1. Deficiency of the Interleukin-1-Receptor Antagonist (DIRA)
DIRA is a autosomal recessive autoinflammatory syndrome, due to the deficiency of the interleukin-1-receptor antagonist (IL1RN). The patients so far described exhibit homozygous truncating mutations in the IL1RN gene.
The cause is a missense-nonsense mutation (or a 175-db deletion) in the IL1RN gene at the long arm of chromosome 2 that encodes the IL-1 receptor antagonist, leading to unopposed IL-1 stimulation [93].
It begins around birth with multifocal osteomyelitis, periostitis, and pustulosis. Persistent elevation of acute phase reactants (ESR and CRP) is observed from birth [52]. The skin manifestations range from groupings of small pustules to a generalized pustulosis. The bone manifestations include osteolytic lesions with a sclerotic rim, epiphyseal ballooning of multiple distal and proximal long bones, widening of ribs and clavicles, heterotopic ossification or periosteal cloaking of the proximal femoral metaphysis and periosteal elevation of the diaphysis [93].
2.2.2. Pyogenic Sterile Arthritis, Pyoderma Gangrenosum, and Acne Syndrome (PAPA)
This autosomal dominant disease is caused by mutations in proline serine threonine phosphatase-interacting protein [PSTPIP1, or CD2-binding protein 1 (CD2BP1)], also interacting with pyrin [94,95]. The arthritis usually has its onset in early childhood. It is pauciarticular in nature, and is characterized by recurring inflammatory episodes that resemble septic arthritis and lead to accumulation of pyogenic, neutrophil-rich material within the affected joints, which ultimately results in significant synovial and cartilage destruction [96,97]. Dermatological manifestations are onset usually during the second decade of life, and are characterized by debilitating, aggressive, ulcerative skin lesions. Cultures of the skin lesions and joint fluid of these patients are sterile. PAPA syndrome treated with oral glucocorticoids or anti TNF/ IL-1 therapy [66].
2.2.3. Early-Onset Sarcoidosis (EOS) (Sporadic Granulomatous Arthritis) and Blau’s Syndrome (Familial Granulomatous Arthritis)
Blau syndrome is a rare autosomal dominant disorder characterized by granulomatous polyarthritis, panuveitis, cranial neuropathies, and exanthema [98,99]. The gene responsible for Blau syndrome, NOD2/CARD15,
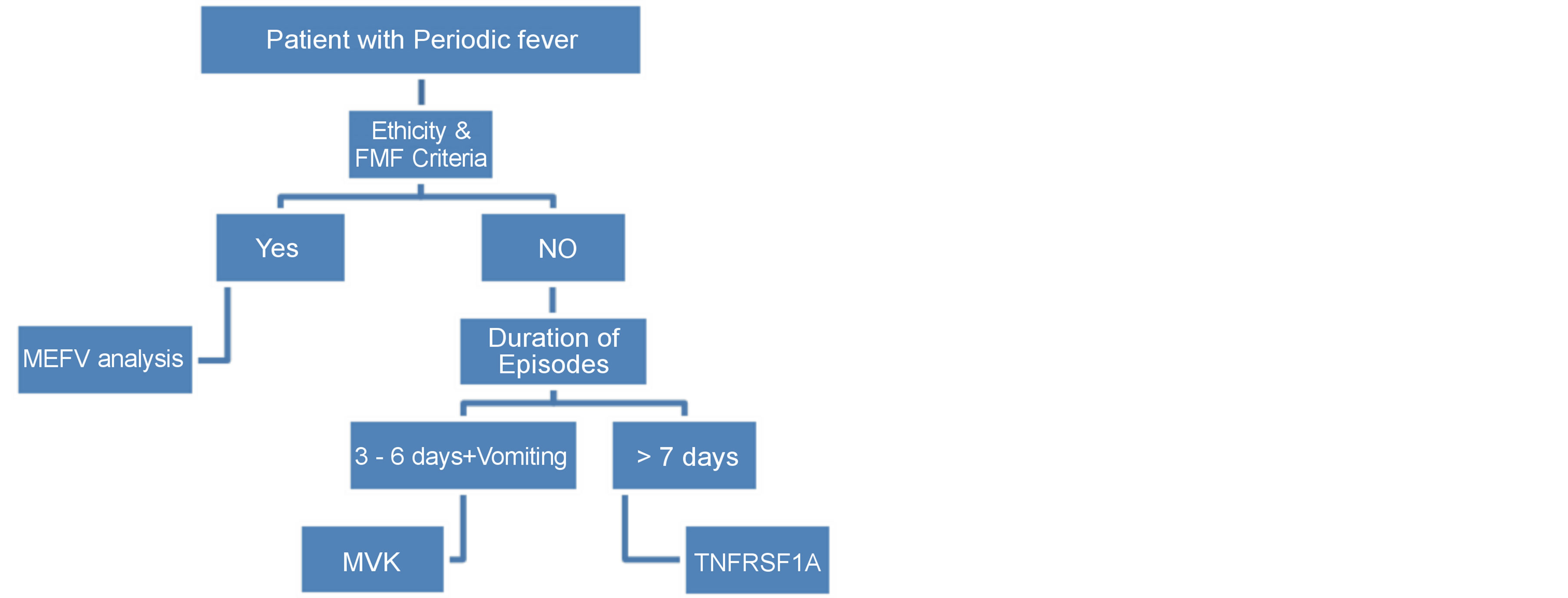
Figure 2. Diagnostic algoritm for autoinflammatory disease.
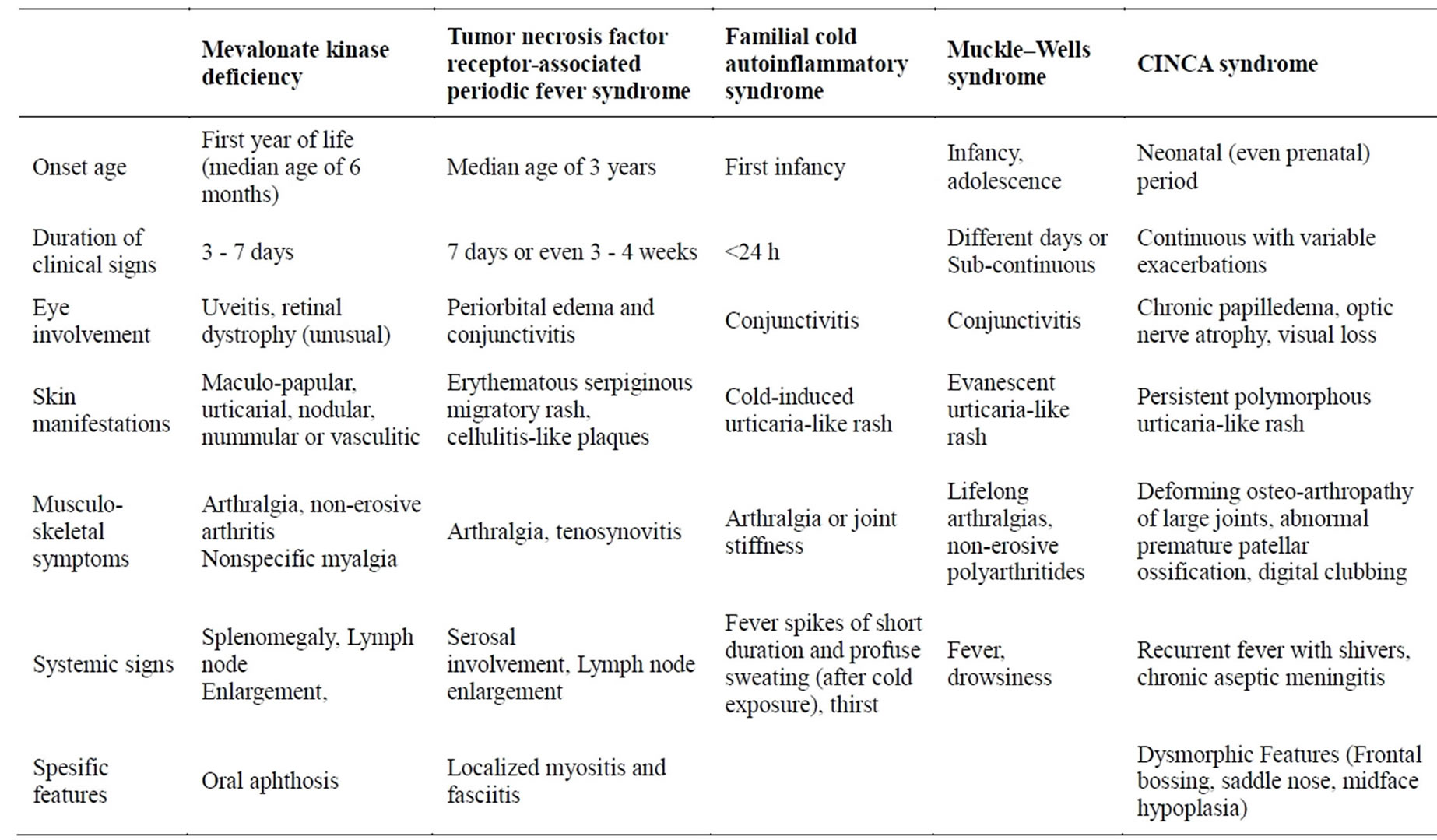
Table 3. Summary of the general clinical signs of other autoinflammatory diseases in childhood.
encodes a protein containing a NACHT domain [100]. In the literature, 10 different genetic mutations leading to substitutions in or near the NACHT domain of NOD2/ CARD15 have been documented in affected patients with either the familial or the sporadic presentation [100-102]. Disease onset is usually observed during the first years of life. Patients are treated with oral steroids and immunosuppressive drugs (methotrexate, cyclosporin A) with variable outcomes. Anti-TNFα (infliximab) and anti-IL-1 treatment suggest a beneficial effect [66].
This familial form needs to be differentiated from early-onset sarcoidosis (OMIM 609464), a sporadic multiple organ disease, histologically defined by noncaseating epithelioid granulomata of lung, lymph node and eye, though are now framed as a unique disorder: histopathologic analysis can reveal proliferating giant cells and epithelioid cells in the lesions, which are commonly observed in adult onset sarcoidosis [103].
2.2.4. Periodic Fever Aphthous Stomatitis, Pharyngitis and Adenopathy Syndrome
The acronym ‘PFAPA’ defines a syndrome in which periodic fever (usually recurring every 3 - 8 weeks, and lasting for 3 - 6 days) is associated with recurrent aphthous stomatitis, pharyngitis with negative throat cultures, and cervical adenitis, starting before the age of 5 in the majority of the patients [104]. The pathogenesis of periodic fever aphthous stomatitis and pharyngitis (PFAPA) remains unclear. An association with a mutant gene has yet to be demonstrated, despite the often positive family history and the possible modifier effects of MEFV mutations on the PFAPA phenotype [105,106]. One third of PFAPA cases may spontaneously resolve after the age of 5, most patients will require medical or surgical treatment. One to three doses of corticosteroids are generally effective in aborting a flare, but may shorten the asymptomatic interval.
3. SUMMARY
Autoinflammatory diseases are genetic or acquired clinical entities globally caused by dysregulated interleukin-1 expression in the absence of causing pathogens, antigen specific T cells or autoantibodies which are very rare except FMF. The diagnosis of most autoinflammatory syndromes relies on clinical history, demonstration of an increased acute-phase response during inflammatory attacks, and, possibly, genetic confirmation, which is still elusive especially for idiopathic febrile syndromes.
Therapeutic strategies that allow the manipulation of danger recognition and the associated signaling pathways will lead not only to the development of new treatments for a number of rare genetic conditions, but also to the exploration of these pathways in more common, genetically complex disorders in which regulation of inflammation is of importance.
DISCLOSURE STATEMENT
There are no financial supports. The authors have declared no conflicts of interest.
REFERENCES
- Hashkes, P.J. and Toker, O. (2012) Autoinflammatory syndromes. Pediatric Clinics of North America, 59, 447- 470. http://dx.doi.org/10.1016/j.pcl.2012.03.005
- Savic, S., Dickie, L.J., Wittmann, M. and McDermott, M.F. (2012) Autoinflammatory syndromes and cellular responses to stress: Pathophysiology, diagnosis and new treatment perspectives. Best Practice & Research Clinical Rheumatology, 26, 505-533. http://dx.doi.org/10.1016/j.berh.2012.07.009
- Masters, S.L., Simon, A., Aksentijevich, I. and Kastner, D.L. (2009) Horror autoinflammaticus: The molecular pathophysiology of autoinflammatory disease. Annual Review of Immunology, 27, 621-668. http://dx.doi.org/10.1146/annurev.immunol.25.022106.141627
- Ben-Chetrit, E. and Levy, M. (1998) Familial Mediterranean fever. Lancet, 351, 659-664. http://dx.doi.org/10.1016/S0140-6736(97)09408-7
- Konstantopoulos, K., Kanta, A., Deltas, C., Atamian, V., Mavrogianni, D., Tzioufas, A.G., et al. (2003) Familial Mediterranean fever associated pyrin mutations in Greece. Annals of the Rheumatic Diseases, 62, 479-481. http://dx.doi.org/10.1136/ard.62.5.479
- La Regina, M., Nucera, G., Diaco, M., Procopio, A., Gasbarrini, G., Notarnicola, C., et al. (2003) Familial Mediterranean fever is no longer a rare disease in Italy. European Journal of Human Genetics, 11, 50-56. http://dx.doi.org/10.1038/sj.ejhg.5200916
- Tunca, M., Akar, S., Onen, F., Ozdogan, H., Kasapcopur, O., Yalcinkaya, F., et al. (2005) Familial Mediterranean fever (FMF) in Turkey: Results of a nationwide multicenter study. Medicine (Baltimore), 84, 1-11. http://dx.doi.org/10.1097/01.md.0000152370.84628.0c
- Gedalia, A., Adar, A. and Gorodischer, R. (1992) Familial Mediterranean fever in children. The Journal of Rheumatology, 35, 1-9.
- Ben-Chetrit, E. and Ben-Chetrit, A. (2001) Familial Mediterranean fever and menstruation. BJOG, 108, 403-407. http://dx.doi.org/10.1111/j.1471-0528.2001.00083.x
- Rigante, D. (2012) The fresco of autoinflammatory diseases from the pediatric perspective. Autoimmunity Reviews, 11, 348-356. http://dx.doi.org/10.1016/j.autrev.2011.10.008
- Brik, R., Shinawi, M., Kasinetz, L. and Gershoni-Baruch, R. (2001) The musculoskeletal manifestations of familial Mediterranean fever in children genetically diagnosed with the disease. Arthritis & Rheumatology, 44, 1416- 1419. http://dx.doi.org/10.1002/1529-0131(200106)44:6<1416::AID-ART236>3.0.CO;2-6
- Lidar, M., Kedem, R., Mor, A., Levartovsky, D., Langevitz, P. and Livneh, A. (2005) Arthritis as the sole episodic manifestation of familial Mediterranean fever. The Journal of Rheumatology, 32, 859-862.
- Okutur, K., Seber, S., Oztekin, E., Bes, C. and Borlu, F. (2008) Recurrent pericarditis as the initial manifestation of Familial Mediterranean fever. Medical Science Monitor, 14, CS139-CS141.
- Tufan, G. and Demir, S. (2010) Uncommon clinical pattern of FMF: Protracted febrile myalgia syndrome. Rheumatology International, 30, 1089-1090. http://dx.doi.org/10.1007/s00296-009-1024-2
- Aksu, K. and Keser, G. (2011) Coexistence of vasculitides with familial Mediterranean fever. Rheumatology International, 31, 1263-1274. http://dx.doi.org/10.1007/s00296-009-1024-2
- Livneh, A., Langevitz, P., Zemer, D., Zaks, N., Kees, S., Lidar, T., et al. (1997) Criteria for the diagnosis of familial Mediterranean fever. Arthritis & Rheumatology, 40, 1879-1885. http://dx.doi.org/10.1002/art.1780401023
- Yalçinkaya, F., Ozen, S., Ozçakar, Z.B., Aktay, N., Cakar, N., Duzova, A., et al. (2009) A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford), 48, 395-398. http://dx.doi.org/10.1093/rheumatology/ken509
- Kondi, A., Hentgen, V., Piram, M., Letierce, A., Guillame-Czitrom, S. and Koné-Paut, I. (2010) Validation of the new pediatric criteria for the diagnosis of familial Mediterranean fever: Data from a mixed population of 100 children from the French reference centre for auto-inflammatory disorders. Rheumatology (Oxford), 49, 2200-2203. http://dx.doi.org/10.1093/rheumatology/keq252
- Ozcakar, Z.B., Yalçinkaya, F., Cakar, N., Acar, B., Bilgiç, A.E., Uncu, N., et al. (2011) Application of the new pediatric criteria and Tel Hashomer criteria in heterozygous patients with clinical features of FMF. European Journal of Pediatrics, 170, 1055-1057. http://dx.doi.org/10.1007/s00431-011-1404-y
- The International FMF Consortium (1997) Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell, 90, 797-807. http://dx.doi.org/10.1016/S0092-8674(00)80539-5
- (1997) A candidate gene for familial mediterranean fever. French FMF Consortium. Nature Genetics, 17, 25-31. http://dx.doi.org/10.1038/ng0997-25
- Chae JJ, Aksentijevich I and Kastner DL. (2009) Advances in the understanding of familial mediterranean fever and possibilities for targeted therapy. British Journal of Haematology, 146, 467-478. http://dx.doi.org/10.1111/j.1365-2141.2009.07733.x
- Milhavet, F., Cuisset, L., Hoffman, H.M., Slim, R., ElShanti, H., Aksentijevich, I., et al. (2008) The infevers autoinflammatory mutation online registry: Update with new genes and functions. Human Mutation, 29, 803-880. http://dx.doi.org/10.1002/humu.20720
- Shinawi, M., Brik, R., Berant, M., et al. (2000) Familial Mediterranean fever: High gene frequency and heterogeneous disease among an Israeli-Arab population. The Journal of Rheumatology, 27, 1492-1495.
- Gershoni-Baruch, R., Shinawi, M., Leah, K., et al. (2001) Familial Mediterranean fever: Prevalence, penetrance and genetic drift. European Journal of Human Genetics, 9, 634-637. http://dx.doi.org/10.1038/sj.ejhg.5200672
- Touitou, I. (2001) The spectrum of familial Mediterranean fever (FMF) mutations. European Journal of Human Genetics, 9, 473-483. http://dx.doi.org/10.1038/sj.ejhg.5200658
- Mattit, H., Joma, M., Al-Cheikh, S., et al. (2006) Familial Mediterranean fever in the Syrian population: Gene mutation frequencies, carrier rates and phenotype genotype correlation. European Journal of Medical Genetics, 49, 481-486. http://dx.doi.org/10.1016/j.ejmg.2006.03.002
- Brik, R., Shinawi, M., Kepten, I., Berant, M. and Gershoni-Baruch, R. (1999) Familial Mediterranean fever: Clinical and genetic characterization in amixed pediatric population of Jewish and Arab patients. Pediatrics, 103, 70- 73. http://dx.doi.org/10.1542/peds.103.5.e70
- Yilmaz, E., Ozen, S., Balci, B., Duzova, A., Topaloglu, R., Besbas, N., et al. (2001) Mutation frequency of Familial Mediterranean fever and evidence for a high carrier rate in the Turkish population. European Journal of Human Genetics, 9, 553-555. http://dx.doi.org/10.1038/sj.ejhg.5200674
- Majeed, H.A., El-Shanti, H., Al-Khateeb, M.S. and Rabaiha, Z.A. (2002) Genotype/phenotype correlations in Arab patients with familial Mediterranean fever. Seminars in Arthritis and Rheumatism, 31, 371-376. http://dx.doi.org/10.1053/sarh.2002.32551
- Daniels, M., Shohat, T., Brenner-Ullman, A. and Shohat, M. (1995) Familial Mediterranean fever: High gene frequency among the non-Ashkenazic and Ashkenazic Jewish populations in Israel. American Journal of Medical Genetics, 55, 311-314. http://dx.doi.org/10.1002/ajmg.1320550313
- Yuval, Y., Hemo-Zisser, M., Zemer, D., Sohar, E. and Pras, M. (1995) Dominant inheritance in two families with familial Mediterranean fever (FMF). American Journal of Medical Genetics, 57, 455-457. http://dx.doi.org/10.1002/ajmg.1320570319
- Booty, M.G., Chae, J.J., Masters, S.L., Remmers, E.F., Barham, B., Le, J.M., Barron, K.S., Holland, S.M., Kastner, D.L. and Aksentijevich, I. (2009) Familial Mediterranean fever with a single MEFV mutation: Where is the second hit? Arthritis & Rheumatism, 60, 1851-1861. http://dx.doi.org/10.1002/art.24569
- Ozen, S. (2009) Changing concepts in familial Mediterranean fever: Is it possible to have an autosomal-recessive disease with only one mutation? Arthritis & Rheumatism, 60, 1575-1577. http://dx.doi.org/10.1002/art.24565
- Singh-Grewal, D., Chaitow, J., Aksentijevich, I. and Christodoulou, J. (2007) Coexistent MEFV and CIAS1 mutations manifesting as familial Mediterranean fever plus deafness [letter]. Annals of the Rheumatic Diseases, 66, 1541. http://dx.doi.org/10.1136/ard.2007.075655
- Fonnesu, C., Cerquaglia, C., Giovinale, M., Curigliano, V., Verrecchia, E., de Socio, G., La Regina, M., Gasbarrini, G. and Manna, R. (2009) Familial Mediterranean fever: A review for clinical management. Joint Bone Spine, 76, 227-233. http://dx.doi.org/10.1016/j.jbspin.2008.08.004
- Onen, F. (2006) Familial mediterranean fever. Rheumatology International, 26, 489-496. http://dx.doi.org/10.1007/s00296-005-0074-3
- Dinarello, C.A., Wolff, S.M., Goldfinger, S.E., Dale, D.C. and Alling, D.W. (1974) Colchicine therapy for familial mediterranean fever. A double-blind trial. New England Journal of Medicine, 291, 934-937. http://dx.doi.org/10.1056/NEJM197410312911804
- Ozturk, M.A., Kanbay, M., Kasapoglu, B., Onat, A.M., Guz, G., Furst, D.E. and Ben-Chetrit, E. (2011) Therapeutic approach to familial Mediterranean fever: A review update. Clinical and Experimental Rheumatology, 29, S77-S86.
- Cronstein, B.N. and Terkeltaub, R. (2006) The inflammatory process of gout and its treatment. Arthritis Research & Therapy, 8, S3. http://dx.doi.org/10.1186/ar1908
- Nuki, G. (2008) Colchicine: A critical appraisal of its mechanism of action and efficacy in crystal-induced inflammation. Current Rheumatology Reports, 10, 218-227. http://dx.doi.org/10.1007/s11926-008-0036-3
- Ben-Chetrit, E., Bergmann, S. and Sood, R. (2006) Mechanism of the anti-inflammatory effect of colchicine in rheumatic diseases: A possible new outlook through microarray analysis. Rheumatology, 45, 274-282. http://dx.doi.org/10.1093/rheumatology/kei140
- Kallinich, T., Haffner, D., Niehues, T., Huss, K., Lainka, E., Neudorf, U., Schaefer, C., Stojanov, S., Timmann, C., Keitzer, R., Ozdogan, H. and Ozen, S. (2007) Colchicine use in children and adolescents with familial Mediterranean fever: Literature review and consensus statement. Pediatrics, 119, e474-e483. http://dx.doi.org/10.1542/peds.2006-1434
- Padeh, S., Gerstein, M. and Berkun, Y. (2012) Colchicine is a safe drug in children with familial Mediterranean fever. Journal of Pediatrics, 161, 1142-1146. http://dx.doi.org/10.1016/j.jpeds.2012.05.047
- Cerquaglia, C., Diaco, M., Nucera, G., La Regina, M., Montalto, M. and Manna, R. (2005) Pharmacological and clinical basis of treatment of familial Mediterranean fever (FMF) with colchicine or analogues: An update. Current Drug Target-Inflammation & Allergy, 4, 117-124. http://dx.doi.org/10.2174/1568010053622984
- Meinzer, U., Quartier, P., Alexandra, J.F., Hentgen, V., Retornaz, F. and Koné-Paut, I. (2011) Interleukin-1 targeting drugs in familial Mediterranean fever: A case series and a review of the literature. Seminars in Arthritis and Rheumatism, 41, 265-271. http://dx.doi.org/10.1016/j.semarthrit.2010.11.003
- Marek-Yagel, D., Berkun, Y., Padeh, S., Abu, A., Reznik-Wolf, H., Livneh, A., Pras, M. and Pras, E. (2009) Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis and Rheumatism, 60, 1862- 1866. http://dx.doi.org/10.1002/art.24570
- Frenkel, J., Houten, S.M., Waterham, H.R., Wanders, R.J., Rijkers, G.T., Kimpen, J.L.L., Duran, R., Poll-The, B.T. and Kuis, W. (2000) Mevalonate kinase deficiency and Dutch type periodic fever. Clinical and Experimental Rheumatology, 18, 525-532.
- Drenth, J.P., van Deuren, M., van der Ven-Jongekrijg, J., Schalkwijk, C.G. and van der Meer, J.W. (1995) Cytokine activation during attacks of the hyperimmunoglobulinemia D and periodic fever syndrome. Blood, 85, 3586- 3593.
- Mandey, S.H., Kuijk, L.M., Frenkel, J. and Waterham, H.R. (2006) A role for geranylgeranylation in interleukin-1beta secretion. Arthritis and Rheumatism, 54, 3690-3695. http://dx.doi.org/10.1002/art.22194
- van der Hilst, J.C., Bodar, E.J., Barron, K.S., Frenkel, J., Drenth, J.P., van der Meer, J.W., Simon, A. and the International HIDS Study Group. (2008) Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine, 87, 301-310. http://dx.doi.org/10.1097/MD.0b013e318190cfb7
- Simon, A., Kremer, H.P., Wevers, R.A., Scheffer, H., De Jong, J.G., van der Meer, J.W. and Drenth, J.P. (2004) Mevalonate kinase deficiency: Evidence for a phenotypic continuum. Neurology, 62, 994-997. http://dx.doi.org/10.1212/01.WNL.0000115390.33405.F7
- D’Osualdo, A., Picco, P., Caroli, F., Gattorno, M., Giacchino, R., Fortini, P., Corona, F., Tommasini, A., Salvi, G., Specchia, F., Obici, L., Meini, A., Ricci, A., Seri, M., Ravazzolo, R., Martini, A. and Ceccherini, I. (2005) MVK mutations and associated clinical features in Italian patients affected with autoinflammatory disorders and recurrent fever. European Journal of Human Genetics, 13, 314-320. http://dx.doi.org/10.1038/sj.ejhg.5201323
- Ammouri, W., Cuisset, L., Rouaghe, S., Rolland, M.O., Delpech, M., Grateau, G. and Ravet, N. (2007) Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology, 46, 1597-1600. http://dx.doi.org/10.1093/rheumatology/kem200
- Siewert, R., Ferber, J., Horstmann, R.D., Specker, C., Heering, P.J. and Timmann, C. (2006) Hereditary periodic fever with systemic amyloidosis: Is hyper-IgD syndrome really a benign disease? American Journal of Kidney Diseases, 48, e41-e45. http://dx.doi.org/10.1053/j.ajkd.2006.05.027
- Touitou, I., Lesage, S., McDermott, M., Cuisset, L., Hoffman, H., Dode, C., Shoham, N., Aganna, E., Hugot, J.P., Wise, C., Waterham, H., Pugnere, D., Demaille, J. and de Menthiere, C.S. (2004) Infevers: An evolving mutation database for auto-inflammatory syndromes. Human Mutation, 24, 194-198. http://dx.doi.org/10.1002/humu.20080
- Houten, S.M., Kuis, W., Duran, M., De Koning, T.J., van Royen-Kerkhof, A., Romeijn, G.J., Frenkel, J., Dorland, L., de Barse, M.M.J., Huijbers, W.A.R., Rijkers, G.T., Waterham, H.R., Wanders, R.J.A. and Poll-The, B.T. (1999) Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nature Genetics, 22, 175-177. http://dx.doi.org/10.1038/9691
- Mandey, S.H., Schneiders, M.S., Koster, J. and Waterham, H.R. (2006) Mutational spectrum and genotype-phenotype correlations in mevalonate kinase deficiency. Human Mutation, 27, 796-802. http://dx.doi.org/10.1002/humu.20361
- Takada, K., Aksentijevich, I., Mahadevan, V., Dean, J.A., Kelley, R.I. and Kastner, D.L. (2003) Favorable preliminary experience with etanercept in two patients with the hyperimunoglobulinemia D and periodic fever syndrome. Arthritis & Rheumatism, 48, 2645-2651. http://dx.doi.org/10.1002/art.11218
- Rigante, D., Ansuini, V., Bertoni, B., Pugliese, A.L., Avallone, L., Federico, G. and Stabile, A. (2006) Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome. Rheumatology International, 27, 97-100. http://dx.doi.org/10.1007/s00296-006-0164-x
- McDermott, M.F., Aksentijevich, I., Galon, J., McDermott, E.M., Ogunkolade, B.W., Centola, M., et al. (1999) Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell, 97, 133-144. http://dx.doi.org/10.1016/S0092-8674(00)80721-7
- Nedjai, B., Hitman, G.A., Church, L.D., Minden, K., Whiteford, M.L., McKee, S., Stjernberg, S., Pettersson, T., Ranki, A., Hawkins, P.N., P Arkwright, D., McDermott, M.F. and Turner, M.D. (2011) Differential cytokine secretion results from p65 and c-Rel NF-κB subunit signaling in peripheral bloodmononuclear cells of TNF receptorassociated periodic syndrome patients. Cellular Immunology, 268, 55-59. http://dx.doi.org/10.1016/j.cellimm.2011.02.007
- Aganna, E., Hammond, L., Hawkins, P.N., Aldea, A., McKee, S.A., van Amstel, H.K., et al. (2003) Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis and Rheumatism, 48, 2632-2644. http://dx.doi.org/10.1002/art.11215
- D’Osualdo, A., Ferlito, F., Prigione, I., Obici, L., Meini, A., Zulian, F., Pontillo, A., Corona, F., Barcellona, R., Di Duca, M., Santamaria, G., Traverso, F., Picco, P., Baldi, M., Plebani, A., Ravazzolo, R., Ceccherini, I., Martini, A. and Gattorno, M. (2006) Neutrophils from patients with TNFRSF1A mutations display resistance to tumor necrosis factor-induced apoptosis: Pathogenetic and clinical implications. Arthritis and Rheumatism, 54, 998-1008. http://dx.doi.org/10.1002/art.21657
- Ravet, N., Rouaghe, S., Dode, C., Bienvenu, J., Stirnemann, J., Levy, P., Delpech, M. and Grateau, G. (2006) Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Annals of the Rheumatic Diseases, 65, 1158-1162. http://dx.doi.org/10.1136/ard.2005.048611
- Federici, S., Caorsi, R. and Gattorno, M. (2012) The autoinflammatory diseases. Swiss Medical Weekly, 142, Article ID: w13602.
- Pelagatti, M.A., Meini, A., Caorsi, R., Cattalini, M., Federici, S., Zulian, F., Calcagno, G., Tommasini, A., Bossi, G., Sormani, M.P., Caroli1, F., Plebani, A., Ceccherini, I., Martini, A. and Gattorno, M. (2011) Long-term clinical profile of children with the low-penetrance R92Q mutation of the TNFRSF1A gene. Arthritis and Rheumatism, 63, 1141-1150. http://dx.doi.org/10.1002/art.30237
- Stojanov, S. and McDermott, M.F. (2005) The tumour necrosis factor receptor-associated periodic syndrome: Current concepts. Expert Reviews in Molecular Medicine, 7, 1-18. http://dx.doi.org/10.1017/S1462399405009749
- Hull, K.M., Drewe, E., Aksentijevich, I., Singh, H.K., Wong, K., McDermott, E.M., Dean, J., Powell, R.J. and Kastner, D.L. (2002) The TNF receptor-associated periodic syndrome (TRAPS): Emerging concepts of an autoinflammatory disorder. Medicine, 81, 349-368. http://dx.doi.org/10.1097/00005792-200209000-00002
- Drewe, E., McDermott, E.M., Powell, P.T., Isaacs, J.D. and Powell, R.J. (2003) Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): Clinical and laboratory findings in a series of seven patients. Rheumatology, 42, 235-239. http://dx.doi.org/10.1093/rheumatology/keg070
- Kallinich, T., Briese, S., Roesler, J., Rudolph, B., Sarioglu, N., Blankenstein, O., Keitzer, R., Querfeld, U. and Haffner, D. (2004) Two familial cases with tumor necrosis factor receptor-associated periodic syndrome caused by a noncysteine mutation (T50M) in the TNFRSF1A gene associated with severe multiorganic amyloidosis. Journal of Rheumatology, 31, 2519-2522.
- Gattorno, M., Pelagatti, M.A., Meini, A., Obici, L., Barcellona, R., Federici, S., Buoncompagni, A., Plebani, A., Merlini, G. and Martini, A. (2008) Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis & Rheumatism, 58, 1516-1520. http://dx.doi.org/10.1002/art.23475
- Hoffman, H., Mueller, J., Brodie, D., Wanderer, A.A. and Kolodner, R.D. (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nature Genetics, 29, 301-305. http://dx.doi.org/10.1038/ng756
- Feldmann, J., Prieur, A.M., Quartier, P., Berquin, P., Certain, S., Cortis, E., Teillac-Hamel, D., Fischer, A. and de Saint Basile, G. (2002) Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. American Society of Human Genetics, 71, 198-203. http://dx.doi.org/10.1086/341357
- Aksentijevich, I., Putnam, C.D., Remmers, E.F., Mueller, J.L., Le, J., Kolodner, R.D., Moak, Z., Chuang, M., Austin, F., Goldbach-Mansky, R., Hoffman, H.M. and Kastner, D.L. (2007) The clinical continuum of cryopyrinopathies: Novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis & Rheumatism, 56, 1273-1285. http://dx.doi.org/10.1002/art.22491
- Henderson, C. and Goldbach-Mansky, R. (2010) Monogenic autoinflammatory diseases: New insights into clinical aspects and pathogenesis. Current Opinion in Rheumatology, 22, 567-578.
- Hoffman, H.M., Wanderer, A.A. and Broide, D.H. (2001) Familial cold autoinflammatory syndrome: Phenotype and genotype of an autosomal dominant periodic fever. Journal of Allergy and Clinical Immunology, 108, 615-620. http://dx.doi.org/10.1067/mai.2001.118790
- Maksimovic, L., Stirnemann, J., Caux, F., Ravet, N., Rouaghe, S., Cuisset, L., Letellier, E., Grateau, G., Morin, A.S. and Fain, O. (2008) New CIAS1 mutation and anakinra efficacy in overlapping of Muckle-Wells and familial cold autoinflammatory syndromes. Rheumatology, 47, 309-310. http://dx.doi.org/10.1093/rheumatology/kem318
- Muckle, T.J. and Wells, M. (1962) Urticaria, deafness, and amyloidosis: A new heredo-familial syndrome. Quarterly Journal of Medicine, 31, 235-248.
- Nazzari, G., Desirello, G. and Crovato, F. (1995) Recurrent urticarial skin eruption since infancy. Muckle-Wells syndrome (MWS). Archives of Dermatology, 131, 81-85. http://dx.doi.org/10.1001/archderm.131.1.81
- Leslie, K.S., Lachmann, H.J., Bruning, E., McGrath, J.A., Bybee, A., Gallimore, J.R., Roberts, P.F., Woo, P., Grattan, C.E. and Hawkins, P.N. (2006) Phenotype, genotype, and sustained response to anakinra in 22 patients with autoinflammatory disease associated with CIAS-1/NALP3 mutations. Archives of Dermatology, 142, 1591-1517. http://dx.doi.org/10.1001/archderm.142.12.1591
- Prieur, A.M. (2001) A recently recognised chronic inflammatory disease of early onset characterised by the triad of rash, central nervous systeminvolvement and arthropathy. Clinical and Experimental Rheumatology, 19, 103-106.
- Prieur, A.M. and Griscelli, C. (1981) Arthropathy with rash, chronic meningitis, eye lesions, and mental retardation. Journal of Pediatrics, 99, 79-83. http://dx.doi.org/10.1016/S0022-3476(81)80961-4
- Prieur, A.M., Griscelli, C., Lampert, F., Truckenbrodt, H., Guggenheim, M.A., Lovell, D.J., Pelkonnen, P., ChevrantBreton, J. and Ansell, B.M. (1987) A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scandinavian Journal of Rheumatology, 66, 57-68. http://dx.doi.org/10.3109/03009748709102523
- Aksentijevich, I., Nowak, M., Mallah, M., Chae, J.J., Watford, W.T., Hofmann, S.R., et al. (2002) De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): A new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis and Rheumatism, 46, 3340-3348. http://dx.doi.org/10.1002/art.10688
- Caroli, F., Pontillo, A., D’Osualdo, A., Travan, L., Ceccherini, I., Crovella, S., Alessio, M., Stabile, A., Gattorno, M., Tommasini, A., Martini, A. and Lepore, L. (2007) Clinical and genetic characterization of Italian patients affected by CINCA yndrome. Rheumatology, 46, 473-478. http://dx.doi.org/10.1093/rheumatology/kel269
- Bilginer, Y., Akpolat, T. and Ozen, S. (2011) Renal amyloidosis in children. Pediatric Nephrology, 26, 1215-1227. http://dx.doi.org/10.1007/s00467-011-1797-x
- Neven, B., Marvillet, I., Terrada, C., Ferster, A., Boddaert, N., Couloignier, V., Pinto, G., Pagnier, A., Bodemer, C., Bodaghi, B., Tardieu, M., Prieur, A.M. and Quartier, P. (2010) Longterm efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis and Rheumatism, 62, 258-267. http://dx.doi.org/10.1002/art.25057
- Goldbach-Mansky, R., Shroff, S.D., Wilson, M., Snyder, C., Plehn, S., Barham, B., Pham, T.H., Pucino, F., Wesley, R.A., Papadopoulos, J.H., Weinstein, S.P., Mellis, S.J. and Kastner, D.L. (2008) A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis and Rheumatism, 58, 2432-2442. http://dx.doi.org/10.1002/art.23620
- Lachmann, H.J., Kone-Paut, I., Kuemmerle-Deschner, J.B., Leslie, K.S., Hachulla, E., Quartier, P., Gitton, X., Widmer, A., Patel, N. and Hawkins, P.N. (2009) Use of canakinumab in the cryopyrin-associated periodic syndrome. New England Journal of Medicine, 360, 2416-2425. http://dx.doi.org/10.1056/NEJMoa0810787
- Jeru, I., Duquesnoy, P., Fernandes-Alnemri, T., Cochet, E., Yu, J.W., Lackmy-Port-Lis, M., Grimprel, E., Landman-Parker, J., Hentgen, V., Marlin, S., McElreavey, K., Sarkisian, T., Grateau, G., Alnemri, E.S. and Amselem, S. (2008) Mutations in NALP12 cause hereditary periodic fever syndromes. Proceedings of the National Academy of Sciences of the United States of America, 105, 1614-1619. http://dx.doi.org/10.1073/pnas.0708616105
- Borghini, S., Tassi, S., Chiesa, S., Caroli, F., Carta, S., Caorsi, R., Fiore, M., Delfino, L., Lasigliè, D., Ferraris, C., Traggiai, E., Di Duca, M., Santamaria, G., D’Osualdo, A., Tosca, M., Martini, A., Ceccherini, I., Rubartelli, A. and Gattorno, M. (2010) Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of a NLRP12 mutation. Arthritis and Rheumatism, 63, 830-839.
- Aksentijevich, I., Masters, S.L., Ferguson, P.J., Dancey, P., Frenkel, J., van Royen-Kerkhoff, A., et al. (2009) An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. New England Journal of Medicine, 360, 2426-2437. http://dx.doi.org/10.1056/NEJMoa0807865
- Lindor, N.M., Arsenault, T.M., Solomon, H., Seidman, C.E. and McEvoy, M.T. (1997) A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome. Mayo Clinic proceedings, 72, 611-615.
- Shoham, N.G., Centola, M., Mansfield, E., Hull, K.M., Wood, G., Wise, C.A. and Kastner, D.L. (2003) Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proceedings of the National Academy of Sciences of the United States of America, 100, 13501-13506. http://dx.doi.org/10.1073/pnas.2135380100
- Wise, C.A., Gillum, J.D., Seidman, C.E., Lindor, N.M., Veile, R., Bashiardes, S. and Lovett, M. (2002) Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Human Molecular Genetics, 11, 961-969. http://dx.doi.org/10.1093/hmg/11.8.961
- Yeon, H.B., Lindor, N.M., Seidman, J.G. and Seidman, C.E. (2000) Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome maps to chromosome 15q. American Journal of Human Genetics, 66, 1443-1448. http://dx.doi.org/10.1086/302866
- Ferrero-Miliani, L., Nielsen, O.H., Andersen, P.S. and Girardin, S.E. (2007) Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1beta generation. Clinical and Experimental Immunology, 147, 227-235.
- Blau, E.B. (1985) Familial granulomatous arthritis, iritis, and rash. Journal of Pediatrics, 107, 689-693. http://dx.doi.org/10.1016/S0022-3476(85)80394-2
- Miceli-Richard, C., Lesage, S., Rybojad, M., Prieur, A.M., Manouvrier-Hanu, S., Hafner, R., Chamaillard, M., Zouali, H., Thomas, G. and Hugot, J.P. (2001) CARD15 mutations in Blau syndrome. Nature Genetics, 29, 19-20. http://dx.doi.org/10.1038/ng720
- Kanazawa, N., Okafuji, I., Kambe, N., Nishikomori, R., Nakata-Hizume, M., Nagai, S., et al. (2005) Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: Common genetic etiology with Blau syndrome. Blood, 105, 1195-1197. http://dx.doi.org/10.1182/blood-2004-07-2972
- van Duist, M.M., Albrecht, M., Podswiadek, M., Giachino, D., Lengauer, T., Punzi, L. and De Marchi, M. (2005) A new CARD15 mutation in Blau syndrome. European Journal of Human Genetics, 13, 742-747. http://dx.doi.org/10.1038/sj.ejhg.5201404
- Fink, C.W. and Cimaz, R. (1997) Early onset sarcoidosis: Not a benign disease. Journal of Rheumatology, 24, 174- 177.
- Caorsi, R., Pelagatti, M.A., Federici, S., Finetti, M., Martini, A. and Gattorno, M. (2010) Periodic fever, apthous stomatitis, pharyngitis and adenitis syndrome. Current Opinion in Rheumatology, 22, 579-584. http://dx.doi.org/10.1097/BOR.0b013e32833cc9cb
- Cochard, M., Clet, J., Le, L., Pillet, P., Onrubia, X., Guéron, T., Faouzi, M. and Hofer, M. (2010) PFAPA syndrome is not a sporadic disease. Rheumatology, 49, 1984-1987. http://dx.doi.org/10.1093/rheumatology/keq187
- Berkun, Y., Levy, R., Hurwitz, A., Meir-Harel, M., Lidar, M., Livneh, A. and Padeh, S. (2011) The familial Mediterranean fever gene as a modifier of periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome. Seminars in Arthritis and Rheumatism, 40, 467-472. http://dx.doi.org/10.1016/j.semarthrit.2010.06.009