Advances in Parkinson's Disease
Vol.04 No.03(2015), Article ID:58392,9 pages
10.4236/apd.2015.43007
LDN-73794 Attenuated LRRK2-Induced Degeneration in a Drosophila Parkinson’s Disease Model
Dejun Yang1,2,3*, Sharmila Das1, Loujing Song1, Tianxia Li1, Jianqun Yan2, Wanli W. Smith1*
1Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, Baltimore, USA
2Department of Physiology and Pathophysiology, Xi’an Jiaotong University School of Medicine, Xi’an, China
3Department of Neurology, University of Massachusetts Medical School, Worcester, USA
Email: *dejun.yang@umassmed.edu, *wsmith@rx.umaryland.edu
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 10 June 2015; accepted 26 July 2015; published 29 July 2015
ABSTRACT
Parkinson’s disease (PD) is a common neurodegenerative disease with unclear pathogenesis. Cur- rently, there are no disease-modifying neuron-protecting drugs to slow down the neuronal degeneration. Mutations in the leucine-rich repeat kinase 2 (LRRK2) cause genetic forms of PD and contribute to sporadic PD as well. Disruption of LRRK2 kinase functions has become one of the potential mechanisms underlying disease-linked mutation-induced neuronal degeneration. To further characterize the pharmacological effects of a reported LRRK2 kinase inhibitor, LDN-73794, in vitro cell models and a LRRK2 Drosophila PD model were used. LDN-73794 reduced LRRK2 kinase activity in vitro and in vivo. Moreover, LDN-73794 increased survival, improved locomotor activity, and suppressed DA neuron loss in LRRK2 transgenic flies. These results suggest that inhibition of LRRK2 kinase activity can be a potential therapeutic strategy for PD intervention and LDN-73794 could be a potential lead compound for developing neuroprotective therapeutics.
Keywords:
LRRK2, Parkinson’s Disease, LDN-73794, Kinase Activity, Neuronal Degeneration, Dopamine Neuron, Drosophila Model

1. Introduction
Parkinson’s disease (PD) is a common age-related progressive neurodegenerative disease with two pathological hallmarks: loss of dopaminergic neurons and the presence of proteinaceous aggregates called Lewy bodies. Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene has been identified to cause a genetic form of PD reported in 2004 [1] [2] . LRRK2 is expressed in the cytosol in neurons, microglia, and astrocytes in brains [3] - [6] and can be detected in Lewy bodies [7] . The physiological function of LRRK2 is not clear but it may play a role in neuronal outgrowth and guidance, as well as regulating gene expression [8] - [13] .
LRRK2 contains a kinase domain and a GTPase domain with two enzymatic activities [1] [2] [14] - [16] . The physiological LRRK2 kinase substrate is not known. LRRK2 can undergo autophosphorylation and phosphorylate a generic substrate, myelin basic protein (MBP) [17] [18] . Moesin, 4E-BP, ezrine, radixin, and s15 have been reported as candidates for LRRK2 substrates [13] [19] - [21] although the physiological relevance of these proteins still requires further investigation. The majority of disease-linked LRRK2 mutations are in the GTPase domain or the kinase domain and alter the LRRK2 catalytic activity related to neuronal degeneration [12] [18] [22] - [24] . The most common PD mutation, G2019S-LRRK2, has been reported to increase kinase activity [17] - [19] [25] [26] and induce neuronal degeneration. Reduction of G2019S-LRRK2 kinase activity protects against neuronal degeneration [18] [23] [27] . Thus, inhibition of LRRK2 kinase activity may have therapeutic potential for PD intervention. Increasing efforts on developing LRRK2 kinase inhibitors lead to the discovery of several LRRK2 kinase inhibitors as “probes” to pharmacologically dissect the LRRK2 biological functions [23] [28] - [32] . Currently, most of the reported LRRK2 inhibitors, such as LDN-73794 [30] , are in the early stages of evaluation using in vitro models. Liu et al. identified LDN-73794, which can inhibit LRRK2 with an IC50 of 3.5 ± 0.3 µM by screen with a peptide (PLK-derived peptide with a motif of RRRSLLE) as a substrate [30] . LDN-73794 is a competitive inhibitor of the binding of ATP and a noncompetitive inhibitor of PLK-pep- tide. The In vitro studies showed that LDN-73794 inhibits LRRK2 kinase activity but has no effect on its GTPase activity. However, in pharmacological effects of LDN-73794 on LRRK2 in cells and in vivo models are not clear.
We recently developed a LRRK2 transgenic Drosophila model with severe locomotor impairment and robust DA neuron degeneration [34] . The human G2019S-LRRK2 is expressed under a dopaminergic neuron promoter in this model that becomes a useful in vivo animal model to test neuroprotective compounds targeting LRRK2 [28] . In this study, in vitro cell culture models and this LRRK2 Drosophila PD model were used to characterize the pharmacological effects of LDN-73794. Our results provided a proof of principle that LRRK2 kinase inhibition can be a novel therapeutic strategy for PD intervention and LDN-73794 can be a potential lead compound for further drug development.
2. Materials and Methods
2.1. Fly Stocks and Reagents
The dopa decarboxylase (ddc)-GAL4, UAS-LRRK2-1, and UAS-G2019S-2 fly stocks were maintained in our lab on standard fly food at 25˚C as described previously [34] .
Anti-tyrosine hydroxylase (TH) antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Anti-actin and anti-Flag antibodies were ordered from Sigma (St. Louis, MO, USA). LDN was purchased from Timtec. Fetal bovine serum, penicillin and streptomycin, LipofectAMINE Plus reagent and cell culture media were from Invitrogen (Carlsbad, CA).
2.2. Cell Culture and Transfection
Human HEK293T (human embryonic kidney) cells were from ATCC grown in DMEM with 10% fetal bovine serum and antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin) at 37˚C under 5% CO2/95% air. The Flag tagged wild type LRRK2 and mutant G2019S constructs were transfected into HEK293T cells using Lipofectamine™ and PLUS™ Reagents (Invitrogen) followed the manufacturer’s protocol as described previously [18] .
2.3. Mouse Primary Cortical Neuronal Cultures and Cell Toxicity Assay
Primary cortical neuronal cultures were from C57BL mice at embryonic day 15 as described previously [4] . Neurons were grown in the poly-d-lysine- and laminin-coated plates (BD Biosource, San Diego) using neurobasal medium with B-27 supplement, Glutamax, and antibiotics. To overcome the low trasfection efficiency of cultured neurons, various LRRK2 constructs were co-transfected along with GFP at 15:1 ratio into primary neurons by Lipofectamine 2000 according to the manufacturer protocol. After 48 h transfection, neurons were fixed with 4% paraformaldehyde and subjected to immunostaining using anti-Flag antibodies followed by 0.8 μg/mL of bisbenzimide (Hoechst 33,342, Sigma) labeling. Most of the GFP positive cells up to 95% expressed LRRK2 as described previously [4] [28] . Cell images were taken from 40 fields in each experimental group. The healthy GFP-positive cells were automatically counted under a fluorescence microscope (Axiovert 200, Zeiss). Cell viability was measured as described previously [28] , only the GFP positive cells were quantified.
2.4. Immunoprecipitation (IP), Western Blot, and Autophosphorylation (Kinase) Assays
Cell lysates or fly head homogenates were subjected to IP using anti-FLAG-agarose (Sigma) as described previously [4] . For western blot analysis, 4% - 12% NuPAGE Bis-Tris gels were used to separate the proteins that were then transferred into polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were probed with various primary antibodes followed by incubation with secondary detection antibodies. Proteins were detected by enhanced chemiluminescence (ECL) reagents. In vitro autophosphorylation (kinase) assay was described previously [18] . Briefly, kinase reactions were performed in kinase assay buffer with 500 µM ATP, 10 µCi of [γ-32P] ATP (3000 Ci/mmol) and 50 mM MgCl2 for 90 min. The reaction was stopped by adding Laemmli sample buffer, and boiled for 5 min. The samples were subjected to 4% - 12% SDS/PAGE and blotted onto PVDF membranes. The membranes were quantified using a phosphoimager (Bio-Rad Molecular Imager FX). For some experiments, LRRK2 autophosphorylation was performed using anti-Flag IP followed by western blot analysis using anti-phosphorylated LRRK2 antibody as described previously [33] [35] .
2.5. Survival and Climbing Assay
Cohorts of 60 flies from each experimental group were subjected to survival counting weekly. Vehicle and LDN-73794 was added into fly food from day 1 of eclosion throughout the fly lifetime. Fresh food with LDN- 73794 was changed every 3 days. Mortality was analyzed by using Kaplan-Meier survival analysis. A p value < 0.05 was considered significant. Climbing assays (negative geotaxis assay) were employed to determine locomotor activity weekly as described previously [34] . Briefly, flies were placed in a vertical plastic cylinder (diameter, 1.5 cm; length, 25 cm) and tapped to the bottom. There were 60 flies from each experimental group. The flies that could climb to or above the median line of the cylinder within 10 seconds were counted.
2.6. Whole-Mount Brain Immunostaining and Dopamine (DA) Neuron Quantification
Adult fly brains at 6 weeks of age were dissected and subjected to whole-mount immunostaining using anti-TH antibodies as described previously [34] . Cohorts of eight flies per group were subjected to immunostaining with mouse monoclonal anti-TH (Immunostar) or rabbit polyclonal anti-TH (Chemicon) antibodies as the primary antibodies. Alexa Fluor 568 or Alexa Fluor 488 goat anti-mouse IgG (Invitrogen) were used as secondary antibodies. The number of the neurons in paired anterolateral medial (PAM) results in high fluorescent density and cannot be quantified precisely. Except PAM, the numbers of TH-positive neurons in all other DA clusters were counted under fluorescent microscopy (Zeiss LSM 250).
2.7. Data Analysis
Quantitative data were expressed as arithmetic means ± SEM based on at least three separate experiments. Statistically significant differences among groups were analyzed by ANOVA using Sigmastart 3.1 statistical software (Aspire Software International, VA). A p value < 0.05 was considered significant.
3. Results
3.1. LDN-73794 Reduced LRRK2 Toxicity in Primary Neurons
To further characterize the effects of LDN-73794 [30] , HEK 293T cells were used to study its effect on LRRK2 autophosphorylation. Cells were transfected with Flag-LRRK2 for 36 hours and serum was withdrawn for an additional 10 hours, followed by addition of LDN-73794 at a 50 µM concentration for 1 hour. Cell lysates were subjected to an in vitro LRRK2 phosphorylation assay. LDN-73794 reduced LRRK2 autophosphorylation at the Ser2032 residue up to 82% compared with vehicle treated control cells (Figure 1(b) and Figure 1(c)). This is consistent with the previously findings that LDN-73794 reduced LRRK2 kinase activity using a PLK-peptide as a substrate [30] .
To assess the effect of LDN-73794 on LRRK2 induced cell toxicity, mouse primary cortical neurons at 7 days in vitro were co-transfected with wild type LRRK2 or G2019S-LRRK2 and GFP at a 15:1 ratio and treated with LDN-73794 for 48 h. LDN-73794 reduced LRRK2-induced neuron toxicity compared with vehicle treated cells expressing GFP/G2019S-LRRK2 (Figure 1(d)) at a dose dependent manner (data not shown). 5 µM LDN- 73794 significantly attenuated LRRK2-induced neural loss, and have no effects on the survival of cells expressing GFP and control vector.
3.2. LDN-73794 Increased Survival and Improved Locomotor Impairment in G2019S-LRRK2 Transgenic Flies
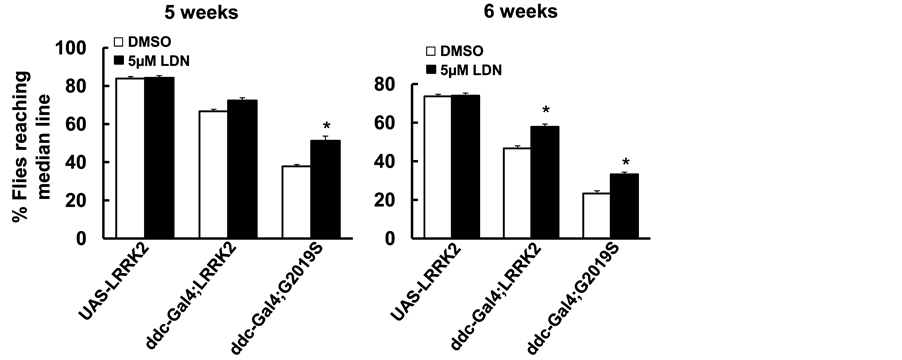
The UAS-WT-LRRK2 and UAS-G2019S-LRRK2 flies crossed with the ddc-GAL4 driver flies resulted in LRRK2 variant expression in DA neurons as described previously [34] . LDN-73794 (5 µM) was added to fly food from day 1 post-eclosion throughout lifetime. The life span of non-pathogenic control flies (UAS-LRRK2 or ddc- GAL4 flies) were about 12 - 13 weeks [34] . Flies expressing wild type and G2019S-LRRK2 significantly reduced survival rate (Figure 2) as consistent with previous findings [34] . LDN-73794 did not alter the lifespan of non-pathogenic control flies (Figure 2). However, LDN-73794 significantly increased the survival of ddc- GAL4;LRRK2 and ddc:GAL4;G2019S flies compared with vehicle treated transgenic flies (Figure 2(a) and Figure 2(b)). Flies expressing wild type or G2019S-LRRK2 in DA neurons displayed severe locomotor impairment as previously described [34] . LDN-73794 did not alter the locomotor activity of non-pathogenic flies (Figure 3(a) and Figure 3(b)). Treatment with LDN73794 significantly improved the locomotor impairment of LRRK2 and G2019S transgenic flies compared with untreated transgenic flies (Figure 3(a) and Figure 3(b)).
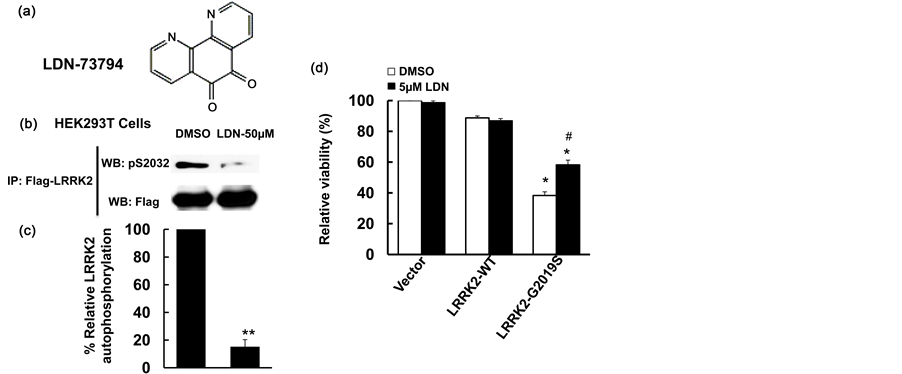
Figure 1. LDN-73794 reduced LRRK2 phosphorylation and protected against LRRK2 toxicity. (a) Che- mical structure of LDN-73794 [31] ; (b) LDN-73794 reduced LRRK2 autophosphorylation. HEK293T cells were transfected with Flag-LRRK2-G2019Sfor 36 h and followed by serum withdrawal for 10 h, then treated with LDN-73794 (0 and 50 µM) for 1 h. Cell lysates were subjected to immunoprecipitation using anti-Flag antibodies and followed by western blot analysis using anti-phospho-LRRK2 antibodies at Ser2032 residue. Shown are representative images from three independent experiments; (c) Quantitation of B. **p < 0.01 by ANOVA; (d) LDN-73794 attenuated LRRK2-induced neuronal degeneration in mouse cortical primary neurons. Mouse primary cortical neurons were co-transfected pCDNA-GFP with LRRK2-WT or LRRK2-G2019S at 1:15 ratio. After 4 h transfection, cells were treated with LDN-73794 for 44 h. The average intensity of DAPI stained nuclei of transfected cells were measured for cell viability assays as described previously [28] . Data are means ± s.d. for three separated experiments, *p < 0.05 by ANOVA, vs vehicle or LDN-73794 treated groups. #p < 0.05 by ANOVA, vs untreated cells expressing LRRK2 or LRRK2-G2019S.
 (a) (b)
(a) (b)
Figure 2. LDN-73794 increased survival of LRRK2 transgenic flies. The ddc-GAL4; LRRK2 (a) or ddc-GAL4; G2019S (b) flies were left untreated or treated with 5 µM LDN-73794 from day 1 post-eclosion throughout lifetime. Cohorts of 60 flies were in each experimental group. Survival data were analyzed by Kaplan-Meier log Rank survival analysis. p < 0.05, statistically significant differences between non-pathogenic and LRRK2 transgenic lines; between vehicle and LDN-73794 treated transgenic flies.
 (a) (b)
(a) (b)
Figure 3. LDN-73794 improved locomotor impairment of G2019S-LRRK2 transgenic flies. The ddc-GAL4; LRRK2 or ddc-GAL4; G2019S flies were left untreated or treated with 5 µM LDN-73794 from day 1 post-eclo- sion until 6 weeks of age. Cohorts of 60 flies (5 - 6 weeks of age) from each experimental group were subjected to climbing assays. (a) The climb assay result at 5 weeks; (b) The climb assay result at 6 weeks. Values are mean ± s.e.m. *: p < 0.05 by ANOVA, vs vehicle treated LRRK2 transgenic flies.
3.3. LDN-73794 Suppressed DA Neuron Loss in G2019S-LRRK2 Transgenic Flies
To assess the effect of LDN-73794 on LRRK2-induced DA neuron degeneration in flies, whole-mount brain immunostaining was employed to detect the DA neurons using anti-TH antibodies. Expression of both wild type and G2019S-LRRK2 reduced the TH-positive neurons compared with non-pathogenic flies (Figure 4), consistent with previous reports [34] . Mutant G2019S transgenic flies resulted in severe loss of TH-positive neurons. Treatment with LDN-73794 suppressed G2019S-LRRK2-induced loss of DA neurons in all clusters (Figure 4). LDN-73794 significantly improved the TH-positive neuron loss in the PPM1/2 clusters (Figure 4(b) and Figure 4(c)). To further assess the effects of LDN-73794 in vivo, flies expressing mutant G2019S-LRRK2 were treated LDN-73794 for 2 weeks. Fly head homogenates were subjected to phosphorylation and kinase activity assays. LDN-73794 significantly attenuated LRRK2 phosphorylation in both wild type and G2019S-LRRK2 flies compared with vehicle treated transgenic flies (Figure 5).

Figure 4. LDN-73794 attenuated LRRK2-induced DA neuron loss. Flies were left untreated or treated with 5 µM LDN-73794 from day 1 post-eclosion until 6 weeks of age. The fly brains were dissected at 6 weeks of age and subjected to immunostaining using anti-TH antibodies. The DA neurons were counted in all clusters except PAM. (a) Quantification of total DA neurons in all clusters except PAM; (b) Quantification of TH-positive neurons in PPM1/2 clusters; (c) Representative images of DA neurons in PPM1 and PPM2 clusters at 6 weeks of age. *p < 0.05 by ANOVA, vs non-transgenic flies; #p < 0.05 by ANOVA, vs vehicle treated LRRK2 transgenic flies.

Figure 5. LDN-73794 reduced LRRK2 kinase activity in transgenic flies. G2019S-LRRK2 transgenic flies were left untreated or treated with 5 µM LDN-73794 for 2 weeks. Fly head homogenates were subjected to LRRK2 phosphorylation ((a) and (c)) or LRRK2 in vitro kinase ((b) and (d)) assays. (a) and (c), Fly head homogenates were immunoprecipitated using anti-Flag antibodies. The resulting immunoprecipitates were subjected to Western blot analysis using anti-phosphorylated LRRK2 antibodies against S2032 residue. (a) shown are representative blots from three independent experiments; (c) The quantitation of (a). Values are mean ± s.e.m. *p < 0.05 by ANOVA as indicated; (b) and (d). Fly head homogenates were subjected to in vitro LRRK2 kinase assays using p32 incorporation as described previously [28] [34] ; (b) Shown are representative image from three independent kinase assays; (d) The quantitation of (b), **p < 0.01 by ANOVA.
4. Discussion
In this study, characterization of a LRRK2 kinase inhibitor, LDN-73794, demonstrated that it reduced LRRK2 autophosphorylation at Ser2032 in HEK293T cells and protected against mutant G2019S-LRRK2 toxicity in mouse primary neurons. Moreover, LDN-73794 significantly increased survival, improved locomotor activity, and suppressed DA neuron loss in a LRRK2 Drosophila model. LDN-73794 also reduced G2019S-LRRK2 phosphorylation in brains of transgenic flies. These results suggest that inhibition of LRRK2 kinase activity can be a potential therapeutic strategy for PD intervention and LDN-73794 could be a potential lead compound for developing novel therapeutics.
LRRK2 kinase activity is altered by several disease-linked LRRK2 mutations in the GTPase or kinase domain including R1441C, R1441H, R1441G, Y1699C, G2019S, and I2020T [12] [18] [36] . The most common PD- linked mutation, G2019S, contributes to about 40% of genetic PD cases and some of the sporadic PD cases as well [37] . G2019S consistently increased LRRK2 protein kinase activity and caused severe neuronal degeneration in cultured neurons [4] [18] [24] [38] . Genetic alterations reducing kinase activity suppress LRRK2-toxicity [18] [25] . Our data showed that LDN-73794 could cross the cell membrane and inhibit LRRK2 phosphoryation in HEK293T cells expressing recombinant LRRK2 proteins. Treatment of LDN-73794 in primary neurons expressing mutant LRRK2 significantly reduced the neuronal degeneration. These results are consistent with recent findings that inhibition of LRRK2 kinase activity by the Raf kinase inhibitors, GW 5074 and sorafenib, displays neuron protective effects against LRRK2 toxicity [22] [28] [38] .
DA neuronal degeneration is the early event in PD pathogenesis. To date, there are still no neuron-protecting drugs for PD treatment. One of the barriers to developing protective compounds is that rodent in vivo genetic testing models require a very long experimental time period. Moreover, LRRK2 transgenic rodent models [36] lack DA neuron degeneration or only display subtle degeneration, which hinders the drug testing [36] .
Our Drosophila PD model expressing human mutant G2019S-LRRK2 provides the unique whole animal testing system for identifying neuronal protection compounds [34] . Unlike LRRK2 transgenic mouse models [36] , flies expressing mutant LRRK2 displays severe motor dysfunction and selective DA neuron degeneration [34] . The Drosophila model provides a critical testing tool linked between in vitro model testing and clinical trials for developing PD-related compounds. In current study, we found that LDN-73794 significantly suppressed LRRK2-induced toxicity in cell and Drosophila models via reducing LRRK2 kinase activity. Although more detailed characterization(including the potential side effects) in animal models are remain to be examined, the Our findings provided the in vivo evidence that LDN-73794 inhibits LRRK2’s kinase activity and suppresses the PD-like phenotype that can become the starting point for drug development to treat PD.
Acknowledgements
We thank Dr. Ted D. Dawson for providing the anti-phosphorylated LRRK2 antibodies.
Cite this paper
DejunYang,11,11,SharmilaDas,LoujingSong,TianxiaLi,JianqunYan,Wanli W.Smith, (2015) LDN-73794 Attenuated LRRK2-Induced Degeneration in a Drosophila Parkinson’s Disease Model. Advances in Parkinson's Disease,04,49-58. doi: 10.4236/apd.2015.43007
References
- 1. Paisan-Ruiz, C., Jain, S., Evans, E.W., Gilks, W.P., Simon, J., van der, B.M., Lopez, D.M., Aparicio, S., Gil, A.M., Khan, N., Johnson, J., Martinez, J.R., Nicholl, D., Carrera, I.M., Pena, A.S., de Silva, R., Lees, A., Marti-Masso, J.F., Perez-Tur, J., Wood, N.W. and Singleton, A.B. (2004) Cloning of the Gene Containing Mutations That Cause PARK8-Linked Parkinson’s Disease. Neuron, 44, 595-600. http://dx.doi.org/10.1016/j.neuron.2004.10.023
- 2. Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., Kachergus, J., Hulihan, M., Uitti, R.J., Calne, D.B., Stoessl, A.J., Pfeiffer, R.F., Patenge, N., Carbajal, I.C., Vieregge, P., Asmus, F., Muller-Myhsok, B., Dickson, D.W., Meitinger, T., Strom, T.M., Wszolek, Z.K. and Gasser, T. (2004) Mutations in LRRK2 Cause Autosomal-Domi- nant Parkinsonism with Pleomorphic Pathology. Neuron, 44, 601-607.
http://dx.doi.org/10.1016/j.neuron.2004.11.005 - 3. Biskup, S., Moore, D.J., Rea, A., Lorenz-Deperieux, B., Coombes, C.E., Dawson, V.L., Dawson, T.M. and West, A.B. (2007) Dynamic and Redundant Regulation of LRRK2 and LRRK1 Expression. BMC Neuroscience, 8, 102. http://dx.doi.org/10.1186/1471-2202-8-102
- 4. Smith, W.W., Pei, Z., Jiang, H., Moore, D.J., Liang, Y., West, A.B., Dawson, V.L., Dawson, T.M. and Ross, C.A. (2005) Leucine-Rich Repeat Kinase 2 (LRRK2) Interacts with Parkin, and Mutant LRRK2 Induces Neuronal Degeneration. Proceedings of the National Academy of Sciences of the USA, 102, 18676-18681. http://dx.doi.org/10.1073/pnas.0508052102
- 5. Biskup, S., Moore, D.J., Celsi, F., Higashi, S., West, A.B., Andrabi, S.A., Kurkinen, K., Yu, S.W., Savitt, J.M., Waldvogel, H.J., Faull, R.L., Emson, P.C., Torp, R., Ottersen, O.P., Dawson, T.M. and Dawson, V.L. (2006) Localization of LRRK2 to Membranous and Vesicular Structures in Mammalian Brain. Annals of Neurology, 60, 557-569.
http://dx.doi.org/10.1002/ana.21019 - 6. Moehle, M.S., Webber, P.J., Tse, T., Sukar, N., Standaert, D.G., De Silva, T.M., Cowell, R.M. and West, A.B. (2012) LRRK2 Inhibition Attenuates Microglial Inflammatory Responses. Journal of Neuroscience, 32, 1602-1611.
http://dx.doi.org/10.1523/JNEUROSCI.5601-11.2012 - 7. Zhu, X., Siedlak, S.L., Smith, M.A., Perry, G. and Chen, S.G. (2006) LRRK2 Protein Is a Component of Lewy Bodies. Annals of Neurology, 60, 617-618.
http://dx.doi.org/10.1002/ana.20928 - 8. Mac Leod, D., Dowman, J., Hammond, R., Leete, T., Inoue, K. and Abeliovich, A. (2006) The Familial Parkinsonism Gene LRRK2 Regulates Neurite Process Morphology. Neuron, 52, 587-593. http://dx.doi.org/10.1016/j.neuron.2006.10.008
- 9. Sakaguchi-Nakashima, A., Meir, J.Y., Jin, Y., Matsumoto, K. and Hisamoto, N. (2007) LRK-1, a C. elegans PARK8- Related Kinase, Regulates Axonal-Dendritic Polarity of SV Proteins. Current Biology, 17, 592-598. http://dx.doi.org/10.1016/j.cub.2007.01.074
- 10. Sheng, D., Qu, D., Kwok, K.H., Ng, S.S., Lim, A.Y., Aw, S.S., Lee, C.W., Sung, W.K., Tan, E.K., Lufkin, T., Jesuthasan, S., Sinnakaruppan, M. and Liu, J. (2010) Deletion of the WD40 Domain of LRRK2 in Zebrafish Causes Parkinsonism-Like Loss of Neurons and Locomotive Defect. PLOS Genetics, 6, e1000914. http://dx.doi.org/10.1371/journal.pgen.1000914
- 11. Lee, S.B., Kim, W., Lee, S. and Chung, J. (2007) Loss of LRRK2/PARK8 Induces Degeneration of Dopaminergic Neurons in Drosophila. Biochemical and Biophysical Research Communications, 358, 534-539. http://dx.doi.org/10.1016/j.bbrc.2007.04.156
- 12. Cookson, M.R. (2010) The Role of Leucine-Rich Repeat Kinase 2 (LRRK2) in Parkinson’s Disease. Nature Reviews Neuroscience, 11, 791-797. http://dx.doi.org/10.1038/nrn2935
- 13. Martin, I., Kim, J.W., Lee, B.D., Kang, H.C., Xu, J.C., Jia, H., Stankowski, J., Kim, M.S., Zhong, J., Kumar, M., Andrabi, S.A., Xiong, Y., Dickson, D.W., Wszolek, Z.K., Pandey, A., Dawson, T.M. and Dawson, V.L. (2014) Ribosomal Protein s15 Phosphorylation Mediates LRRK2 Neurodegeneration in Parkinson’s Disease. Cell, 157, 472-485.
http://dx.doi.org/10.1016/j.cell.2014.01.064 - 14. Cookson, M.R., Dauer, W., Dawson, T., Fon, E.A., Guo, M. and Shen, J. (2007) The Roles of Kinases in Familial Parkinson’s Disease. The Journal of Neuroscience, 27, 11865-11868.
http://dx.doi.org/10.1523/JNEUROSCI.3695-07.2007 - 15. Moore, D.J. (2008) The Biology and Pathobiology of LRRK2: Implications for Parkinson’s Disease. Parkinsonism & Related Disorders, 14, S92-S98.
http://dx.doi.org/10.1016/j.parkreldis.2008.04.010 - 16. Gandhi, P.N., Chen, S.G. and Wilson-Delfosse, A.L. (2009) Leucine-Rich Repeat Kinase 2 (LRRK2): A Key Player in the Pathogenesis of Parkinson’s Disease. Journal of Neuroscience Research, 87, 1283-1295. http://dx.doi.org/10.1002/jnr.21949
- 17. West, A.B., Moore, D.J., Biskup, S., Bugayenko, A., Smith, W.W., Ross, C.A., Dawson, V.L. and Dawson, T.M. (2005) Parkinson’s Disease-Associated Mutations in Leucine-Rich Repeat Kinase 2 Augment Kinase Activity. Proceedings of the National Academy of Sciences of the United States of America, 102, 16842-16847. http://dx.doi.org/10.1073/pnas.0507360102
- 18. Smith, W.W., Pei, Z., Jiang, H., Dawson, V.L., Dawson, T.M. and Ross, C.A. (2006) Kinase Activity of Mutant LRRK2 Mediates Neuronal Toxicity. Nature Neuroscience, 9, 1231-1233.
http://dx.doi.org/10.1038/nn1776 - 19. Jaleel, M., Nichols, R.J., Deak, M., Campbell, D.G., Gillardon, F., Knebel, A. and Alessi, D.R. (2007) LRRK2 Phosphorylates Moesin at Threonine-558: Characterization of How Parkinson’s Disease Mutants Affect Kinase Activity. Biochemical Journal, 405, 307-317.
http://dx.doi.org/10.1042/BJ20070209 - 20. Imai, Y., Gehrke, S., Wang, H.Q., Takahashi, R., Hasegawa, K., Oota, E. and Lu, B. (2008) Phosphorylation of 4E-BP by LRRK2 Affects the Maintenance of Dopaminergic Neurons in Drosophila. The EMBO Journal, 27, 2432-2443. http://dx.doi.org/10.1038/emboj.2008.163
- 21. Gloeckner, C.J., Schumacher, A., Boldt, K. and Ueffing, M. (2009) The Parkinson Disease-Associated Protein Kinase LRRK2 Exhibits MAPKKK Activity and Phosphorylates MKK3/6 and MKK4/7, in Vitro. Journal of Neurochemistry, 109, 959-968.
http://dx.doi.org/10.1111/j.1471-4159.2009.06024.x - 22. Tan, E.K. and Schapira, A.H. (2010) LRRK2 as a Therapeutic Target in Parkinson’s Disease. European Journal of Neurology, 18, 545-546.
- 23. Lee, B.D., Shin, J.H., Van Kampen, J., Petrucelli, L., West, A.B., Ko, H.S., Lee, Y.I., Maguire-Zeiss, K.A., Bowers, W.J., Federoff, H.J., Dawson, V.L. and Dawson, T.M. (2010) Inhibitors of Leucine-Rich Repeat Kinase-2 Protect against Models of Parkinson’s Disease. Nature Medicine, 16, 998-1000. http://dx.doi.org/10.1038/nm.2199
- 24. Xiong, Y., Coombes, C.E., Kilaru, A., Li, X., Gitler, A.D., Bowers, W.J., Dawson, V.L., Dawson, T.M. and Moore, D.J. (2010) GTPase Activity Plays a Key Role in the Pathobiology of LRRK2. PLoS Genetics, 6, e1000902. http://dx.doi.org/10.1371/journal.pgen.1000902
- 25. Greggio, E., Jain, S., Kingsbury, A., Bandopadhyay, R., Lewis, P., Kaganovich, A., van der Brug, M.P., Beilina, A., Blackinton, J., Thomas, K.J., Ahmad, R., Miller, D.W., Kesavapany, S., Singleton, A., Lees, A., Harvey, R.J., Harvey, K. and Cookson, M.R. (2006) Kinase Activity Is Required for the Toxic Effects of Mutant LRRK2/Dardarin. Neurobiology of Disease, 23, 329-341. http://dx.doi.org/10.1016/j.nbd.2006.04.001
- 26. Luzon-Toro, B., de la Torre, E.R., Delgado, A., Perez-Tur, J. and Hilfiker, S. (2007) Mechanistic Insight into the Dominant Mode of the Parkinson’s Disease-Associated G2019S LRRK2 Mutation. Human Molecular Genetics, 16, 2031-2039.
http://dx.doi.org/10.1093/hmg/ddm151 - 27. Liu, Z., Hamamichi, S., Dae, L.B., Yang, D., Ray, A., Caldwell, G.A., Caldwell, K.A., Dawson, T.M., Smith, W.W. and Dawson, V.L. (2011) Inhibitors of LRRK2 Kinase Attenuate Neurodegeneration and Parkinson-Like Phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s Disease Models. Human Molecular Genetics, 20, 3933-3942.
http://dx.doi.org/10.1093/hmg/ddr312 - 28. Yang, D., Li, T., Liu, Z., Arbez, N., Yan, J., Moran, T.H., Ross, C.A. and Smith, W.W. (2012) LRRK2 Kinase Activity Mediates Toxic Interactions between Genetic Mutation and Oxidative Stress in a Drosophila Model: Suppression by Curcumin. Neurobiology of Disease, 47, 385-392. http://dx.doi.org/10.1016/j.nbd.2012.05.020
- 29. Deng, X., Dzamko, N., Prescott, A., Davies, P., Liu, Q., Yang, Q., Lee, J.D., Patricelli, M.P., Nomanbhoy, T.K., Alessi, D.R. and Gray, N.S. (2011) Characterization of a Selective Inhibitor of the Parkinson’s Disease Kinase LRRK2. Nature Chemical Biology, 7, 203-205.
http://dx.doi.org/10.1038/nchembio.538 - 30. Liu, M., Dobson, B., Glicksman, M.A., Yue, Z. and Stein, R.L. (2010) Kinetic Mechanistic Studies of Wild-Type Leucine-Rich Repeat Kinase 2: Characterization of the Kinase and GTPase Activities. Biochemistry, 49, 2008-2017. http://dx.doi.org/10.1021/bi901851y
- 31. Liu, M., Poulose, S., Schuman, E., Zaitsev, A.D., Dobson, B., Auerbach, K., Seyb, K., Cuny, G.D., Glicksman, M.A., Stein, R.L. and Yue, Z. (2010) Development of a Mechanism-Based High-Throughput Screen Assay for Leucine-Rich Repeat Kinase 2—Discovery of LRRK2 Inhibitors. Analytical Biochemistry, 404, 186-192.
http://dx.doi.org/10.1016/j.ab.2010.05.033 - 32. Deng, X., Choi, H.G., Buhrlage, S.J. and Gray, N.S. (2012) Leucine-Rich Repeat Kinase 2 Inhibitors: A Patent Review. Expert Opinion on Therapeutic Patents, 22, 1415-1426.
http://dx.doi.org/10.1517/13543776.2012.729041 - 33. Li, T., Yang, D., Zhong, S., Thomas, J.M., Xue, F., Liu, J., Kong, L., Voulalas, P., Hassan, H.E., Park, J.S., MacKerell Jr., A.D. and Smith, W.W. (2014) Novel LRRK2 GTP-Binding Inhibitors Reduced Degeneration in Parkinson’s Disease Cell and Mouse Models. Human Molecular Genetics, 23, 6212-6222.
- 34. Liu, Z., Wang, X., Yu, Y., Li, X., Wang, T., Jiang, H., Ren, Q., Jiao, Y., Sawa, A., Moran, T., Ross, C.A., Montell, C. and Smith, W.W. (2008) A Drosophila Model for LRRK2-Linked Parkinsonism. Proceedings of the National Academy of Sciences of the United States of America, 105, 2693-2698. http://dx.doi.org/10.1073/pnas.0708452105
- 35. Li, X., Moore, D.J., Xiong, Y., Dawson, T.M. and Dawson, V.L. (2010) Reevaluation of Phosphorylation Sites in the Parkinson Disease-Associated Leucine-Rich Repeat Kinase 2. The Journal of Biological Chemistry, 285, 29569-29576.
http://dx.doi.org/10.1074/jbc.M110.127639 - 36. Li, T., Yang, D., Sushchky, S., Liu, Z. and Smith, W.W. (2011) Models for LRRK2-Linked Parkinsonism. Parkinson’s Disease, 2011, Article ID: 942412.
http://dx.doi.org/10.4061/2011/942412 - 37. Kumari, U. and Tan, E.K. (2009) LRRK2 in Parkinson’s Disease: Genetic and Clinical Studies from Patients. FEBS Journal, 276, 6455-6463.
http://dx.doi.org/10.1111/j.1742-4658.2009.07344.x - 38. Iaccarino, C., Crosio, C., Vitale, C., Sanna, G., Carri, M.T. and Barone, P. (2007) Apoptotic Mechanisms in Mutant LRRK2-Mediated Cell Death. Human Molecular Genetics, 16, 1319-1326. http://dx.doi.org/10.1093/hmg/ddm080
Abbreviations
PD, Parkinson Disease;
LRRK2, Leucine-Rich Repeat Kinase 2;
TH, Tyrosine Hydroxylase;
PAL, Protocerebral Antero Lateral;
PAM, Paired Anterolateral Medial;
PPM, Protocerebral Posterior Medial;
DA, Dopamine;
IP, Immunoprecipitation;
ddc, The Dopa Decarboxylase.
NOTES

*Corresponding authors.