American Journal of Plant Sciences
Vol.05 No.21(2014), Article ID:50398,10 pages
10.4236/ajps.2014.521329
Rapid and Reliable Method of High-Quality RNA Extraction from Diverse Plants
Saroj Kumar Sah*, Gurwinder Kaur, Amandeep Kaur
School of Agricultural Biotechnology, Punjab Agricultural University, Ludhiana, India
Email: *saroj-biotec@pau.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 19 August 2014; revised 19 September 2014; accepted 2 October 2014
ABSTRACT
The isolation of high quality RNA is a crucial technique in plant molecular biology. The quality of RNA determines the reliability of downstream process like real time PCR. In this paper, we re- ported a high quality RNA extraction protocol for a variety of plant species. Our protocol is time effective than traditional RNA extraction methods. The method takes only an hour to complete the procedure. Spectral measurement and electrophoresis were used to demonstrate RNA quality and quantity. The extracted RNA was further used for cDNA synthesis, expression analysis and copy number determination through Real Time PCR. The results indicate that RNA was of good quality and fit for real time PCR. This high throughput plant RNA extraction protocol can be used to isolate high quality RNA from diverse plants for real time PCR and other downstream applications.
Keywords:
RNA Extraction, Diverse Plants, Trizol, High Quality Protocol, Real Time PCR

1. Introduction
High quality RNA extraction is an important step for gene expression studies. As a nucleic acid, RNA is used for protein synthesis, also widely employed in studies of gene expression pattern in different plants. Types and quantity of RNA in plants depends upon expression of particular genes, which leads to a particular phenotype. Obtaining a sufficient quantity of pure RNA is more challenging for next generation sequencing of transcrip- tomes and Quantitative Real Time PCR (qPCR) analysis [1] . RNA is mostly single stranded, often contains ri- bose sugar that carries 2’ hydroxyl group that makes the RNA more subjected to hydrolysis than genomic DNA. It is more complicated to obtain high quality RNA especially when plant samples contain high level of RNAase, large quantities of polysaccharides, low concentration of nucleic acid (high water content), different types of phenolics like tannis and fibrous tissues such as lignin (wood), that are difficult to remove and lysis [2] . The phenolic substances (i.e. Polysachharides and polyphenols) bind irreversibly with proteins and nucleic acids, leading to oxidation and degradation that makes it unsuitable for downstream applications [3] - [6] . RNA extrac- tion becomes difficult due to the presence of these secondary metabolites, that’s why RNA extraction protocols needs modification. Number of methods for RNA extraction in various plant species has been developed, most of these are plant specific, many of these are practised for particular model organisms. So the use of these pro- tocols for other non model species is somewhat limited [1] . Method for total RNA isolation from durian tissues has been devised by Ky et al. [7] . Ghawana et al. [8] described a phenol-based method for RNA isolation in plant species i.e. rich in secondary metabolites such as rheum (Rheum austral D. Don) and arnebia (Arnebia euchroma (Royle) I. M. Johnst.). Vasanthaiah et al. [9] devised the efficient protocol for functional RNA isola- tion in different grape tissue. Gehrig et al. [10] proposed a polyethylene glycol (PEG) based method to extract RNA from tissues with high presence of polysaccharides and polyphenols (e.g., Aloe L., Ananas Mill., Clusia L., Euphorbia L.). Many protocols have been proposed for RNA extraction from specific type of tissue, such as the trizol-based methods proposed for RNA extraction from siliques and seeds of Arabidopsis (DC) Heynh. [11] or the seeds of Davidia involucrata Baill. [12] . Many methods focus on rapid RNA isolation, but these are special- ly proposed for model organisms (e.g., Arabidopsis leaves) and are limited to PCR-based downstream applica- tions (e.g., Berendzen et al. [13] ). More than 50 references have been published for plant RNA extraction me- thods from last 3 years. Approximately 90% have been tested in only single plant lineage [14] [15] and mostly are modification of the cetyltrimethylammonium bromide (CTAB) method with polyvinylpyrrolidone [16] [17] . Recently, Yockteng et al. [1] published a high quality RNA extraction method for high quality RNA for diverse types of plants for gene expression analysis and next generation sequencing.
Because of extraction techniques variability and the quality of their products, the comparative analysis of ge- nomes or transcriptomes across plant lineages and tissue types remains a challenge for plant evolution research- ers. Most isolation methods are complex and have low throughputs due to presence of polysaccharides and po- lyphenol. Thus, these methods produce low RNA yields. Real time PCR has been used as a major analytical platform for quantification of nucleic acid, due to its greater sensitivity and accuracy and better quantitative performance as compared to conventional PCR [18] . For real time PCR, it is important to isolate high quality RNA because impurities in RNA affect PCR amplification and influence the reliability of real time [19] . Al- though real time PCR provides increased high throughput and speed but high quality RNA isolation has lagged. Therefore a cost effective and high throughput method is needed to produce high quality RNA.
Keeping in view the above facts, we have standardised a high throughput protocol which can be utilized for wide range of plant tissues across a broad range of taxa. We tested this protocol with different explants of dif- ferent crops like cereals crops (rice, wheat, maize), vegetable crops (onion), commercial crop (sugarcane). Our protocol is safer, easiest and cost-efficient than other RNA extraction methods. The purpose of this study is to evaluate the efficiency of Trizol to isolate high yield and quality of RNA from different types of tissues. Our motive is to introduce this protocol and not to discount other protocols that work well in particular cases, but the aim is to present a single protocol that works well across a broad variety of plant tissue types and plant species within an hour.
2. Materials and Methods
2.1. Plant Materials
Fresh leaves and callus of rice (Oryza sativa), leaf samples of wheat (Triticum aestivum), Wheat kernels (Triti- cum aestivum), roots of maize (Zea mays), leaf samples of onion (Allium cepa) and spindles of sugarcane (Sac- charum officinalis) were collected from glasshouse of “School of Agricultural Biotechnology, Punjab Agricul- tural University, Ludhiana” and used for RNA extraction with following method.
2.2. Reagents
Trizol (Life technologies Cat No. # 15596-018), Tri reagent (Sigma-Aldrich®, USA, Cat No. # T9424), Tri extract, Fast RNA® Green kit (MP biomedicals, USA, Cat No. # 116045-050)
Liquid nitrogen and dry ice
Chloroform (Sisco Research Laboratories Pvt. Ltd., India, Cat No. # 032967)
Isopropanol (2-propanol) (Sisco Research Laboratories Pvt. Ltd., India, Cat No. # 092956)
Diethyl pyrocarbonate (DEPC) (Amresco, USA, Cat No. # E174-100G)
Ethanol (Changshu Yangyuan Chemical, China, Cat No. # XK-13011-00009)
70% DEPC treated Ethanol
Sodium dodecyl sulphate (SDS) (Amresco, USA, Cat No. # 0227-1KG)
Nuclease free water (Promega, USA, Cat No. # P1193)
3-(N-morphorinio) propane sulphonic acid (MOPS) buffer (Hi-media, India, Cat No. # TC067)
2X RNA loading dye (Thermo scientific, USA, Cat No. # R0641)/Glycerol (Hi-media, India Cat No. # MB- 060-500 ML)
Formaldehyde (Hi-media, India, Cat No. # AS017-5L)
Ribo Ruler High Range RNA ladder (Thermo scientific, USA, Cat No. # SM1823)
Agarose (Hi-media, India, Cat No. # MB002-500G)
RNAseZap® (Ambion®/Life technologies, USA Cat No. # AM9780)
2.3. Equipments
Fume hood (Optional)
Centrifuge (Eppendrof, Germany)
Pre-cooled pestle and mortar
Vortex mixture (Labnet, India)
Gel electrophoresis system (Bio-Rad, USA)
Nano Drop ND-1000 Spectrophotometer (Thermoscientific)
Spectrophotometer (Hitachi, Japan)
Gel documentation system (Avegene, USA)
2.4. Preparation for RNA Isolation
The preparation for total RNA isolation is quite critical than DNA isolation as in RNA isolation the risk of RNase (catalyzes degradation of RNA) contamination is always there, that unlike DNase does not require any cofactor such as Mg2+ ion for its activity. Therefore, to remove RNAse contamination, all the glasswares and plasticwares should be sterilized.
2.5. Preparation of 0.1% DEPC Water
Add 1 ml of DEPC into 1 litre autoclaved distilled water and allow it to stand for overnight (12 hr) at 37˚C.
2.6. Sterilization with DEPC Water
For DEPC water treatment microtubes (1.5 and 2 ml) and tips (10 μl, 200 μl and 1 ml) were fully immersed in DEPC water overnight at 37˚C. DEPC water was also added into the mortars in which pestles were immersed. After overnight treatment the tips and microtubes were filled in tip boxes and jam jars respectively. The tip boxes and jam jars were kept in hot air oven at 80˚C for overnight baking, which results in complete drying of mois- ture. Thereafter, all the material was autoclaved at 121˚C for 20 min. The pestles-mortars were baked at 300˚C for 4 h incubation before their use for samples grinding.
2.7. Non-Disposable Plastic Ware
Plastic ware should be thoroughly rinsed with a solution of 0.1 M NaOH and 1mM EDTA, followed by rinsing with RNase free water. Chloroform resistant plasticwares should be rinsed with chloroform to inactivate RNases. Glasswares should be treated with DEPC (0.1%), allow it to stand for overnight (12 hrs) at 37˚C and then autoc- laved to eliminate residual DEPC.
2.8. Electrophoresis Tank
Electrophoresis tank should be cleaned with a detergent solution (0.5% SDS) thoroughly rinsed with RNase free water, then rinsed with ethanol and allow it to dry.
2.9. Sample Collection
The most critical step in RNA isolation procedure is sample collection, because the common sources of RNAse contamination are human contact, environmental exposure and aqueous solutions. For sample collection, aseptic scissor, autoclaved falcon tubes, tissue paper, gloves, RNAse Zap and liquid nitrogen are required. Gloves were swabbed with RNAse Zap solution to avoid RNAse contamination. Then young leaves were cleaned with tissue paper to remove dust, cut with scissor and put in autoclaved falcon tubes and then kept in liquid nitrogen. But in case of root samples, we need more precautions while collecting the samples. For this first of all take out the roots from soil, then remove the soils with the help of tissue paper or cotton after that kept it in liquid nitrogen. In case of callus, excised 2 - 3 mm size callus and crush immediately without storing it in liquid nitrogen.
2.10. Basic Protocol
The Trizol reagent was developed by Chomczynski and Sacchi [20] is a mixture of phenol and guanidine iso- thiocyanate. Trizol maintain RNA integrity during homogenization, solubilising or precipitating other cellular components. For the effective use of the plant RNA reagents from different manufacturers, we modified the fol- lowing protocol with some additional steps apart from manufacturers’ instructions. As indicated all solutions are prepared with sterile RNase free water, and all supplies and handling materials are cleaned with RNaseZap (Ambion) prior to storage. This protocol is optimized to isolate RNA from approximately 50 mg of plant tissues. If the amount of plant tissue is increased, reagent volumes must be increased accordingly.
2.10.1. Homogenization of Tissues
To obtain high yield of RNA, grinding is a critical point in the extraction of genetic material. So, grinding should be fine as much as possible and temperature should be low as much as possible to avoid degradation. Take 50 mg of sample and grind it with the help of liquid nitrogen in mortar and pestle that was sterilized and baked, then add 1 ml of Trizol/Tri reagent/Tri extract per 50 - 100 mg of tissue in 2 ml microcentrifuge tube. Sample volume should be less than 100 µl. Mix the sample by vigorously shaking by hand or by briefly vortex- ing until the sample is thoroughly resuspended.
2.10.2. Phase Separation
In this step, incubate the samples for 5 minutes at room temperature. After that add 0.2 ml of chloroform to each tube and shake vigorously by hand for 15 seconds, then incubate the samples for 5 minutes at room temperature. After the incubation centrifuge for 15 minutes at 12,000 ×g (RCF) at 4˚C or room temperature.
2.10.3. RNA Precipitation
Transfer the upper aqueous phase to a fresh tube. Then add 0.5 ml of isopropyl alcohol to precipitate RNA (If this RNA will be used for RT-PCR, first add 50 µl isopropyl alcohol to precipitate RNA, mix it and incubate samples at room temperature for 5 min and centrifuge at 12,000 ×g (RCF) for 10 minutes at 4˚C or room tem- perature), Mix it gently, incubate the samples at room temperature for 10 minutes and centrifuge at 12,000 ×g (RCF) for 10 minutes at 4˚C or room temperature. The RNA will form a white minute pellet on side or bottom of the tube.
2.10.4. RNA Washing
Discard the supernatant gently and wash the pellet with 1 ml of chilled 75% ethanol (Freshly prepared in DEPC water). Mix samples by vortexing for 15 secs, then centrifuge at 12,000 ×g (RCF) for 5 minutes at 4˚C or room temperature. Repeat this step twice.
2.10.5. Disolving the RNA
Remove the supernatant gently, then air dry the pellet not more than 5 minutes. Dissolve the pellet in 60 µl RNase free water (commerical) or autoclaved DEPC treated water.
2.10.6. Storage
For the short time storage we can put it in −20˚C and for long time storage put it in −80˚C.
2.11. Determination of RNA Concentration and Purity
RNA concentration and purity can be assessed by two ways, i.e. agarose gel electrophoresis and spectrophoto- meter.
The presence and overall quality of RNA preparation was determined by electrophoresis on denaturing 1.2% Agarose gel at 80 V for 45 min in 1× MOPS buffer [21] (Table 1). The gel electrophoresis tank, combs, mould, measuring cylinder should be cleaned properly with 0.5% SDS followed by ethanol.
The concentration of RNA was quantified in ND-1000 UV/VIS spectrophotometer (Hitachi, Japan). The RNA was diluted in autoclaved double distilled water to make dilution factor of 100, i.e. 1 µl RNA sample + 99 µl autoclaved double distilled water. The quantification of RNA was done in duplicates. The O.D values were tak- en at 260 and 280 nm. The RNA concentration was determined as per the formula given below.
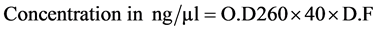
2.12. Optional Cleanup
If the sample is not clean, the following modified cleanup procedure will help to purify the total RNA. This protocol is adapted from that published for DNA cleanup by Rohland and Reich [22] , using magnetic beads to capture nucleotide material and permit additional washing steps that aid in the removal of undesirable metabo- lites. All stock solutions and reagents must be prepared with RNase-free water.
2.13. cDNA Synthesis
One μg of total RNA was used to synthesize cDNA with 1st strand cDNA synthesis kit (TakaRa Prime scriptTM, Japan) according to manufacturer’s instructions. The authenticity of cDNA was confirmed using 26S rRNA gene specific primers 26SF and 26SR (Table 2, primer set 1; Singh et al. [23] ). The reaction mixture (20 μl) con- tained 30 ng cDNA (2 μl), 10 μM of each primer (1 μl), 1 mM dNTPs (3 μl), 25 μM MgCl2 (1.2 μl), 5× PCR buffer (4 μl) and 5 units/μl TaqDNA polymerase (0.25 μl) (Promega, USA) and sterile water (7.55 μl). The PCR amplification profile consisted of an initial denaturation at 95˚C for 5 min, followed by 30 cycles of 95˚C for 1 min, 55˚C for 2 min and 72˚C for 2 min and a final extension at 72˚C for 5 min. The amplicons were electro-

Table 1. Composition of 10× MOPS buffer.
Preparation of 1.2% denaturing gel: Take 1.2 gm agarose in 250 ml Conical flask and dissolved in 100 ml 1× MOPS buffer and heat in microwave oven for 2 min. After cooling, add 37% formaldehyde 1.78 ml and add 2 µl EtBr and pour it in casting tray. After solidification use it for RNA quantification. Preparation of RNA sample: Add 2 volumes of 2× RNA loading buffer to 1 volume of RNA sample and denature at 70˚C for 10 minutes. Followed by cooling. Rinse well with buffer and load the RNA samples into lanes. Run the gel for approximately 1.5 hr at 80 V.
Table 2. Primers used for PCR amplification.
phoretically separated on 1.0 per cent agarose gel at 80 V for 2 h, stained with ethidium bromide, visualized un- der UV light and photographed using photo gel documentation system (Avegene, USA).
2.14. Quantitiative Real Time PCR
The gene expression was analyzed by qRT-PCR using iQ™ SYBR® Green supermix (BioRad, USA). The qRT- PCR primers were designed for the reference genes, i.e. actin-11 for rice (Table 2, Primer Set 2, Jain et al. [24] ) and β-tubulin for sugarcane (Table 2, Primer Set 3, Iskandar et al. [25] ) seperately using oligo analysis tools (http://eu.idtdna.com/pages). The qRT-PCR analyses were performed on iQ5 (BioRad, USA) in 20 μl volume containing 2 μl of cDNA (30 ng) template and 18 μl of PCR master-mixture. The PCR master-mixture contained 10 µM of each qPCR primer (1 µl), 2× PCR SYBR® Green supermix (10 µl) and sterile water (6 µl). The actin genes were analyzed using the following profile: 95˚C for 3 min, then 40 cycles of 95˚C for 10 s, 60˚C for 30 s, and 72˚C for 30 s and 81 cycles for melt curve at 55˚C - 95˚C for 30 s. The assays were performed in tripli- cates.
3. Results and Discussion
3.1. Measuring RNA Concentration, Purity and Yield by Spectrophotometry
To assess the presence and purity of extracted RNA, use the ratio of absorbance at 230 nm, 260 nm, and 280 nm. If the RNA is pure, we expect a 260/280 ratio around 2, although this ratio does not guarantee pure RNA. If the ratio is appreciably lower, it is an indication of the presence of protein, phenol, or other contaminants that absorb strongly at or near 280 nm. The ratio 260/230 is expected to be around 2 - 2.2. If this value is appreciably lower, it is an indication that contaminants such as carbohydrates, EDTA, guanidine isothiocyanate, and phenol that absorb at 230 nm are present in the sample. Ratios lower than expected could indicate that additional cleaning is necessary and the optional cleanup should be followed. In this experiment, all the extracted RNA showed a 260/280 nm absorbance ratio of 1.98 - 2.14 and 260/230 ratio of 2.05 - 2.24 (Table 3).
3.2. Analysis of RNA Quality by Electrophoresis
Total RNA from different crop samples was analysed on 1.2% denaturing formaldehyde agarose gel electrophoresis [21] and high quality RNA was obtained (Figure 1). There are three distinct bands were visible on gels, the first band represents 28S, 2nd represents 18S and 3rd band represents 5S RNA, indicate that our method provides high quality RNA without degradation. If RNA is not of good quality there will be smearing and distinct band will not be visible (Figure 2).

Figure 1. Total RNA isolated from different explants (S1 and S2: Leave sample of rice; S3 and S4: Leave sample of wheat; S5 and S6: Kernels of wheat; S7 and S8: Root sample of maize; S9 and S10: Leave sample of onion; S11 and S12: Leave sample of sugarcane).

Figure 2. Smearing representing poor quality RNA.
3.3. Critical Step
The major steps before and during RNA isolation is sterilization. We have to do proper sterilization as men- tioned above. Working area should be properly cleaned and sterile. Tissue should be immediately homogenized in Trizol after crushing. During layer separation, aqueous phase should be free of organic contamination. RNA pellet should not be air dry for more than 5 minutes otherwise it will be very difficult to dissolve.
3.4. cDNA Synthesis and Its Confirmation
The primary cDNA was synthesized from approximately 1 μg of total
Table 3. Concentration, purity and yield of RNA.
Data are reported as means ± standard deviation for N = 2 in each group.

Figure 3. Confirmation of cDNA using 26S rRNA primers (M: Ladder, NC: Negative Control; S1 and S2: Leave sample of rice; S3 and S4: Leave sample of wheat; S5 and S6: Kernels of wheat; S7 and S8: Root sample of maize; S9 and S10: Leave sample of onion; S11 and S12: Leave sample of sugarcane).

Figure 4. Amplification of cDNA using 26S rRNA primers with genomic DNA contamination.
3.5. Amplication of cDNA with Real Time PCR
The expression of actin and β-tubulin reference gene in the four samples (two from rice and two from sugarcane) were estimated in triplicates by qRT-PCR (Figure 5). The CT values ranged from 26.81 to 27.56 and the stan- dard deviation (SD) values ranged from 0.0223 to 0.392 for the reference gene (Figure 5(a) and Figure 5(b)). The coefficient of variation for reference gene was less than 1, indicating good reliability of CT values and qRT- PCR assay was stable and reliable. To check the specificity, melt curve analysis was carried out. In this result showed single peak, which determine the specificity of the reaction (Figure 6). These results indicated that RNA isolated by our protocol was of high quality and suitable for real time PCR with no interference in PCR amplification.
 (a)
(a) (b)
(b)
Figure 5. (a) Real time PCR analysis of cDNA with actin gene (rice sample); (b) Real time PCR analysis of cDNA with tubulin gene (Sugarcane sample).

Figure 6. Melt curve analysis of reference genes.
3.6. Evaluation of the RNA Extraction Protocol
In this study, evaluation of quality and quantity of total RNA isolated from different tissues was done with spec- trophotometer and formaldehyde denatured gel. This protocol does not give a significant difference in the yield of total RNA. This method gave the highest yield for all kind of tissues ranging from 2834 to 3246 ng/µl RNA. This method gave A260/A280 range of 1.98 - 2.14 and abundant band for 28SrRNA than 18SrRNA, indicating that the RNA was not degraded and free from protein and polysaccharide contamination. Expression of 26SrRNA was monitored with all samples. Expression of this gene was represented as a single band without any smearing; indicating that the RNA was intact.
Real Time PCR permits monitoring of the progression of cDNA amplification after each cycle through fluo- rescence [27] . qRT-PCR has been applied to copy number variation, alleilic discrimination, gene expression, and molecular diagnostics [28] . The purity of nucleic acid is critical to obtain reproducible results in real time PCR [29] . Sensitive and reproducible real time PCR amplification indicated that RNA isolated by our procedure is of high quality and free from polysaccharide and polyphenols contamination.
4. Conclusion
Our RNA extraction protocol is a high-throughput protocol and the RNA purity is sufficiently high to meet the requirements of real time PCR. In conclusion, we have developed a rapid and reliable method of high-quality RNA extraction, i.e. widely used to isolate RNA from different types of plant tissues. It is highly significant to wider plant science community and also fruitful to other users wishing to conduct high throughput transcript profiling in a time and cost effective manner. We have used extracted RNA for copy number determination and expression analysis of transgenes in rice and sugarcane. We have also used extracted RNA for transcriptome analysis in maize roots under water logging stress.
Acknowledgements
We are thankful to Dr. Kuldeep Singh, Director, School of Agricultural Biotechnology, Punjab Agricultural University for providing the research facilities.
References
- Yockteng, R., Almeida, A.M.R., Yee, S., Andre, T., Hill, C. and Specht, C.D. (2013) A Method for Extracting High- Quality RNA from Diverse Plants for Next Generation Sequencing and Gene Expression Analysis. Application in Plant Science, 1, Article ID: 13000700. http://dx.doi.org/10.3732/apps.1300070
- MacRae, E. (2007) Extraction of Plant RNA. In: Hilario, E. and Mackay, J., Eds., Methods in Molecular Biology, 353, 15-24. Protocols for Nucleic Acid Analysis by Nonradioactive Probes, 2nd Edition, Humana Press, Totowa.
- Loomis, W.D. (1974) Overcoming Problems of Phenolics and Quinines in the Isolation of Plant Enzymes and Organelles. Methods in Enzymology, 131, 528-544. http://dx.doi.org/10.1016/0076-6879(74)31057-9
- Logemann, J., Schell, J. and Wilmitzer, L. (1987) Improvement Method for the Isolation of RNA from Plant Tissues. Analytical Biochemistry, 163, 45-50. http://dx.doi.org/10.1016/0003-2697(87)90086-8
- Dabo, S.M., Michell Jr., E.D. and Melcher, U.A. (1993) A Method for the Isolation of Nuclear DNA from Cotton (Gossypium) Leaves. Analytical Biochemistry, 210, 34-38. http://dx.doi.org/10.1006/abio.1993.1146
- Asif, M.H., Dhawan, P. and Nath, P. (2000) A Simple Procedure for Isolation of High Quality RNA from Ripening Banana Fruit. Plant Molecular Biology Reporter, 18, 109-115. http://dx.doi.org/10.1007/BF02824018
- Ky, H., Yeap, S.K. and Napis, S.B. (2012) The Best Method for Isolated Total RNA from Durian Tissues. Interna- tional Food Research Journal, 19, 1181-1183.
- Ghawana, S., Paul, A., Kumar, H., Kumar, A., Singh, H., Bhardwaj, P.K. and Rani, A., et al. (2011) An RNA Isolation System for Plant Tissues Rich in Secondary Metabolites. BMC Research Notes, 4, 85. http://dx.doi.org/10.1186/1756-0500-4-85
- Vasanthaiah, H.K.N., Ketam, R. and Sheikh, M.B. (2008) Efficient Protocol for Isolation of Functional RNA from Different Grape Tissue Rich in Polyphenols and Polysaccharides for Gene Expression Studies. Electronic Journal of Biotechnology, 11. http://dx.doi.org/10.2225/vol11-issue3-fulltext-5 http://www.ejbiotechnology.cl/content/vol11/issue3/full/5/index.html. ISSN 0717-3458
- Gehrig, H.H., Winter, K., Cushman, J., Borland, A. and Taybi, T. (2000) An Improved RNA Isolation Method for Succulent Plant Species Rich in Polyphenols and Polysaccharides. Plant Molecular Biology Reporter, 18, 369-376. http://dx.doi.org/10.1007/BF02825065
- Meng, L. and Feldman, L. (2010) A Rapid TRIzol-Based Two-Step Method for DNA-Free RNA Extraction from Arabidopsis Siliques and Dry Seeds. Biotechnology Journal, 5, 183-186. http://dx.doi.org/10.1002/biot.200900211
- Qi, G., Li, J.T., Ruan, Q.P., Yang, J. and Su, Z.X. (2009) An Optimized, Small-Scale Preparation of High-Quality RNA from Dry Seeds of Davidia involucrata. Phytochemical Analysis, 20, 139-142. http://dx.doi.org/10.1002/pca.1108
- Berendzen, K., Searle, I., Ravenscroft, D., Koncz, C., Batschauer, A., Coupland, G., Somssich, I.E. and Ulker, B. (2005) A Rapid and Versatile Combined DNA/RNA Extraction Protocol and Its Application to the Analysis of a Novel DNA Marker Set Polymorphic between Arabidopsis thaliana Ecotypes Col-0 and Landsberg erecta. Plant Methods, 1, 4. http://dx.doi.org/10.1186/1746-4811-1-4
- Kumar, A. and Singh, K. (2012) Isolation of High Quality RNA from Phyllanthus emblica and Its Evaluation by Downstream Applications. Molecular Biotechnology, 52, 269-275. http://dx.doi.org/10.1007/s12033-011-9492-5
- Gudenschwager, O., González-Agüero, M. and Defilippi, B.G. (2012) A General Method for High-Quality RNA Isolation from Metabolite-Rich Fruits. South African Journal of Botany, 83, 186-192. http://dx.doi.org/10.1016/j.sajb.2012.08.004
- Japelaghi, R.H., Haddad, R. and Garoosi, G.A. (2011) Rapid and Efficient Isolation of High Quality Nucleic Acids from Plant Tissues Rich in Polyphenols and Polysaccharides. Molecular Biotechnology, 49, 129-137. http://dx.doi.org/10.1007/s12033-011-9384-8
- Yin, D., Liu, H., Zhang, X. and Cui, D. (2011) A Protocol for Extraction of High-Quality RNA and DNA from Peanut Plant Tissues. Molecular Biotechnology, 49, 187-191. http://dx.doi.org/10.1007/s12033-011-9391-9
- Box, M.S., Coustham, V., Dean, C. and Mylne, J.S. (2011) Protocol: A Simple Phenol-Based Method for 96 Well Extraction of High Quality RNA from Arabidopsis. Plant Methods, 7, 7. http://dx.doi.org/10.1186/1746-4811-7-7
- Maciel, B.M., Dias, J.C., Romano, C.C., Sriranganathan, N., Brendel, M. and Rezende, R.P. (2011) Detection of Salmonella enteritidis Is Asymptomatic Carrier Animals: Comparison of Quantitative Real Time PCR and Bacteriological Culture Methods. Genetics and Molecular Research, 10, 2578-2588.
- Chomczynski, P. and Sacchi, N. (1987) Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate- Phenol-Chloroform Extraction. Analytical Biochemistry, 162, 156-159. http://dx.doi.org/10.1016/0003-2697(87)90021-2
- Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual. 2nd Edition, Cold Spring Harbor Lab., Cold Spring Harbor, New York.
- Rohland, N. and Reich, D. (2012) Cost-Effective, High-Throughput DNA Sequencing Libraries for Multiplexed Target Capture. Genome Research, 22, 939-946. http://dx.doi.org/10.1101/gr.128124.111
- Singh, K., Raizada, J., Bhardwaj, P., Ghawana, S., Rani, A., Singh, H., Kaul, K. and Kumar, S. (2004) 26S rRNA- Based Internal Control Gene Primer Pair for Reverse Transcription-Polymerase Chain Reaction-Based Quantitative Expression Studies in Diverse Plant Species. Analytical Biochemistry, 335, 330-333. http://dx.doi.org/10.1016/j.ab.2004.08.030
- Jain, M., Tyagi, A.K. and Khurana, J.P. (2006) Molecular Characterization and Differential Expression of Cytokinin- Responsive Type—A Response Regulators in Rice (Oryza sativa). BMC Plant Biology, 6, 1. http://dx.doi.org/10.1186/1471-2229-6-1
- Iskandar, H.M., Simpson, R.S., Casu, R.E., Bonnett, G.D., Maclean, D.J. and Manners, J.M. (2004) Comparison of Reference Genes for Quantitative Real-Time Polymerase Chain Reaction Analysis of Gene Expression in Sugarcane. Plant Molecular Biology Reporter, 22, 325-337. http://dx.doi.org/10.1007/BF02772676
- Goidin, D., Mamessier, A., Staquet, M.J., Schmitt, D. and Berthier-Vergnes, O. (2001) Ribosomal 18S RNA Prevails over Glyceraldehydes-3-Phosphate Dehydrogenase and Beta-Actin Genes as Internal Standard for Quantitative Comparison of mRNA Levels in Invasive and Noninvasive Human Melanoma Cell Subpopulations. Analytical Biochemistry, 295, 17-21. http://dx.doi.org/10.1006/abio.2001.5171
- Ginzinger, D.G. (2002) Gene Quantification Using Real-Time Quantitative PCR: An Emerging Technology Hits the Mainstream. Experimental Hematology, 30, 503-512. http://dx.doi.org/10.1016/S0301-472X(02)00806-8
- Hindson, B.J., Ness, K.D., Masquelier, D.A., Belgrader, P., Heredia, N.J., Colston, B.W., et al. (2011) High-Through- put Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Analytical Chemistry, 83, 8604- 8610. http://dx.doi.org/10.1021/ac202028g
- Cankar, K., Stebih, D., Dreo, T., Zel, J. and Gruden, K. (2006) Critical Points of DNA Quantification by Real-Time PCR-Effects of DNA Extraction Method and Sample Matrix on Quantification of Genetically Modified Organisms. BMC Biotechnology, 6, 37. http://dx.doi.org/10.1186/1472-6750-6-37
NOTES

*Corresponding author.