Modern Research in Inflammation
Vol. 1 No. 1 (2012) , Article ID: 21863 , 4 pages DOI:10.4236/mri.2012.11002
Hydroperoxides and cytokines as biomarkers in detecting atherosclerosis predisposition in cigarette smokers
![]()
1Fondazione G. Monasterio CNR-Regione Toscana, Pisa, Italy; *Corresponding Author: walterl@ifc.cnr.it
2Department of Human and Environmental Sciences, University of Pisa, Pisa, Italy
3Institute of Agricultural Biology and Biotechnology, CNR, Pisa, Italy
4Department of Pharmacology, University of Bologna, Bologna, Italy
Received 25 June 2012; revised 20 July 2012; accepted 17 August 2012
Keywords: IL-6; TNFα; CRP; Hydroperoxides; Ossidative Stress; CAD
ABSTRACT
Objectives: Smoking increases oxidative modification of LDL, associated with lower HDL plasma levels, systemic inflammatory response and endothelial dysfunction. We tested the hypothesis that the risk status for coronary atherosclerosis disease (CAD) of cigarettes smokers might be identified by means of serum oxidative levels and vascular inflammation determination. Design and Methods: Oxidative stress levels, cytokines, and the metabolic status were investigated on 499 subjects admitted to our institute. The association between biomarkers and smoking habits in the presence/absence of disease and with the number of vessel affected, was studied. Results: Oxidative stress and inflammatory levels (p < 0.001) were strongly induced by smoking habits. Serum values of the subjects categorised as CAD, non CAD and healthy subjects differed significantly (p < 0.001) only for the degree of oxidative stress. Glycaemia was able to affect C-reactive protein serum levels with a positive association (p < 0.05). The analysis of the study population indicated that serum oxidative stress levels significantly increased with increasing number of vessels affected (p < 0.01). When statistical analysis was performed separately in both smoking groups, smokers did not show any particular difference for both oxidative stress and inflammation markers between the two groups of cardiovascular patients (CAD and non CAD) and the control group, while for non smokers, the differences were evident. Conclusion: These findings indicate that the considered biomarkers, especially oxidative stress, can be useful to predict the biological damage caused by cigarette smoking, as well as to identify subjects characterised by a higher risk of cardiovascular event, but cannot evaluate the presence of disease in subjects with smoking habit.
1. INTRODUCTION
Exposure to environmental tobacco smoke has been linked to an increased risk of many adverse health outcomes, including lung cancer, asthma onset and exacerbation, acute respiratory illness and atherosclerosis. Tobacco smoke comprises a large number of chemical constituents, partitioned to various degrees between the gas and condensed phases. Among the constituents of environmental tobacco smoke are chemical compounds that are regulated as hazardous air pollutants by the US federal government. Polycyclic aromatic hydrocarbons (PAHs) and volatile N-nitrosamines, and also tobacco specific N-nitrosamines are considered to be the major carcinogens in tobacco smoke [1]. Epidemiological and experimental studies showed the presence of the dose-effect relationship between the number of cigarettes smoked and lung cancer risk, exposure to tar or tobacco smoke and skin cancers or squamous cell carcinoma of the trachea and lung [1]. Other studies indicate that acetaldehyde, acrolein, acrylonitrile, benzene, 1,3-butadiene, and formaldehyde should be of particular concern as contributors to health risk from chronic, residential environmental tobacco smoke exposure [2].
Smoking causes acute hemodynamic alterations such as increase in heart rate, systematic and coronary vascular resistance, myocardial contractility, and myocardial oxygen demand. These short-term effects could lower the ischemic threshold in smokers with coronary artery disease and contribute to the increased risk for acute cardiovascular events [3].
The exact toxic components of cigarette smoke and the mechanisms involved in cigarette smoking-related cardiovascular dysfunction are largely unknown, but cigarette smoking increases inflammation, thrombosis, and oxidation of low-density lipoprotein cholesterol. Recent experimental and clinical data support the hypothesis that cigarette smoke exposure increases oxidative stress as a potential mechanism for initiating cardiovascular dysfunction [4].
Smoking is one of the most important risk factors for both atherosclerosis and ischemic heart disease. Endothelial dysfunction can be considered a pathological outcome of smoking and many studies [3] showed that this biological damage is directly associated to the duration of exposure [5]. Since atherosclerosis is characterised by early structural change of the endothelial cells with loss of the regulatory vascular tone, by platelet-endothelial interaction and leukocyte adhesion control [3], endothelial damage is thought to be a leading event of atherosclerosis [6].
There is strong evidence that smoking leads to endothelial dysfunction mainly by increased inactivation of nitric oxide (NO) determined by oxygen-derived free radicals; smoking also increases oxidative modification of LDL, is associated with lower HDL plasma levels and induces a systemic inflammatory response with elevation of the C-reactive protein (CRP) and interleukin-6 serum concentration [7].
8-isoprostane (8-epi-PGF2 alpha), a lipid peroxidation product of non enzymatic origin, has been proposed as a marker for oxidative stress, and it has been shown to have potent pulmonary, renal and coronary vasoconstrictor activity [8-10]. 8-isoprostane is produced by the random oxidation of tissue phospholipids by oxygen free radicals, and elevated levels of this molecule have been found in plasma and urine of heavy smokers [11]. The vasoconstriction effect induced by 8-isoprostane seems to be linked to the overproduction of endotheline-1 (ET-1) by endothelial cells [12].
The presence of specific binding sites for F2-isoprostanes on endothelial cells (TXA2 receptor) suggests that this eicosanoid, released during oxidant injury, could alter normal endothelial cells physiology [12]. The release of ET-1 by 8-epi-PGF2 alpha may help to explain the large increases in plasma and urinary concentrations for both ET-1 and 8-epi-PGF2 alpha in patients with high oxidative stress levels [12,13]. These findings seem to indicate an association between cigarette smoking and oxidative damage, endothelial dysfunction, or vascular inflammation.
The aim of this investigation was to study whether the risk status for coronary atherosclerosis disease (CAD) of cigarettes smokers might be identified by means of serum oxidative levels and vascular inflammation determination. For this purpose, the levels of oxygen free radicals (dROMs), interleukine-6 (IL-6), C-reactive protein (CRP), and tumour necrosis α (TNF α) were analysed in a general population consisting of people affected by cardiovascular disease.
2. MATERIALS AND METHODS
2.1. Study Population
The study includes 499 subjects admitted to the Fondazione Gabriele Monasterio. The characteristics of the entire population, stratified for smoking habit, are summarised in Table 1. First group: patients with documented or suspected ischemia who underwent coronary angiography and that were affected by CAD did not suffer from any acute cardiovascular event during the last
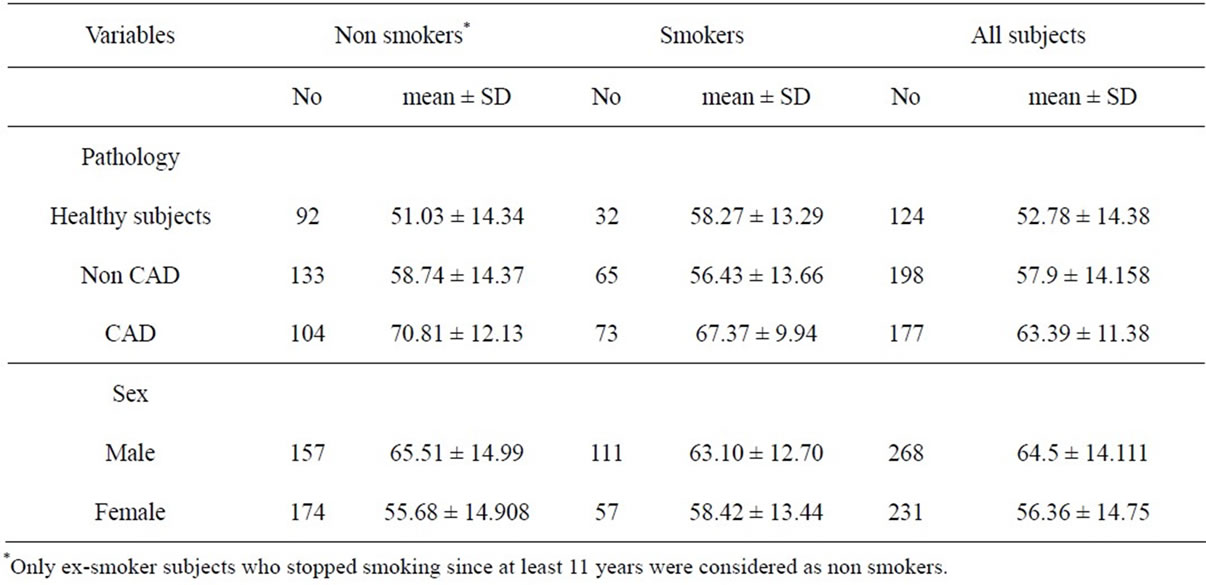
Table 1. Composition and mean age (years) of the study population stratified for smoking habit.
three months; in each case the mean reduction lumen of coronary arteries was at least about 50% (affected vessels). The second group consisted of patients affected by other heart diseases (non CAD) established on the basis of physical examination and routine laboratory tests. The third group (healthy controls) consisted of people not affected by any disease and not underwent pharmacological treatment. Patients treated with statins or other hypolipemic or hypoglicemic therapy were excluded from the study. Patients were treated with antianginal and antiplatelets, but none of them made use of sublingual nitrates. Any other medications were suspended seven days prior to blood drawing. The disease was diagnosed for the first time, so that no therapy was carried out before. Healthy subjects did not undergo coronary angiography.
Written informed consent was obtained from all subjects before they entered the study. The study adheres to the principles outlined in the declaration of Helsinki.
All blood samples were collected at the same time, placed on ice at 4˚C and immediately centrifuged at 3000 rpm for 10 minutes; serum was stored at −80˚C for not more than 15 days. Collection of blood samples from patients affected by CAD was performed the day before the cardiac catheterization.
2.2. Analytical Methods
Plasma concentrations of total cholesterol (TC), HDL-C and Tryglicerides (Tg) were determined by standard laboratory methods. The concentration of LDL-C was calculated using the Friedewald equation [14], and expressed as mg/dl.
Recently a new colorimetric method has been proposed to measure the generation of peroxy radicals (D-Roms test, Diacron international, Italy) first products of the reaction between free radicals and oxygen [15,16]. This test is based on the ability of transition metals to catalyse in the presence of peroxides with generation of free radicals that are trapped by an alchilamine; reaction of alchilamine then yields a coloured radical detectable at 505 nm. Results are expressed as Carratelli Units (U.C.) (1 U.C. = 0.08 mg H2O2 dl).
The Biosource high sensitive-IL-6 (hs-IL-6) and high sensitive-TNFα (hs-TNFα) were evaluated by an ELISA technique employing a solid-phase sandwich, enzymelinked immuno-sorbent assay (Cayman, Ann Arbor MI, USA; Biosource International, Inc. Camarillo, California, USA).
High sensitive CRP (Hs-CRP) was measured by Immulite System (Diagnostic Products Corporation [DPC]), a methods based on a chemiluminescent immunoreaction. During immunoreaction for CRP detection, the enzymatic conjugate, alkaline phosphatase, is bound. After washes with buffer a chemiluminescent substrate (adamantildioxetane) is added to the complex and, after 10 minutes at 37˚C incubation, the light produced from the reaction is measured by a photomultiplier tube. During the reaction alkaline phosphatase dephosphorizes the adamantildioxetane phosphate producing an unstable anion that emits photons.
2.3. Statistical Analysis
dROMs, IL-6, CRP and TNFα serum level values were log transformed to normalise the distributions [17,18]. The obtained results were analysed by parametric tests. Multifactor analysis of variance (MANOVA) was employed to evaluate jointly the effect of smoking habit, pathological condition, number of affected vessels and some metabolic parameters on dROMs, IL-6, CRP and TNFα serum concentration after adjusting for age and sex. The categorical variable describing affected vessels classify the study population into three groups: subjects with no affected vessels; CAD patients with one or two affected vessels, and CAD patients with three or four affected vessels. For each categorical factors included in the main analysis, differences in serum levels of these markers between subject groups and the reference group were evaluated by the Dunnet’s test. The association between variables and the number of cigarettes smoked was estimated by regression analysis. A p level lower than 0.05 was considered statistically significant.
3. RESULTS
Table 2 reports the results of MANOVA analysis on serum levels of the four markers assessed in the study population. Smoking habit affected greatly all markers: in fact, smokers showed highly significant increases of their levels with respect to non smokers (dROMs 264.21 ± 3.92 and 3.16 ± 5.96 U.C. p < 0,001; IL-6 1.97 ± 0.18 and 3.05 ± 0.27 pg/ml, p < 0.001; TNFα 1.97 ± 0.18 and 3.05 ± 0.27 pg/ml, p < 0.001; CRP 0.84 ± 0.22 and 1.95 ± 0.68 mg/dl; p < 0.001). In addition, serum values of the study population classified according to their pathological condition differed significantly (p < 0.001) only in the case of dROMs. In fact, compared to the group of either non CAD patients or controls, the group affected by CAD had significantly increased dROMs levels (317.8 ± 6.8 vs. 263.98 ± 5.79 or 261.31 ± 3.99, respectively). At variance, no difference among the same groups was observed for IL-6, TNFα and CRP levels. Among the metabolic parameters evaluated, only glycaemia was able to influence CRP serum levels showing a statistically significant association (p < 0.05). With regards to confounding factors, IL-6 or CRP was positively correlated to chronological age of the study population (p < 0.001 or p < 0.01). Further MANOVA analysis
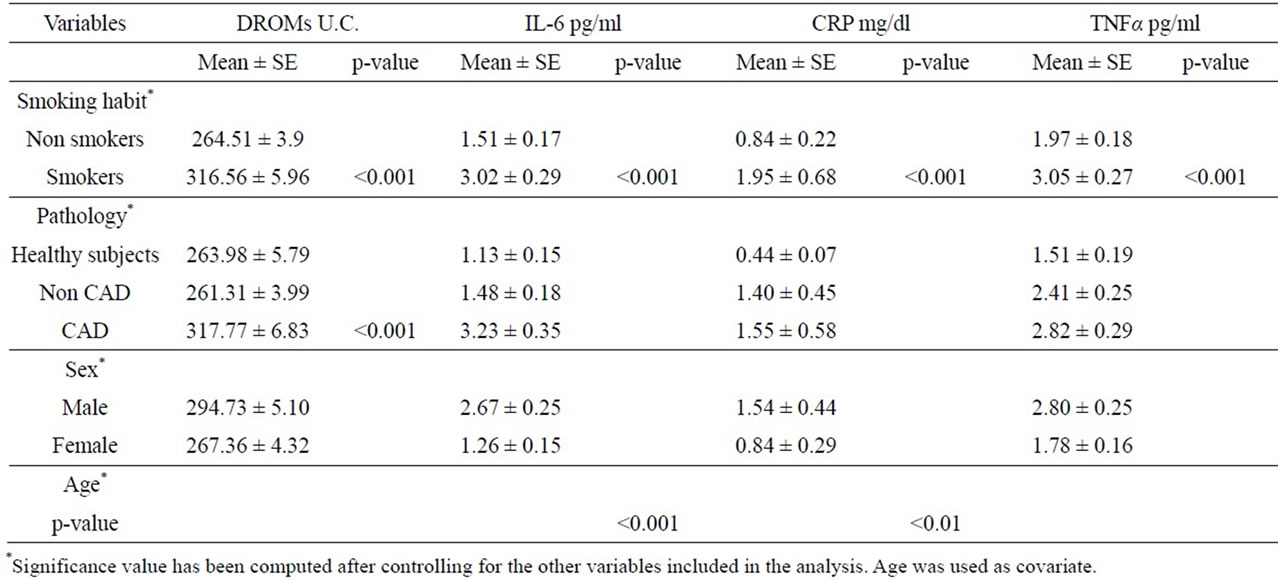
Table 2. Results of MANOVA analysis on biomarkers of oxidative stress and inflammation.
of the study population as a whole indicated that serum dROMs levels significantly increased with increasing number of vessels affected (Figure 1), whereas markers of inflammation showed, in general, minor or no correlation. Linear regression analysis showed a significant association between the number of cigarettes smoked per day and dROMs (p < 0.05) or IL-6 (p < 0.01) serum concentration (data not shown).
However, when statistical analysis was examined separately in both smoking groups, smokers did not show any particular difference for both oxidative stress and inflammation markers between the two groups of cardiovascular patients (CAD and non CAD) and the control group. Interestingly, among non smokers, patients suffering from CAD showed mean dROM levels (306 ± 9.02) significantly higher (p < 0.001) than those of non CAD (245.08 ± 3.44) and controls (265.7 ± 5.6). The same trend (p < 0.05) was seen for IL-6 (2.73 ± 0.42 the mean value of CAD patients vs. 1.78 ± 0.24 and 0.61 ± 0.1 pg/ml for non CAD and controls, respectively), for TNFα (2.97 ± 0.45, on average, for the CAD group vs. 1.75 ± 0.22 and 1.16 ± 0.23 pg/ml for the non CAD and control group, respectively), and for CRP (1.57 ± 0.28 for CAD patients vs. 1.05 ± 0.22 and 0.67 ± 0.18 mg/dl for non CAD and controls, respectively (see Figure 2).
4. DISCUSSION
Smoking is strongly associated with chronic obstructive pulmonary disease, cancer, and atherosclerosis [19,20]. As cigarette smoke contains different kinds of free radicals, it can cause biological damage via oxidative stress [21], resulting in the induction of modified lipids, proteins and DNA bases, mainly 8-oxodG. A direct and controlled evidence of this feature is supported by the finding that smoking cessation is able to significantly reduce the urinary excretion rate of oxidized guanosines [22].
In addition, oxidation of LDL by cigarette smoke may contribute to the link between cigarette smoking and atherogenesis. Cigarette smoke extract alters LDL into a form recognized and incorporated by macrophages; this could explain the increased incidence of atherosclerosis and coronary heart disease observed in smokers [23], as confirmed by the formation of lipid hydroperoxides after plasma exposure to the gas phase of cigarette smoke [24].
Other evidences support the view that smokers undergo oxidative stress. For example, the risk of severe hypovitaminosis C is increased in smokers with respect to non-smokers [25], and smokers' risk of coronary artery disease correlates inversely with their intake of the antioxidants vitamin E and beta carotene [26,27]. Cigarette smoke is replete with a number of oxidants [24] and consistent with this, smokers have increased F2-isoprostane levels in both plasma and urine. However, F2-isoprostane levels do seem to depend on the age and sex of smokers or on the number of pack-year or cigarettes per day smoked [28]. In animal models, it has been shown that a brief exposure to cigarette smoke is able to promote atherosclerotic plaque development [29], and the combination of second hand smoke and hypercholesterolemia resulted, to a greater extent, in mtDNA (mitochondrial DNA) damage and atherosclerosis [30]. In addition, it has been documented that benzo[a]pyrene, a polycyclic aromatic hydrocarbon present in tobacco smoke, can modulate VSMCs from a quiescent to a proliferative phenotype both in vitro and in vivo [31,32].
The present work aimed to assess whether or not markers of oxidative stress and vascular inflammation
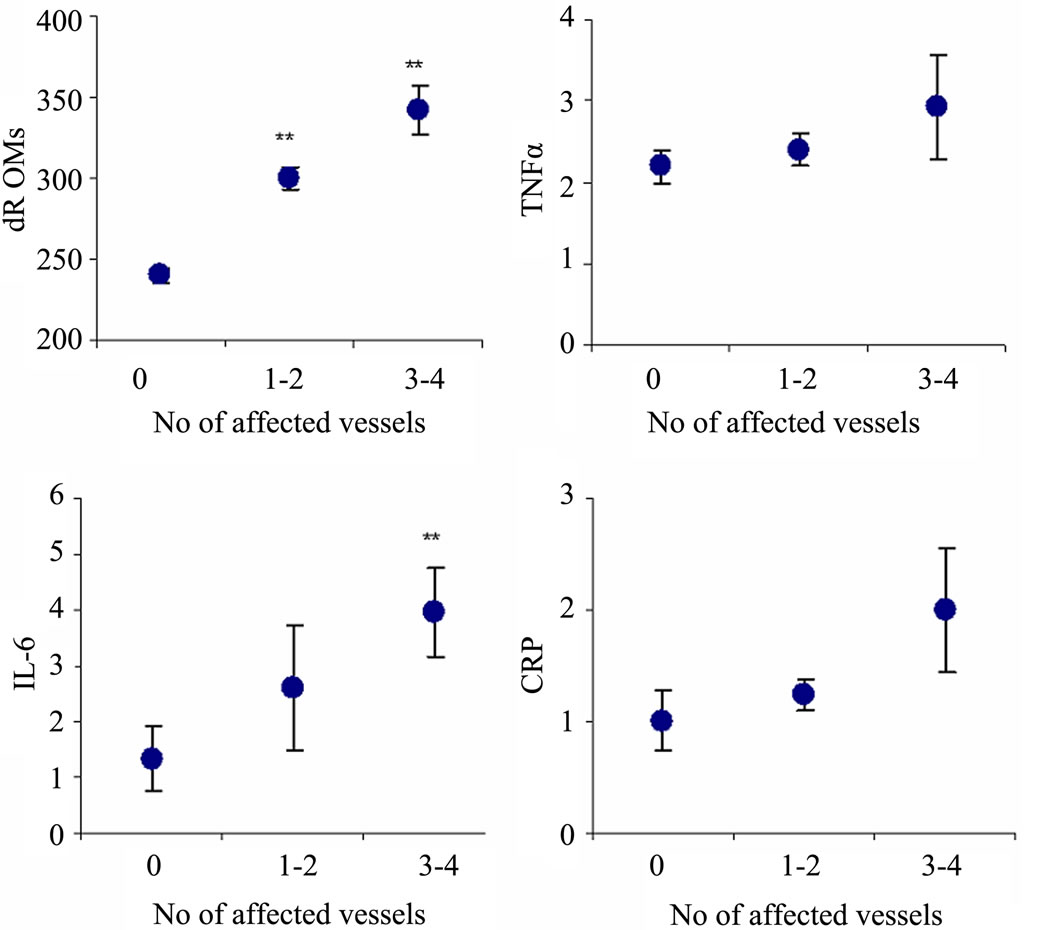
**p < 0.01 dROMs (patients group with 1 - 2 and 3 - 4 affected vessels), **p < 0.01 IL-6 (patients group with 3 - 4 affected vessels).
Figure 1. MANOVA analysis of the study population indicated that serum dROMs and IL-6 levels significantly increased with increasing number of vessels affected. The other markers of inflammation showed, in general, minor or no correlation.
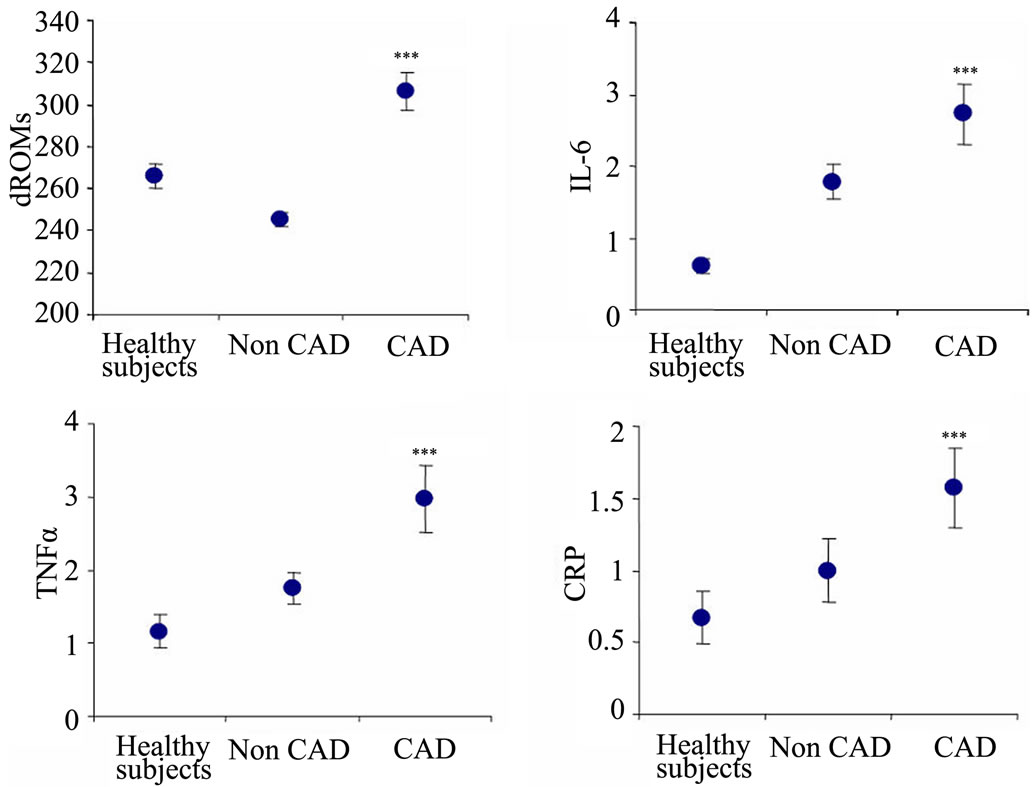
***p < 0.001.
Figure 2. Among non smokers, patients suffering from CAD showed mean levels of free radicals and inflammation significantly higher than those of non CAD and controls.
can provide a reliable estimate of the risk for cardiovascular diseases in a general population, especially in the case of smokers [16,33]. The results of our study demonstrated that dROMs, IL-6, CRP and TNF serum concentrations markedly increased in cigarette smokers and, except for TNF and CRP, correlated with the number of cigarettes smoked. In addition, we showed that the presence of CAD was able to influence the levels of free radicals, where the concentration was directly associated with the severity of the disease (i.e. the number of vessel affected). We also found that CRP resulted influenced by glycemia and that both CRP and IL-6 values increased with increasing the age of the subjects. When the analysis were performed in the group of smokers no difference for both oxidative stress and inflammation markers among the groups were observed, while in the case of non smokers the group affected by CAD showed a high significant increase of the levels of each markers. Thus, our findings suggest that smoking habit represent the major risk factor for cardiovascular disease and its effect on vascular inflammation and oxidative stress is so strong to not discriminate controls from patient affected by CAD in the group of smokers.
In conclusion, the present study reinforce the hypothesis that oxidative stress and inflammatory serum levels might be considered as useful markers to predict the biological damage caused by cigarette smoking and to identify subjects characterised by a higher risk of cardiovascular event, but cannot evaluate the presence of disease in subjects with smoking habit.
5. ACKNOWLEDGEMENTS
The authors are grateful to Dr. Lucrecia Mota for her English editing support.
REFERENCES
- Starek, A. and Podolak, I. (2009) Carcinogenic effect of tobacco smoke. Roczniki Państwowego Zakładu Higieny, 60, 299-310.
- Nazaroff, W.W. and Singer, B.C. (2004) Inhalation of hazardous air pollutants from environmental tobacco smoke in US residences. Journal of Exposure Analysis and Environmental Epidemiology, 14, S71-S77. doi:10.1038/sj.jea.7500361
- Heitzer, T. and Meinertz, T. (2000) Prevention of coronary heart disease: smoking. Zeitschrift fur Kardiologie, 94, 30-42.
- Ambrose, J.A. and Barua, R.S. (2004) The pathophysiology of cigarette smoking and cardiovascular disease: an update. Journal of the American College of Cardiology, 43, 1731-1737. doi:10.1016/j.jacc.2003.12.047
- Czernin, J., Sun, K., Brunken, R., et al. (1995) Effect of acute and long-term smoking on myocardial blood flow and flow reserve. Circulation, 91, 2891-2897. doi:10.1161/01.CIR.91.12.2891
- Ip, J.H., Fuster, V., Badimon, L., et al. (1990) Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle cell proliferation. Journal of the American College of Cardiology, 15, 1667-1687. doi:10.1016/0735-1097(90)92845-S
- Poreba, R., Skoczynska, A. and Derkacz, A. (2004) Effect of tobacco smoking on endothekial function in patients with coronary arteriosclerosis. Polskie Archiwum Medycyny Wewnetrznej, 111, 27-36.
- Palombo, C., Lubrano, V. and Sampietro, T. (1999) Oxidative stress, F2-isoprostanes and endothelial dysfunction in hypercholesterolemia. Cardiovascular Research, 44, 474-476. doi:10.1016/S0008-6363(99)00367-3
- Banerjee, M., Kang, K.H., Morrow, J.D., et al. (1992) Effects of a novel prostaglandin, 8-epi-PGF2 alpha, in rabbit lung in situ. The American Journal of Physiology, 263, H660-H663.
- Wilson, S.H., Best, P.J., Lerman, L.O., et al. (1999) Enhanced coronary vasoconstriction to oxidative stress product, 8-epi-prostaglandinF2 alpha, in experimental hypercholesterolemia. Cardiovascular Research, 44, 601- 607. doi:10.1016/S0008-6363(99)00225-4
- Morrow, J.D., Frei, B., Longmire, A.W. et al. (1995) Increase in circulating products of lipid peroxidation (F2- isoprostanes) in smokers. Smoking as a cause of oxidative damage. The New England Journal of Medicine, 332, 1198-1203. doi:10.1056/NEJM199505043321804
- Yura, T., Fukunaga, M., Khan, R. et al. (1999) Freeradical-generated F2-isoprostane stimulates cell proliferation and endothelin-1 expression on endothelial cells. Kidney International, 56, 471-478. doi:10.1046/j.1523-1755.1999.00596.x
- Fukunaga, M., Yura, T. and Badr, K.F. (1995) Stimulatory effect of 8-Epi-PGF2 alpha, an F2-isoprostane, on endothelin-1 release. Journal of Cardiovascular Pharmacology, 26, S51-S52.
- Friedewald, W.T., Levy, R.I. and Fredrickson, D.S. (1972) Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clinical Chemistry, 18, 499-502.
- Alberti, A., Bolognini, L., Macciantelli, D., et al. (2000) The radical cation of N,N-diethyl-para-phenylendiamine: a possible indicator of oxidative stress in biological samples. Research on Chemical Intermediates, 26, 253-267. doi:10.1163/156856700X00769
- Lubrano, V., Vassalle, C., L’Abbate, A., et al. (2002) A new method to evaluate oxidative stress in humans. Immunoanalyse & Biologie Spécialisée, 17, 172-175. doi:10.1016/S0923-2532(02)01188-2
- De Sutter, J., De Buyzere, M., Gheeraert, P., et al. (2001) Fibrogen and C-reactive protein on admission as markers of final infart size after primary angioplasty for acute myocardial infarctio. Atherosclerosis, 157, 189-196. doi:10.1016/S0021-9150(00)00703-6
- Buffon, A., Liuzzo, G., Biasucci, M.L., et al. (1999) A preprocedural serum levels of C-reactive protein predict early complications and late restenosis after coronary angioplasty. Journal of the American College of Cardiology, 34, 1512-1521. doi:10.1016/S0735-1097(99)00348-4
- Hoidal, J.R. and Niewoehner, D.E. (1983) Pathogenesis of emphysema. Chest, 83, 679-685. doi:10.1378/chest.83.4.679
- Kannel, W.B. (1981) Update on the role of cigarette smoking in coronary artery disease. American Heart Journal, 101, 319-328. doi:10.1016/0002-8703(81)90197-6
- Church, D.F. and Pryor, W.A. (1985) Free-radical chemistry of cigarette smoke and its toxicological implications. Environmental Health Perspectives, 64, 111-126. doi:10.1289/ehp.8564111
- Prieme, H., Loft, S., Klarlund, M., et al. (1998) Effect of smoking cessation on oxidative DNA modification estimated by 8-oxo-7,8-dihydro-2’-deoxyguanosine excretion Carcinogenesis, 19, 347-351. doi:10.1093/carcin/19.2.347
- Yokode, M., Kita, T., Arai, H., et al. (1988) Cholesteryl ester accumulation in macrophages incubated with low density lipoprotein pretreated with cigarette smoke extract. Proceedings of the National Academy of Sciences of the United States of America, 85, 2344-2348. doi:10.1073/pnas.85.7.2344
- Frei, B., Forte, T.M., Ames, B.N., et al. (1991) Gas phase oxidants of cigarette smoke induce lipid peroxidation and changes in lipoprotein properties in human blood plasma: protective effects of ascorbic acid. Biochemical journal, 277, 133-138.
- Schectman, G., Byrd, J.C. and Gruchow, H.W. (1989) The influence of smoking on vitamin C status in adults. American Journal of Public Health, 79, 158-162. doi:10.2105/AJPH.79.2.158
- Rimm, E.B., Stampfer, M.J., Ascherio, A., et al. (1993) Vitamin E consumption and the risk of coronary heart disease in men. The New England Journal of Medicine, 328, 1450-1456. doi:10.1056/NEJM199305203282004
- Singh, R.B., Niazma, M.A., Bishnoi, I., et al. (1994) Diet antioxidant vitamins, oxidative stress and risk of coronary artery disease: the peerzada prospective study. Acta Cardiologica, 49, 453-497.
- Morrow, J., Frei, B., Longmire, A., et al. (1995) Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. The New England Journal of Medicine, 332, 1198-1203. doi:10.1056/NEJM199505043321804
- Penn, A. and Snyder, C.A. (1993) Inhalation of sidestream cigarette smoke accelerates development of arteriosclerotic plaques. Circulation, 88, 1820-1825. doi:10.1161/01.CIR.88.4.1820
- Knight-Lozano, C.A., Young, C.G., Burow, D.L., et al. (2002) Cigarette smoke exposure and hypercholesterolemia increase mitochondrial damage in cardiovascular tissues. Circulation, 105, 849-854. doi:10.1161/hc0702.103977
- Ramos, K.S., Zhang, Y., Sadhu, D.N., et al. (1996) The induction of proliferative vascular smooth muscle cell phenotypes by benzo(a)pyrene is characterized by upregulation of inositol phospholipid metabolism and c-Ha-ras gene expression. Archives of Biochemistry and Biophysics, 332, 213-222. doi:10.1006/abbi.1996.0335
- Ou, X. and Ramos, K.S. (1992) Proliferative responses of quail aortic smooth muscle cells to benzo[a]pyrene: Implications in PAH-induced atherogenesis. Toxicology, 74, 243-258. doi:10.1016/0300-483X(92)90143-3
- Lubrano, V., Cocci, F., Battaglia, D., et al. (2005) Usefulness of high-sensitivity IL-6 measurement for clinical characterization of patients with coronary artery disease. Journal of Clinical Laboratory Analysis, 19, 110-114. doi:10.1002/jcla.20061