American Journal of Plant Sciences
Vol.07 No.03(2016), Article ID:64401,9 pages
10.4236/ajps.2016.73037
Standardization of DNA Extraction Method from Mature Dried Leaves and ISSR-PCR Conditions for Melia dubia Cav. ―A Fast Growing Multipurpose Tree Species
Swati Rawat1, Geeta Joshi1*, D. Annapurna1, A. N. Arunkumar1, Nataraja N. Karaba2
1Tree Improvement and Genetics Division, Institute of Wood Science and Technology, Bengaluru, India
2Department of Crop Physiology, University of Agricultural Sciences GKVK, Bengaluru, India

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 19 January 2016; accepted 6 March 2016; published 10 March 2016
ABSTRACT
Melia dubia Cav. of family Meliaceae is a fast growing, high value tree species native to India. Isolating DNA from matured dried leaves of M. dubia was difficult due to accumulation of secondary metabolites, majorly polyphenolics, which resulted in dark brown to black colour of the pellet. In this study, a modified STE-(Sucrose, Tris-HCl and Ethylene Diamine Tetra Acetic Acid) CTAB (hexadecyltrimethylammonium bromide) method was standardized for removal of polyphenolics. The protocol developed yielded 200 - 1000 ng/µl of quality DNA without any impurities as evident by A260/280 ratio ranging from 1.75 - 2.0. It was also suitable for extracting quality DNA from other members of Meliaceae like Azadirachta indica and Melia azedarach. In downstream applications, the extracted DNA was used for PCR amplification by using ISSR and SSR markers. ISSR PCR conditions were optimized in a reaction volume of 25 µl, consisting of 30 ng of template DNA, 1.5 mM MgCl2, 200 µM of each of dNTPs and 2 U of Taq polymerase. The best amplification was observed and the same was applicable for SSR markers.
Keywords:
DNA Extraction, Downstream Applications, ISSR, Mature Dried Leaves, Melia dubia, SSR

1. Introduction
Melia dubia Cav. (Meliaceae) is a large deciduous fast growing tree species, commonly known as Malabar Neem. It is an indigenous multipurpose tree species, naturally distributed in Sikkim Himalayas, North Bengal, upper Assam, Khasi hills of Orissa, Deccan and Westran Ghats at altitudes of 1500 - 1800 m [1] [2] . This species is an excellent raw material for wood based industries like paper and plywood due to its increased pulp recovery and strength [2] . The wood is also used for packing cases, ceiling planks, construction, agricultural implements, pencils, match boxes, splints and furniture [1] . M. dubia has medicinal properties and the extract from different parts of the plant has antiviral activity [3] . The leaf oil is reported to exhibit bacteriostatic, fungistatic [4] and antifeedant activity [5] . The fruits of M. dubia are used in folk medicine as an antihelmintic, astringent and in the treatment of colic [6] . Paste made out of the green fruits is used in treatment of scabies and maggot- infested sores [7] . Despite of its high value and wide spread use, there is no information available on its existing germplasm diversity. Therefore it becomes very essential to catalogue its natural genetic diversity to raise quality planting stock as well as to preserve this valuable species for long term.
The most crucial step for any molecular study is isolation of pure, intact and high-quality DNA [8] . However, due to excessive presence of secondary metabolites, isolating pure DNA from plants is very difficult as compared to animals and microorganisms [9] . Various factors are responsible for degrading DNA during extraction. One such problem is the presence of endonucleases which directly or indirectly interferes with enzymatic reactions [10] . Presence of polysaccharides makes DNA more viscous [11] , inhibits Taq polymerase activity [12] and restriction enzyme activity [13] . Another major problem encountered with most of the fully developed and mature leaves is accumulation of polyphenolics and tannins. These, when in oxidized, form covalently binds with DNA and make it resistant to restriction enzymes and give DNA a brown colour [14] . Various methods have been developed for plants in order to encounter all these problems and isolate high quality DNA [12] [15] -[21] .
The biochemical composition of each plant and tree species varies considerably. So it is quite impossible to develop a single isolation protocol which suits every plant species. It has been found that even closely related species require different protocol [10] . In addition to a reliable DNA extraction protocol, the storage of leaf tissues is also very important [22] . Most of the reported DNA extraction protocols use young leaves for DNA extraction. However, when the samples are to be collected from distant places it is always not possible to cryogenically store the leaf till extraction. Unlike evergreen species, in deciduous species young leaves are not available throughout the year. So, drying and storage of leaf tissues is very important. Therefore, there is a need of such protocols which could extract DNA from matured dried leaves. Moreover, the DNA isolated must be useful to several downstream protocols such as cloning and other related molecular applications.
The aim of present study was to standardize protocol for extraction of quality DNA from mature dried leaves of M. dubia. We also report the optimization of ISSR-PCR which could be useful for diversity analysis of this species.
2. Materials and Methods
Fresh mature green leaf samples were collected from different locations of Karnataka, India. Leaves were cleaned with distilled water, dried and put in zip lock covers containing silica gel for complete removal of moisture. Upon dehydration, dry leaves were separated from silica gel and stored at −20˚C until use.
2.1. DNA Extraction and Quantification
2.1.1. DNA Isolation
Two genomic DNA extraction kits were employed 1) GF-1 Plant DNA extraction Kit (Vivantis technologies Sdn. Bhd., Malaysia) and 2) DNAsure plant Mini Kit (Genetix Biotech Asia Pvt. Ltd., New Delhi). Different methods employed to extract DNA were CTAB method [23] , CTAB method [23] with varying concentration of PVPP and β-mercaptoethanol, protocol developed for extracting DNA from mature dried leaves of Quercus humboldtii and Colombobalanus excelsa [17] , DNA isolation from four angiospermic plants [24] and for plants from mangroves and salt marshes [18] .
In addition to the above protocols, a new modified CTAB method was developed and standardized to isolate high quality DNA from mature dried leaves of M. dubia. The standardized protocol was as follows:
Incubation with Extraction buffer-I: Leaf sample (400 mg) was ground to a fine powder in a ceramic mortar and pestle using liquid nitrogen with 25 mg of PVPP. The powdered leaf tissues were immediately transferred to a 20 ml centrifuge tube containing 4 ml of prewarmed extraction buffer-I (0.5 M Sucrose, 120 mM TrisHCl, 50 mM EDTA and 1.7 M NaCl) and 100 µl of β-mercaptoethanol was added to it and vortexed well. Freshly prepared extraction buffer-I was used. Tubes were incubated at 65˚C for 60 minutes. Then centrifuged at 10,000 rpm for 10 minutes and supernatant was transferred to a new tube.
Incubation with Extraction buffer-II: Prewarmed 4 ml extraction buffer-II (100 mM TrisHCl, 20 mM EDTA, 1.7 M NaCl, 2% CTAB) was added to the supernatant and tubes were kept on incubation for 30 minutes at 65˚C. After incubation an equal volume of chloroform: isoamylalcohol (24:1) was added and mixed vigorously by inverting 15 - 20 times. Samples were centrifuged at 12,000 rpm for 10 minutes. Aqueous phase was transferred to a new tube. 25 µl of RNase (20 mg/ml) was added to the tubes and incubated at 37˚C on dry bath for an hour. 20 µl Proteinase K (20 mg/ml) treatment was also given for the same time. Chloroform: Isoamylalcohol separations were done until clear solution was obtained. Clear supernatant was taken and 1/4th volume of 5 M NaCl and equal volume of chilled isopropanol was added and gently inverted for several times and kept at −20˚C overnight. Next day samples were centrifuged at 12,000 rpm for 10 minutes. DNA pellet was given 70% (v/v) ethanol washes for 3 times, then air dried at room temperature for 2 - 3 hours and dissolved in 50 - 100 µl of TE buffer (10 mM TrisHCl and 1mM EDTA pH-8.0) and stored at −20˚C for further use.
2.1.2. Quantitative and Qualitative Analysis of Isolated DNA
To check the purity, the purified DNA was separated on 0.8% (w/v) agarose gel prepared using 1X TAE buffer and visualized on Gel Documentation system (Herolab, Germany). DNA was quantified using Biospectrophotometer (Eppendorf, Germany) at a wavelength of 260 and 280 nm. Purity was checked from ratio of absorbance 260/280 [25] .
2.2. Optimization of ISSR-PCR Conditions
For standardization of PCR condition, in 25 µl reaction mixture, varying concentration of DNA (10 to 70 ng), dNTPs (100 to 300 µM), Taq polymerase (0.5 to 2.5 U) and MgCl2 (0.0 to 3.5 mM) (Table 1) were tried in DNA thermocycler (Eppendorf mastercycler gradient, Germany). Different concentration of agarose gel (1.5 to 3.0 %) was also tried. PCR condition was set at initial denaturation of 94˚C for 3 minutes, followed by 94˚C for 30 seconds, specific annealing temperature of primer for 30 seconds, elongation for 1 minute at 72˚C and the cycle was repeated 39 cycles and a final extension step at 72˚C for 10 minutes and a hold temperature of 4˚C at the end. The PCR product was analyzed on 2% (w/v) agarose gel and visualized under gel documentation system (Herolab, Germany). Standardized conditions were used for SSR markers also.
3. Results and Discussion
3.1. DNA Extraction and Quantification
The isolation of genomic DNA from mature dried leaves of M. dubia was complicated due to high contamination of polyphenolics, polysaccharides and proteins. Accumulation of polyphenolics increases with leaf development [26] , and it reduces DNA quality in mature leaves than in young leaves. For DNA isolation, fresh young leaves could not be used all the time as the locations were in distant areas and collections were done at different periods of time. To avoid oxidation leaves were dried in Silica Gel.

Table 1. Optimization of ISSR-PCR reaction parameters for M. dubia.
DNA extracted using commercially available kits contained polyphenolic impurities (Figure 1, Lane 1 and 2) with poor yield and A260/280 value less than 1.6 which did not meet optimal limit of 1.8 [25] .
Genomic DNA extracted using CTAB protocol [23] yielded dark brown to black colour pellet showing very high contamination of polyphenolics (Figure 1, Lane 3). Next, we attempted to modify the CTAB protocol [23] with varying quantities of PVPP and β-mercaptoethanol to remove polyphenols released during cell lysis [27] . Addition of antioxidants did not help much as the pellet obtained was brown in colour. Later a protocol containing LiCl in extraction buffer for extracting DNA from mature leaves was employed [17] . The pellet was light brown in colour, the quantity of DNA obtained was very low and absorbance ratio was between 0.99 and 1.33 (Table 2), without any band (Figure 1, Lane 4). CTAB protocol was further modified by adding STE as described
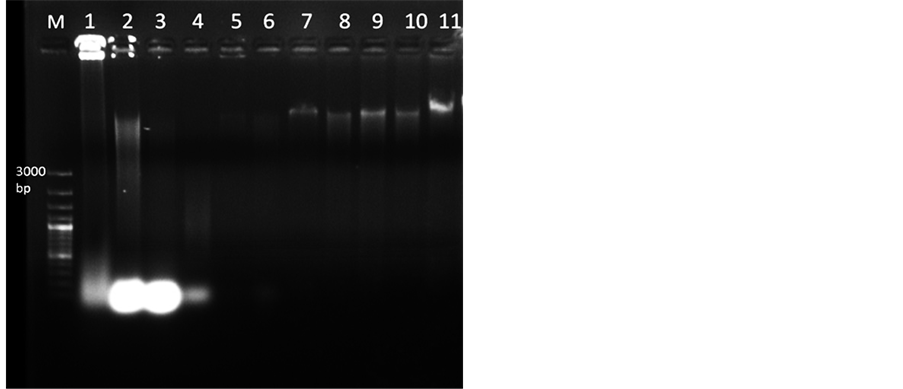
Figure 1. Genomic DNA isolated from mature and dried M. dubia leaves (Lane 1 to 9) resolved under 0.8% agarose. Lane M, 100 bp plus DNA marker, Lane 1-2 DNA isolated through GF-1 Plant DNA extraction Kit (Vivantis) and DNAsure Plant Mini Kit (Genetix) respectively. Lane 3, DNA isolated by CTAB protocola. Lane 4, DNA extracted by protocol for Quercus humboldtii and Colombobalanus excelsab. In Lane 5 DNA extracted by protocol for plants from mangroves and salt marshesc. Lane 6 shows DNA isolated through protocol for four angiospermic plantsd. Lane 7 shows DNA from supernatant by STE buffer having 1.0 M NaCl. Lane 8 shows DNA from pellet by STE buffer with 1.7 M NaCl. Lane 9, DNA isolated through standardized protocol from supernatant through STE buffer with 1.7 M NaCl. Lanes 10-11 DNA isolated from dried leaves of Azadirachta indica and Melia azedarach respectively using standardized protocol. Note: a reference [20] , breference [17] , creference [18] , dreference [21] .
Table 2. Comparison of different protocols opted for DNA extraction.
Note: areference [20] , breference [17] , creference [21] , dreference [18] .
in protocol used for four angiosperms [24] . Pellet obtained in this case was brown in colour with absorbance ratio 1.1 - 1.3 (Figure 1, Lane 6). As per protocol for mangrove plants [18] , by incubating with STE buffer (Sucrose, TrisHCl and EDTA) with 1.0 M NaCl, further processing of the pellet did not yield good DNA (Figure 1, Lane 5). However, incubating with STE buffer along with 1.0 M NaCl and taking supernatant for further processing resulted in quality DNA (Figure 1, Lane 7). Further increasing the concentration of NaCl to 1.7 M and taking the pellet also did not improve the quality of DNA much (Figure 1, Lane 8). The DNA quantity and quality was further improved by incubating the sample with STE buffer containing 1.7 M NaCl (extraction buffer -I) and taking the supernatant, as observed by spectrophotometer readings (1.75 - 2.0) and ethidium bromide stained agarose gel image (Figure 1, Lane 9). Incubation time with extraction buffer-I was increased from 40 to 60 minutes which improved the process of breakage of cells and nuclear membrane thereby improving the quality of DNA [28] . Increased concentrations of NaCl help in breakage of cell membrane and inhibit co-precipitation of polysaccharides with DNA by increasing polysaccharide solubility in ethanol [29] . Although sucrose has been used in extraction buffer for total DNA isolation from plant cells, its role is not very specific, but if omitted it can adversely affect DNA yield in 50 % of the cases [24] .
The other factors which helped in extracting quality DNA are use of liquid nitrogen, addition of PVPP, β- mercaptoethanol and higher concentrations of NaCl (extraction buffer-2). Liquid nitrogen was used for grinding the tissues which helped in creating non-oxidative environment and also thick cell wall was easy to grind as it was tough to homogenize dry leaves in buffer. During homogenization, polyphenols are released from vacuoles and they then react rapidly with cytoplasmic enzymes so PVP purges and forms complex hydrogen bonds with polyphenols and gets precipitated, which can easily be separated from DNA by centrifugation [30] . β-mercap- toethanol acts as strong reducing agent in higher concentrations and helps in reducing the polyphenols [31] .
The standardized CTAB protocol yielded good quality of DNA, as depicted by A260/280 ratio ranging from 1.75 - 2.0 indicating insignificant levels of impurities in comparison to other protocols (Table 2). The quantity of DNA ranged from 200 - 1000 ng/µl. The agarose gel electrophoresis (0.8%) of genomic DNA isolated through various protocols showed that the standardized CTAB protocol yielded high quantity of DNA (Figure 1, Lane 9). Standardized protocol was also used to extract DNA from other members of Meliaceae, Azadiracta indica and Melia azedarach and it worked for these two species. DNA quality and quantity ranged from 1.87 to 2.0 and 300 to 310 ng/µl respectively (Table 2), with intact band (Figure 1, Lane 10 and 11).
3.2. Optimization of ISSR-PCR Conditions
DNA samples isolated through standardized DNA extraction protocol were used as template for ISSR-PCR which showed good amplification. Experiments were further carried out for optimizing the parameters of the ISSR- PCR. The optimal content of DNA depends on the kind of material and purity of DNA [32] . Studies have shown that for good amplification template DNA ranges between 5 - 500 ng for most of the tree species [33] . We tested template DNA (10 - 70 ng), and the target amplification did not differ for 10, 20 and 30 ng of DNA. Using the three concentrations, we carried out PCR analysis with three different sets of primers i.e. UBC 855, UBC 891 and UBC 847. The template DNA of 30 ng was found to be optimum (Figure 2(a), Lane 9). Other researchers have also reported that 30 ng DNA as optimal quantity for these types of PCR [32] . Concentration of 200 µM for each of dNTPs gave the best results (Figure 2(b), Lane 3) when tested with range of 100, 150, 200, 250, and 300 µM with primer UBC 813. dNTPs concentration had great influence on ISSR-PCR amplified results [34] . Taq polymerase activity was tested with the gradient concentrations between 0.5 U to 2.5 U with primer UBC 870 and 2.0 U produced clear bands (Figure 2(c), Lane 4). In the earlier reports it has been shown that Taq polymerase, a thermo-stable enzyme, which assembles a new DNA strand from the nucleotides, with lower concentrations did not amplify and increased concentrations showed good amplification with difference in specificity [35] [36] .
Since Mg2+ ion influenced PCR, we attempted to vary MgCl2 concentrations starting from 0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5 mM with primer UBC 888. The optimum concentration for ISSR was found to be 1.5 mM (Figure 2(d), Lane 4). As the concentration of MgCl2 was increased, the bands were not clear and formation of smear was observed. Mg2+ concentration have been an important factor in opening the double helix of primer and template DNA and stimulating Taq polymerase activity [32] . In an earlier study, excess of MgCl2 concentrations has shown non-specific amplifications or poor yield of amplicons [36] .
DNA fragment analysis depends on the pore size of the agarose gel. The lower the concentration of the agarose,

Figure 2. Optimization of ISSR-PCR parameters for M. dubia. (a) Optimization of template DNA, lane 1-3 contains 10 ng of DNA lane 4-6 contains 20 ng of DNA and lane 7-9 contains 30 ng of DNA with 3 primers UBC 855, UBC 891 and UBC 847 respectively. (b) dNTPs concentration from 100, 150, 200, 250, 300 µM with primer UBC 813 (lane 1-5). (c) Grading concentration of Taq polymerase (0.5, 1.0, 1.5, 2.0, 2.5 U) with primer UBC 870 from lane 1 to 5. (d) Varying MgCl2 concentration, Lane 1 to 8 showing 0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5 mM of MgCl2 with primer UBC 888. (e) Concentration of agarose from 1.5% to 3.0% with primer UBC 813 and UBC 845.
the larger the pore size and hence larger the DNA that can be sieved. However, low-concentration gel (0.1% - 0.2%) are fragile, difficult to handle and in the higher concentration of gels, electrophoresis of large DNA molecules can take several days. Therefore, varying concentrations of agarose were tested (1.5%, 2%, 2.5%, 3.0%) with two primers (UBC 813 and UBC 845) and 2% gel was found optimum for running the PCR products that we generated in our experiment (Figure 2(e)).
DNA extracted from samples collected from different locations were run using the optimized PCR parameters by using ISSR markers (Table 1) and it showed very clear amplification with primer UBC 864 (Figure 3). The standardized conditions of ISSR-PCR (Table 1) were used for SSR marker MSSR7 with 1.5 µl of each forward (F: ACGCAAAGCTTCGAGAACCTTCAA) and reverse primer (10 pm/µl) (R: ACGATGTGGGCGTTCTAC- GCA) at annealing temperature (Ta) 62˚C and the amplified products showed very clear bands in 2.5% agarose gel (Figure 4).
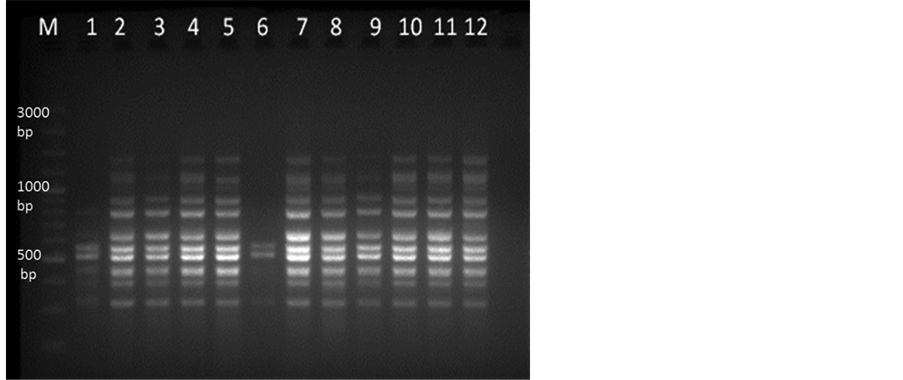
Figure 3. ISSR-PCR amplification in 2% Agarose gel. Lane M represent 100 bp plus DNA marker and lane 1-12 showing M. dubia DNA samples collected from different locations of Karnataka with primer UBC 864.
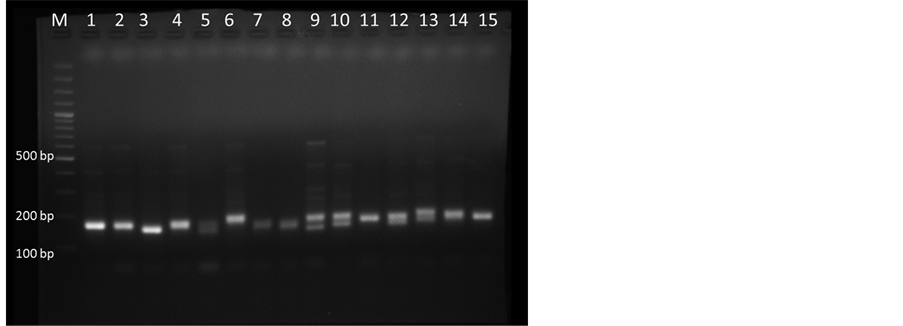
Figure 4. SSR-PCR amplification in 2.5% Agarose gel. Lane M, 100 bp plus DNA marker and lane 1-15 representing M. dubia DNA samples collected from different locations of Karnataka with primer MSSR7.
In this study, a fast inexpensive DNA extraction protocol was standardized for M. dubia, which enabled to extract highly pure DNA from mature dried leaves containing high content of polyphenolics and secondary metabolites. This protocol was also applicable for extracting quality DNA in other Meliaceae members. Extracted DNA showed very good amplification with ISSR and SSR markers which would be useful for molecular studies in M. dubia.
Acknowledgements
Authors gratefully acknowledge the financial support provided by Karnataka Forest Department to carry out this study (APCCF (RU)A1/RAC/CR-39/2012-13).
Cite this paper
SwatiRawat,GeetaJoshi,D.Annapurna,A. N.Arunkumar,NatarajaN. Karaba, (2016) Standardization of DNA Extraction Method from Mature Dried Leaves and ISSR-PCR Conditions for Melia dubia Cav. —A Fast Growing Multipurpose Tree Species. American Journal of Plant Sciences,07,437-445. doi: 10.4236/ajps.2016.73037
References
- 1. Anon (1966) The Wealth of India. Raw Materials, Vol. VI. L-M, CSIR, Delhi.
- 2. Parthiban, K.T., Bharathi, A.K., Seenivasan, R., Kamala, K. and Rao, M.G. (2009) Integrating Melia dubia in Agroforestry Farms as an Alternate Pulpwood Species. Asia Pacific Agroforestry Newsletter, 34, 3-4.
- 3. Vijayan, P., Raghu, C., Ashok, G., Dhanaraj, S.A. and Suresh, B. (2004) Antiviral Activity of Medicinal Plants of Nilgiris. Indian Journal of Medical Research, 120, 24-29.
- 4. Nagalakshmi, M.A.H., Thangadurai, D. and Pullaiah, T. (2003) In Vitro Antimicrobial Efficacy of Leaf Essential Oils of Chukrasia tabularis Adr. Juss. and Melia dubia Cav. (Meliaceae). Phytotherapy Research, 17, 414-416.
http://dx.doi.org/10.1002/ptr.1147 - 5. Koul, O., Singh, G., Singh, R., Singh, J., Daniewski, W.M. and Berlozecki, S. (2004) Bioefficacy and Mode-of-Action of Some Limonoids of Salannin Group from Azadirachta indica A. Juss and Their Role in a Multicomponent System against Lepidopteran Larvae. American Journal of Bioscience, 29, 409-416.
http://dx.doi.org/10.1007/BF02712112 - 6. Kiritkar, K.R. and Basu, B.D (1999) Indian Medicinal Plants, Vol. 1. International Book Distributors, Dehradun.
- 7. Susheela, T., Balaravi, P., Theophilus, J., Reddy, N.T. and Reddy, P.U.M. (2008) Evaluation of Hypoglycaemic and Antidiabetic Effect of Melia dubia Cav. Fruits in Mice. Current Science, 94, 1191-1195.
- 8. Tan, S.C. and Yiap, B.C. (2009) DNA, RNA, and Protein Extraction: The Past and the Present. Journal of Biomedicine and Biotechnology, 2009, Article ID: 574398.
- 9. Sangwan, N.S., Sangwan, R.S. and Kumar, S. (1998) Isolation of Genomic DNA from the Antimalarial Plant Artemisia annua. Plant Molecular Biology Reporter, 16, 365.
http://dx.doi.org/10.1023/A:1007545102889 - 10. Weising, K., Nybom, H., Wolff, K. and Meyer, W. (1995) DNA Isolation and Purification. In: Editor’s name, Ed., DNA Fingerprinting in Plants and Fungi, CRC Press, Boca Raton, 44-59.
- 11. Do, N. and Adams, R.P. (1991) A Simple Technique for Removing Polysaccharide Contaminants from DNA. BioTechniques, 10, 162-166.
- 12. Fang, G., Hammar, S. and Grumet, R. (1992) A Quick and Inexpensive Method for Removing Polysaccharides from Plant Genome DNA. BioTechniques, 13, 52-56.
- 13. Pandey, R.N., Adams, R.P. and Flournoy, L.E. (1996) Inhibition of Random Amplified Polymorphic DNAs (RAPDs) by Plant Polysaccharides. Plant Molecular Biology Reporter, 14, 17-22.
http://dx.doi.org/10.1007/BF02671898 - 14. Katterman, F.R.H. and Shattuck, V.I. (1983) An Effective Method of DNA Isolation from Mature Leaves of Gossypium Species That Contain Large Amounts of Phenolic Terpinoids and Tannins. Preparative Biochemistry, 13, 347-359.
http://dx.doi.org/10.1080/00327488308068177 - 15. Porebski, S., Bailey, L.G. and Baum, B.R. (1997) Modification of a CTAB DNA Extraction Protocol for Plants Containing High Polysaccharides and Polyphenol Component. Plant Molecular Biology Reporter, 15, 8-15.
http://dx.doi.org/10.1007/BF02772108 - 16. Lefort, F. and Douglas, G.C. (1999) An Efficient Micro-Method of DNA Isolation from Mature Leaves of Four Hardwood Tree Species Acer, Fraxinus, Prunus, and Quercus. Annals of Forest Science, 56, 259-263.
http://dx.doi.org/10.1051/forest:19990308 - 17. Fernandez, J.F., Sork, V.L., Gallego, G., Lopez, J., Bohorques, A. and Tomhe, J. (2000) Cross-Amplification of Microsatellite Loci in a Neotropical Quercus Species and Standardization of DNA Extraction from Mature Leaves Dried in Silica Gel. Plant Molecular Biology Reporter, 18, 397.
http://dx.doi.org/10.1007/BF02825070 - 18. Sahu, S.K., Thangaraj, M. and Kathiresan, K. (2012) DNA Extraction Protocol for Plants with High Levels of Secondary Metabolites and Polysaccharides without Using Liquid Nitrogen and Phenol. ISRN Molecular Biology, 2012, Article ID: 205049.
http://dx.doi.org/10.5402/2012/205049 - 19. Das, S.S., Das, S. and Ghosh, P. (2013) Optimization of DNA Isolation and RAPD-PCR Protocol of Acanthus volubilis wall., Arare Mangrove Plant from Indian Sundarban, for Conservation Concern. European Journal of Experimental Biology, 3, 33-38.
- 20. Bhandari, A., Lamichhane, N., Lamichhane, S. and Shakya, P. (2014) An Efficient Method for Isolation of Genomic DNA from Silica Gel Dried Leaves of Taxus sps. International Journal of Biosciences, 5, 147-152.
http://dx.doi.org/10.12692/ijb/5.5.147-152 - 21. Pandey, A. and Tamta, S. (2015) High-Molecular-Weight DNA Extraction from Six Quercus Species of Himalaya, India. International Journal of Advanced Research, 3, 30-34.
- 22. Khanuja, S.P.S., Shasany, A.K., Darokar, M.P. and Kumar, S. (1999) Rapid Isolation of DNA from Dry and Fresh Samples of Plants Producing Large Amounts of Secondary Metabolites and Essential Oil. Plant Molecular Biology Reporter, 17, 74.
http://dx.doi.org/10.1023/A:1007528101452 - 23. Doyle, J.J. and Doyle, J.L. (1987) A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochemical Bulletin, 19, 11-15.
- 24. Ahmed, S.M., Ganaie, M.M., Qazi, P.H., Vijeshwar, V., Basir, S.F. and Qazi, G.N. (2004) Rapid DNA Isolation Protocol for Angiospermic Plants. Bulgarian Journal of Plant Physiology, 30, 25-33.
- 25. Sambrook, J., Maccallum, P. and Russell, D. (2001) Molecular Cloning: A Laboratory Manual. 3rd Edition, Cold Spring Harbor Press, Cold Spring Harbor, 2344 p.
- 26. Coley, P.D. and Barone, J.A. (1996) Herbivory and Plant Defences in Tropical Forests. Annual Review of Ecology and Systematics, 27, 305-335.
http://dx.doi.org/10.1146/annurev.ecolsys.27.1.305 - 27. Lodhi, M.A., Ye, G.N., Weeden, N.F. and Reisch, B.I. (1994) A Simple and Efficient Method of DNA Extraction from Grapevine Cultivars and Vitis Species and Ampelopsis. Plant Molecular Biology Reporter, 12, 6-13.
http://dx.doi.org/10.1007/BF02668658 - 28. Ginwal, H.S. and Maurya, S.S. (2010) Evaluation and Optimization of DNA Extraction Method for Dalbergia sissoo Leaf. Indian Journal of Biotechnology, 9, 69-73.
- 29. Moreira, P.A. and Oliveira, D.A. (2011) Leaf Age Affects the Quality of DNA Extracted from Dimorphandra mollis (Fabaceae), a Tropical Tree Species from the Cerrado Region of Brazil. Genetics and Molecular Research, 10, 353-358.
http://dx.doi.org/10.4238/vol10-1gmr1030 - 30. Maliyakal, E.J. (1992) An Efficient Method for Isolation of RNA and DNA from Plants Containing Polyphenolics. Nucleic Acids Research, 20, 2381.
http://dx.doi.org/10.1093/nar/20.9.2381 - 31. Suman, P.S.K., Ajit, K.S., Darokar, M.P. and Sushil, K. (1999) Rapid Isolation of DNA from Dry and Fresh Samples of Plants Producing Large Amounts of Secondary Metabolites and Essential Oils. Plant Molecular Biology Reporter, 17, 1-5.
http://dx.doi.org/10.1023/A:1017213630972 - 32. Pan, H., Yang, C.-P., Wei, Z.-G. and Jiang, J. (2006) DNA Extraction of Birch Leaves by Improved CTAB Method and Optimization of Its ISSR System. Journal of Forestry Research, 17, 298-300.
http://dx.doi.org/10.1007/s11676-006-0068-3 - 33. Huang, J.C. and Sun, M. (2000) Genetic Diversity and Relationship of Sweet Potato and Its Wild Relatives in Pomoea Series Batatas as Revealed by Inter-Simple Sequence Repeat (ISSR) and Restriction Analysis of Chloroplast. Theoretical and Applied Genetics, 10, 1050-1060.
http://dx.doi.org/10.1007/s001220051386 - 34. Zhang, H.Q., An, L.J. and Zu, Y.G. (1999) Effect on the Experimental Results of the Content of Compositions in RAPD Experiment of Pinus koraiensis. Bulletin of Botanical Research, 19, 183-186.
- 35. Padmalatha, K. and Prasad, M.N.V. (2006) Optimization of DNA Isolation and PCR Protocol for RAPD Analysis of Selected Medicinal and Aromatic Plants of Conservation Concern from Peninsular India. African Journal of Biotechnology, 5, 230-234.
- 36. Harini, S.S., Leelambika, M., Shiva Kameshwari, M.N. and Sathyanarayana, N. (2008) Optimization of DNA Isolation and PCR-RAPD Methods for Molecular Analysis of Urginea indica Kunth. International Journal of Integrative Biology, 2, 138-144.
NOTES

*Corresponding author.