Advances in Lung Cancer
Vol. 2 No. 2 (2013) , Article ID: 33601 , 22 pages DOI:10.4236/alc.2013.22006
Pre-malignant processes of smoking-induced lung adenocarcinoma development: A conceptual biological model
![]()
1Tobin Consulting LLC, Newtown Square, PA, USA
2Philip Morris International R&D, Philip Morris Products S.A., Neuchâtel, Switzerland; *Corresponding Author: gregory.vuillaume@pmi.com
Copyright © 2013 Frank Tobin et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 5 March 2013; revised 4 April 2013; accepted 12 April 2013
Keywords: Lung Adenocarcinoma; Cigarette Smoke; Field Cancerization
ABSTRACT
Chronic exposure to cigarette smoke is the leading cause of human lung cancer and its most prevalent form, adenocarcinoma. However, the mechanisms by which smoking induces adenocarcinoma are largely inferred from the analysis of fully developed tumors. The current work focuses on the early events that precede the existence of clinically detectable tumors and where the progressive mechanisms are believed to be different from the ones driving established tumor growth. Biological information was drawn from the literature and generalized into a conceptual model, or framework, which describes and integrates the main processes involved in the early stages of smoking-induced lung adenocarcinoma development. No such integrative representation currently exists. The biological framework presented here is based on the “field of injury” of the lung. It covers the smoking-induced stepwise transition of unexposed (naïve) lung tissue to the first appearance of neoplastic cells through defined tissue states referred to as pre-field and field. Each tissue state exhibits its own formalized characteristics (or phenotype properties), which evolve as a result of the combined effects of smoking, the interactions between the different tissue properties, and the local environment represented in the framework as lung inflammation and immune surveillance. The resulting network of influences between the lung tissue states and properties provides a good understanding of the early events involved in lung adenocarcinoma triggered by smoking. The resulting conceptual model—an integrative mechanistic hypothesis—can explain a broad range of cigarette smoking and smoking cessation scenarios.
1. INTRODUCTION
Cigarette smoke (CS) consumption is causally related to a broad spectrum of adverse health effects, especially lung cancer, which is the leading cause of cancer deaths in the US and worldwide [1]. Smoking has been identified as the etiologic agent in approximately 90% of lung cancer cases [2,3]. While the smoking-related risk to develop squamous cell carcinoma of the lung (SCC) has remained stable, adenocarcinoma is now the most prevalent form of lung cancer among smokers [2]. This is most likely due to changes in cigarette design and manufacture during the past several decades [4,5]. While lung adenocarcinomas can arise in non-smokers, the mutagenic pathways between smoking-dependent and smoking-independent lung adenocarcinomas are divergent [6]. Lung adenocarcinoma in non-smokers is largely linked to kinase mutation and amplification of the epidermal growth factor receptor (EGFR) gene, while smoker-specific peripheral adenocarcinoma arises preferentially from sequential K-ras mutation (up to 30%) in combination with aberrant promoter methylation (for example, methylation of p16INK4) and p53 mutations (up to 70%) [7]. This paper will focus solely on the pathophysiology of smoking-dependent lung adenocarcinoma.
Although extensive epidemiological data exist for smoking-induced lung adenocarcinoma incidence and mortality [8], there are few insights into the initiating mechanisms, the sequence of events, or the timeline of events in the development of the tumor. Since lung cancer develops largely asymptomatically in chronic smokers, detailed mechanistic knowledge of early changes is limited. However, the molecular analyses of tumor specimens have indicated a complex, evolution-driven process. Based on numerous clinical and preclinical mechanistic studies using whole genome transcriptome analyses [9- 19], general principles have been identified as feasible disease promoting mechanisms. This paper provides an integrating conceptual model for the biology that underpins a plausible explanatory mechanism that underlies CS-dependent lung tissue remodeling from smokingonset to neoplastic transformation. To our knowledge, such an integrative approach to this disease scenario is unique and has not been published before. The outcome of the resulting biological framework can be used for a variety of subsequent research efforts, including more refined versions.
The aims of the biological framework are to:
• Provide a reasonable and plausible explanation for most of the main phenomena associated with the development of CS-related lung adenocarcinomas.
• Explain smoking cessation related phenomena and the effect of prior smoking.
• Reconcile diverse quantitative experimental data-genetic, genomic, epigenetic, proteomic, physiological, histological, cytological, and animal.
While these goals are certainly challenging, there is enough information available to start building this biological conceptual framework, which can then be used as a testable hypothesis for the design of new experiments.
2. CONCEPTUAL MODELING APPROACH
The systems viewpoint takes into account the known biological changes occurring in the cigarette smoker’s lung—from the naïve state (never-smoker) to the initial malignant cells that may develop into full-blown lung adenocarcinoma. Excluded are all other cancers, including adenocarcinoma in non-smokers, as well as, for the time being, any genetic predisposing factors [20]. Similarly, co-morbidities and their effects are currently not considered.
Building the biological conceptual framework involves triaging the biology and deciding on the appropriate level of granularity in the information used. By including everything, the framework would be as complete as possible, but might be so complex that it would be difficult to use in any practical manner. Consolidating various elements makes the modeling more feasible and may allow easier identification of the underlying mechanisms. All the concepts in this framework, as well as all the properties and influences, are derived from experimental observations in humans or animal models. We have indicated those instances where knowledge gaps exist, or where it is necessary to make assumptions.
Even though the smoking-induced tissue transformations are believed to be continuous, a small number of discrete tissue phenotypes adequately represent almost all pre-neoplastic phenomena occurring in the target tissues [21,22]. Based on the field of injury and cancerization theories [10,23,24], these defined discrete tissue states describe the sequence of events leading to the first malignant cells. The “target tissue” is a never-smoker lung—the specific cells capable of becoming an adenocarcinoma. Pre-field tissue is considered smoking stressed, yet maintaining tissue homeostasis. Field lesions in a chronically inflamed lung are severely stressed and genomically unstable tissues that are no longer homeostatic. The biological model ends just as the first malignant cell appears—a rather fuzzy boundary, but one that can be defined as the late stage boundary of highly perturbed field tissue. SET (Smoking Exposed Tissue)—quasinormal tissues resulting from a very long period of smoking abstinence—is relevant in some cessation scenarios. Figure 1 illustrates how the tissue states evolve during smoking and recover after prolonged cessation.
The change in each tissue state is affected by the state of the lung and that tissue’s specific tissue properties. The alterations in the properties are aggregates of various biological processes that either drive towards or inhibit the malignant progression. These processes change over time and impact the overall tumorigenic nature of the lung as well as other lung properties. These interactions or processes—the influences—change the properties of the lung tissues and the state of the entire lung.
Smoking is the key driver for tumorigenic changes in the lung and in tissue properties. One of the goals in developing the conceptual model is to facilitate a description of these influences and show how cigarette consumption (i.e. dose) influences the sequence of events in tumor development. Figure 2 depicts a simplistic view of the characteristics of cigarette consumption and their subsequent impact on the lungs. Dose alone is insufficient because it is an instantaneous measure of consumption, not a measure of biological effect. Instead, we use a generalization of smoking dose to handle the lung memory (for example, clearance of previously deposited tar, tobacco metabolites, etc.) and its overall state (lung health). This generalization of consumption is called smoking potency and is used as a conceptual surrogate for the overall effective dose delivered to the lungs. See Table 1 (at the end of the text) for the details of the specific smoking potency influences.

Figure 1. Evolution of the tissue phenotypes during smoking and cessation. (a) During smoking, naïve tissue is initially converted into the pre-field phenotype (1) and for a short time both exist simultaneously. After some time of continuous smoking only pre-field tissue exists. It progresses (2) under the influence of the smoking-induced insults. With sustained, heavy smoking the first field lesions (3) form and progress independently (4) of the pre-field tissue. With enough time and sustained, heavy enough smoking, field tissue may become the precursor of an adenocarcinoma (5); (b) During cessation, field and pre-field lesions may coexist. Field lesions may continue to progress (6) for a while, but eventually stop and may die off completely if abstinence lasts long enough (7). In the event, there is too much progression “momentum”, any remaining field cells may continue to progress and still become an incipient adenocarcinoma (11). Meanwhile, the pre-field lesions benefit from the improving inflammatory environment and start to revert to a less perturbed phenotype (8) eventually reaching their least stressed state (9). After decades of abstinence, the overall lung situation becomes quasi-normal and only SET tissue remains (10). Inside both the pre-field and field boxes, is an illustration of how the overall progressive state (2 and 4 in (a) and 7 and 9 in (b)) is advancing (in smoking) or reverting (in cessation).
3. LUNG PROPERTIES
Cigarette smoke exposure is implicated in the etiology of numerous diseases [2], with strong evidence that these diseases are largely activated by skewed immune responses [25]. Since cigarette smoke is a rich source of reactive oxygen and nitrogen species [26], it is reasonable to assume that the inflammatory phenotypes associated with CS-dependent lung diseases is the result of a persistent oxidative environment that causes tissue damage and death [27,28]. Necrotic cell death produces one of the strongest known “danger” signals for tissues, triggering a pronounced innate immune reaction [29], and it is assumed that the smoking-dependent field of injury is composed of local necrotic lesions contributing towards an organ-wide inflammatory environment. Finally, genes involved in xenobiotic metabolism and antioxidant response have been shown to be quickly up-regulated in CS-exposed rodent lung tissue to combat cell injury and death, and to increase cell survival while at the same time inflammation-related genes become gradually up-regulated in a doseand time-dependent manner [17,18]. Continuous smoking may compromise surveillance competencies of the immune system as evidenced for example, by the impairment of natural killer (NK) cell-dependent immune surveillance functions in smokers [30,31]. This may potentially further enhance disease development. Altogether, data from mechanistic preclinical studies on smoking rodent models of CS-in-
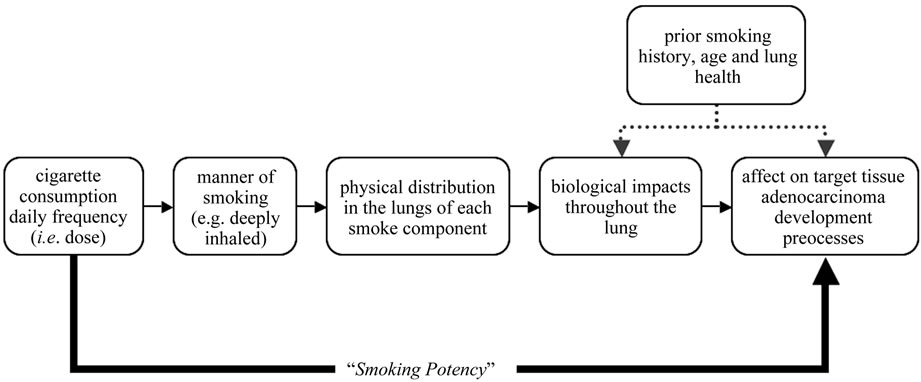
Figure 2. Smoking potency. Smoking potency is a phenomenological modeling approach to subsume the complex multiple phenomena involved in the impact of smoking on adenocarcinoma tumorigenesis into a single generalization. Although based on the smoking dose, it is used instead of dose in the model to be more representative of the actual dose impact as it takes into account both dose and the “smoking memory” of the lung so that clearance effects can be handled.
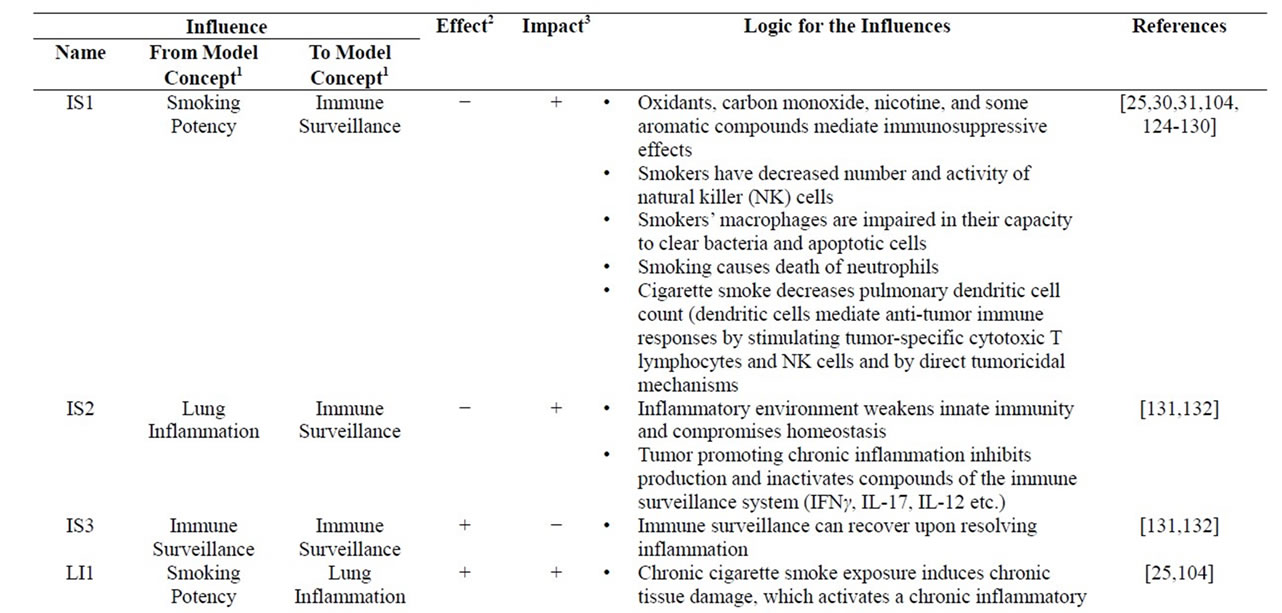


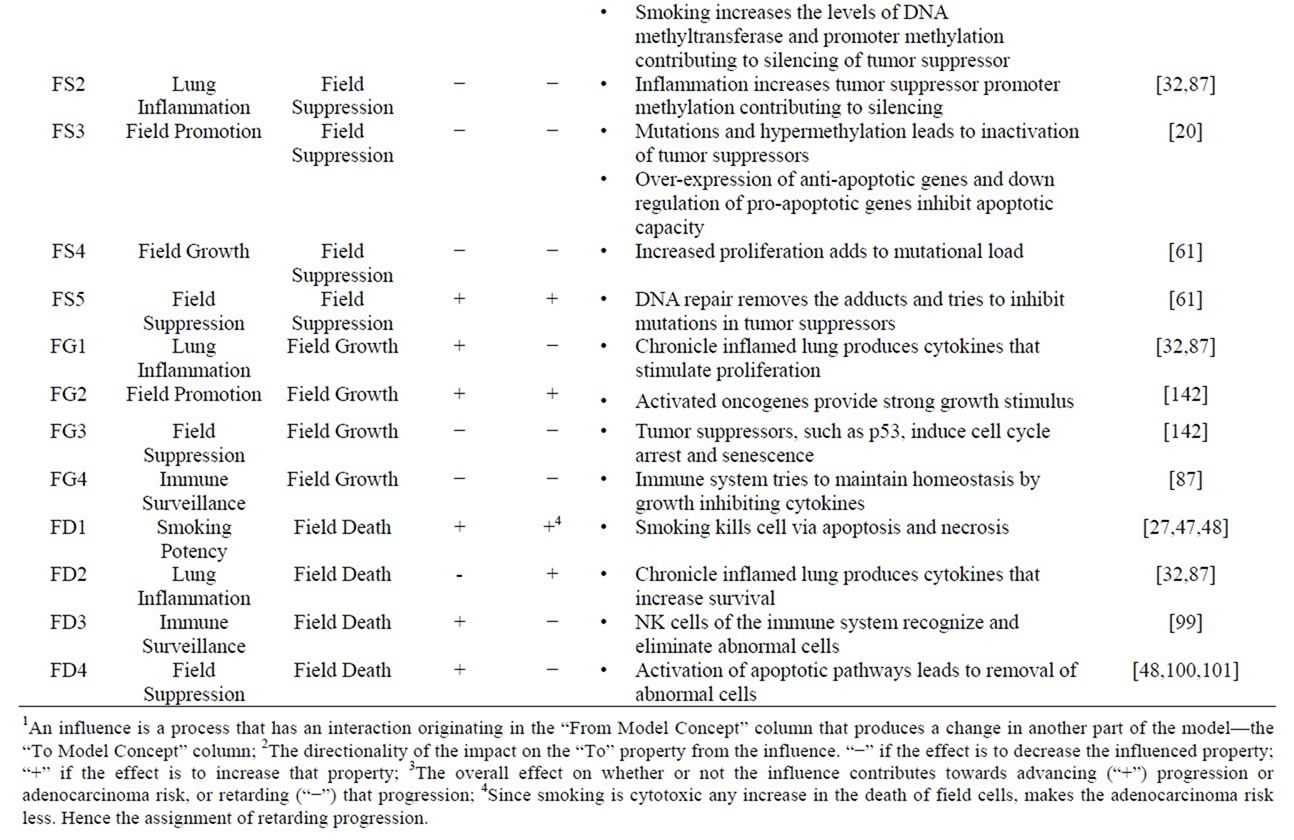
Table 1. Influences. The network of influences that constitute the core of the model is detailed. Each influence consists of three pieces: which entity in the model is causing the influence (the “from” entity), which entity is being influence (the “to” entity), and whether the influence on the value of the “to” entity is being increased (“+”) or decreased (“−”). Also indicated is whether the influence is progression stimulating or inhibiting and the logic and relevant references.
duced lung cancer, along with clinical studies, and transcriptomic data from human smokers’ bronchoscopy specimens, support the notion that chronic exposure to CS primarily targets lung immune functions [25,32].
As the lung attempts to maintain tissue homeostasis during the initial decades of smoking, the damages are reliably repaired through regular healing processes. The effectiveness of this control mechanism however, may become increasingly compromised as smoking continues and as the inflammatory phenotype changes from acute (physiological) to chronic (pathological) [33,25]. While inflammation is not a primary cause of the diseases associated with chronic smoking-induced inflammatory phenotypes, it significantly contributes to their development [33,34]. Hanahan and Weinberg recently extended their original list of the “Hallmarks of Cancer” by including “tumor-promoting inflammation” as an “enabling characteristic” [35]. The impact of inflammatory processes on smoking-dependent lung adenocarcinoma formation was recently shown in a mouse lung cancer model [36]. Following up on the well-accepted concept that chronic oxidative stress results in continuous cellular damage and tissue malfunction, a sustained pro-inflammatory stimulus can be assumed, which, after decades, may eventually lead to a change in the inflammatory phenotype as described by Lu et al. [31]. In long-term heavy smokers, this cause-and-effect feedback is most strongly seen in the progression from oxidatively-stressed pre-field tissue phenotypes to the formation of field lesion phenotypes which contribute further to the inflammatory state of smokers’ lungs [32,37,38].
The chronic inflammatory phenotype and the related skewed immune functions are two key drivers in smoking-dependent lung cancer development. The two critical and counteracting domains of lung function and smoking can be distilled into two mutually interacting concepts (called lung properties) of lung inflammation and immune surveillance. These organ level properties affect the whole lung. Both are generalizations, aggregating (in a lumped model sense) of the many different aspects of the immune system functions and processes that occur throughout the body. These processes can promote or attenuate the tissue state progression or regression, on a whole organ level. Smoking directly causes lung inflammation, and the resulting tissue damage comprises of all the different elements of an inflamed lung. Immune surveillance, on the other hand, is the combined action of the various components of the immune system invoked to counteract smoking-induced changes. In the conceptual model, smoking always worsens inflammation and compromises immune surveillance. The balance between lung inflammation and immune surveillance thus largely dictates the speed of the progression from one tissue state to the next, as well as the functioning of the lung as an organ.
Three key summary influences capture the essence of the complex immune-inflammatory interactions driving the overall tumor development phenomenology of smoking and cessation (within the narrow focus of adenocarcinoma initiation):
1) Lung inflammation degrades immune surveillance (influence IS2 in Figure 3).
2) Immune surveillance resolves lung inflammation (influence LI5 in Figure 3).
3) Immune surveillance heals immune surveillance (influence IS3 in Figure 3).
The feedback loop characteristics of these influences can be seen clearly in Figure 3, as counteracting influences IS2 and LI5. The self-healing capability of the immune response (influence IS3) [31] provides another feedback loop, which represents an anti-tumorigenic aspect. Smoking potency exerts strong pro-tumorigenic influences by simultaneously stimulating lung inflammation (LI1 influence in Figure 3) and degrading immune surveillance capacity (IS1 influence in Figure 3). In essence, lung inflammation and immune surveillance become overall surrogates for lung health (in the narrow context of adenocarcinoma development). These influences are described in more detail in Table 1.
4. TISSUE STATES AND THEIR PROPERTIES
In general, adenocarcinomas of the lung develop in the

Figure 3. Lung inflammation and immune surveillance influences. A diagram of the generalization of the mutual interactions between the immune surveillance and inflammatory processes of the lungs, as well the influences of smoking on them. The mutual feedback effects are clearly visible. Red lines indicate pro-tumorigenic influences; blue ones are anti-tumorigenic influences. Names of the influences (inside rectangular boxes) are cross-references to Table 1. IS1: Smoking degrades the lung’s immune surveillance capacity. IS2: Inflammatory environment weakens innate immunity and compromises homeostasis. IS3: Immune surveillance can recover upon resolving inflammation. LI1: Smoking contributes to the increase of a persistent, chronic inflammatory state. LI5: Innate immunity fights lung inflammation to maintain lung homeostasis.
periphery, originating from alveolar surface epithelial cells or bronchial mucosal glands. These adenocarcinomas can be further histologically discriminated into several subtypes, such as bronchoalveolar carcinoma and acinar or papillary adenocarcinoma, although often a mix of several subtypes often occur (for a review see [39]). Because of a lack of discriminating mechanistic knowledge on differentiation, specific subtypes of adenocarcinoma are ignored. Alveolar type II (AT II) cells are considered the main source for smoking-related adenocarcinoma formation. While the alveolar Type I cells are terminally differentiated, the Type II cells can proliferate, as well as differentiate, into new Type I cells. This is important when the lung epithelium is injured and needs repair [40]. Stereology numbers provided by Ochs et al. were used to estimate the size of the target cell population; in the human lung—there are approximately 24 billion alveolar Type II cells [41].
4.1. Pre-Field Tissue
Once the target tissues experience sustained smoke exposure, they are presumed to be stressed irreversibly and are considered to be early pre-field tissue. We estimate that the transition from “naive” tissue to early pre-field tissue is completed in about 6 months. However, since there are no quantitative data from smokers after such a short time, the estimation is based on rodent studies (Gebel, et al., 2004; 2006) taking into account the differences between the species. The smoking exposure level and the demographic situation of the smoker determine how rapidly non-smoking tissue is converted into pre-field tissue. The non-smoking target tissue is straightforward—the only property needed is the cell count for the target tissue (Ochs, 2006).
The pre-field phenotype is a stressed phenotype that may persist in asymptomatic chronic smokers (“healthy smokers”) for decades. Numerous gene expression profiles have been recorded from the smoke-exposed lung in preclinical models of rodent inhalation [11,17-19,42,43] and from clinical setting (lung brushings and biopsies (as summarized in [10,44]). Both preclinical and clinic data have clearly shown the persistence of this stress response [2,17,18,27,45,46].
In general, cigarette smoke is cytotoxic and at high doses, initiates necrotic cell death of lung epithelial cells. In addition, various reactive oxygen (ROS) and nitrogen species (RNS) contained within the smoke itself, and generated as a result of it, cause damage to lipids (lipid peroxidation), proteins, and DNA (ssDNA breaks) leading to apoptosis [27] as evidenced by in vitro studies [47,48]. It is not known whether CS exposure causes net cell death in a healthy smoker lung. In fact, studies focusing on COPD patients, showed no appreciable cell death in smoker lungs as compared to healthy nonsmoker lungs [49-51]. Nonetheless, even though undetectable by conventional methods, it is reasonable to assume that there is some cell death and tissue damage in smoking-exposed lung tissue [27,52-54]. Moreover, the gradual, but persistent development of a cluster of inflammation-related genes is a clear indication for cytotoxic events which may include necrosis and apoptosis [27].
To balance cell death, highly coordinated tissue repair processes are activated: damaged epithelial cells as well as nearby structural and immune cells release various factors and cytokines that induce cellular proliferation [55-57]. It is assumed that while the overall cell turnover is increased in the pre-field, homeostasis is maintained. Since the overall cell count remains constant, it is not necessary to consider death and growth rates as conceptual model properties for pre-field tissue.
The stressed tissue may become defective in clearing apoptotic cells by macrophages [58,59], which, in turn, can lead to secondary necrosis, a strong immunogenic process [60]. As smoking continues, the sustained effects of the chemical insult and the oxidative stress buildup in the cells result in the accumulation of inflammation in the lungs. This affects the staging of the pre-field tissue propelling it from its early stage characteristics towards its late stage that is the precursor of field tissue.
4.1.1. Pre-Field Stress Property
To handle the expected pre-field behaviors, the framework only needs to have properties that drive and inhibit progression (illustrated in Figure 4). The pre-field stress property is the generalization of the various damaging cell and tissue consequences of CS exposure. It combines the different processes occurring in the stressed-lung phenotype, potentially resulting in severe cell damage or even cell death (influence PFS1 in Figure 4) [27]. The spectrum of CS-dependent reactive compounds also includes carcinogenic intermediates that are known to bind to DNA and form covalent adducts that may provoke nucleotide mis-incorporation as well as replication and transcription errors, if un-repaired [61]. Stress also includes smoking induced mitochondrial damage, which increases the intracellular ROS levels. The resulting diminished energy-producing capacity favors necrotic cell death over apoptosis [62]. Stress in the pre-field is antagonized by tissue integrity activities (influence PFS2 in Figure 4), while it in turn inhibits pre-field integrity (influence PFI3 in Figure 4) forming another feedback interaction.
4.1.2. Pre-Field Integrity Property
Pre-field integrity includes all the tissue and cellular level functions that maintain tissue homeostasis and attempts to repair damages that result from smoking-de-

Figure 4. Influences on the properties of the pre-field phenotype. Names of the influences (inside rectangular boxes) are cross-references to Table 1. Red lines indicate pro-tumorigenic influences; blue ones are anti-tumorigenic influences. PFS1: Smoking causes lipid peroxidation and ssDNA breaks that stress the cell. Stressed cells die through apoptosis or necrosis. PFS2: Antioxidant response and xenobiotic metabolism fight smoking induced damage and increase survival. DNA repair prevents the accumulation of mutations. LI2: Stressed cells secrete inflammatory mediators and cell debris from necrotic (primary or secondary) death promotes inflammation. PFI3: Increased death causes increased proliferation rate and this predisposes to errors, i.e. the cell’s repair capacity is compromised. PFI1: Continuing smoking creates an adaptation and decrease in antioxidant response and the function of xenobiotic metabolizing enzymes. Smoking increases DNA adducts decreasing repair capacity. LI3: When smoking induced wound heals, inflammation can resolve. PF cells secrete anti-inflammatory factors that have negative influence on LI. PFI2: Signals from the IS promote tissue replenishment by the alveolar epithelial cells. PFI4: Integrity can inhibit further damage to itself and heal, presumably via growth factors and matrix proteins secreted by the alveolar epithelial cells.
pendent stress. The immune surveillance provides a signal that boosts the integrity to initiate cell-based tissue repair processes [63], including, for example, antioxidant response and xenobiotic metabolism, which protect from cell damage and increase survival [64]. It also boosts DNA repair activity against smoking induced DNA adducts [61], and tissue repair by the alveolar cells [55,57] as represented by influence PFI2 in Figure 4. When smoking continues over decades, pre-field integrity becomes increasingly compromised, leading to a gradual decay of the tissue-level healing capacity, as well as a decreased antioxidant response and degraded capacity of xenobiotic metabolizing functions [57]. The contributing factors for this decay are continuous smoking (influence PFI1 in Figure 4) and the actions of constant stress (influence PFI3 in Figure 4), which eventually diminish the full integrity capacity. Pre-field tissue is still considered “mildly damaged” and integrity functions can still maintain tissue homeostasis, presumably due to its regenerative (“self-healing”) potential (influence PFI4 in Figure 4) [27].
4.2. Field Tissue
With continued smoking, the pre-field properties evolve in a dose-dependent manner through the smoking influences (influences PFI1 and PFS1in Figure 4). By the time pre-field tissue properties reach their most progressed state, lung inflammation has become chronic, resulting in a new self-perpetuating (“non-resolving”) inflammatory phenotype [33]. As inflammation changes its influence from physiological to growth promoting, the immune surveillance capacity is weakened further and tissue homeostasis is severely compromised. Localized, highly progressed pre-field areas are further damaged, resulting in a few cells changing into a significantly more damaged phenotype. These “progenitor” cells appear as small clonal patches and constitute the emergence of field tissue. According to the concept of “field cancerization”, it is conceivable that these lesions represent an area of the tissue, (the “field”), from which the tumor eventually originates. Meanwhile, the molecular and cellular changes linked with the development of the lesion, as, for example, reflected by the severity of the chronic inflammatory phenotype, are detectable throughout the organ, and thus represent the “field of injury” [24].
It is reasonable to assume that initially the incipient field lesions are multi-focal. Based on clinical studies of pre-neoplastic lesions, we estimate that a lung can harbor several sufficiently progressed pre-field patches that will be converted into the more dangerous field phenotype. Accordingly, only the most progressed pre-field cells— the initial cohort of field cells—need to be followed in order to determine the timing of the formation of the first “true” adenocarcinoma cell. This assumption is motivated by computed tomography (CT) studies following the progression of very small lung nodules [65-74] as well as by observations that the pre-neoplastic lesions preceding lung adenocarcinoma are always multifocal [75-77]. The process of the creation of field lesion is shown in Figure 1 step 3.
These newly formed field lesions are no longer bound by the same homeostatic control mechanisms, as was the situation with pre-field tissue. Coupled with the effects of an ever worsening inflammatory state, they are becoming more susceptible to a slew of genetic, genomic and epigenetic alterations [32] that move the phenotype closer towards a more neoplastic variant (Figure 1 steps 4 or 6). Field tissue is more complex than pre-field tissue for two main reasons: crucial tumorigenic promoting genomic instability is now manifested and tissue homeostasis is no longer under control. Inflammation is chronic and responsible for the immune surveillance functions becoming more compromised. Moreover, the gene expression profile of “field tissue” already shows a precancerous fingerprint [15].
During prolonged active smoking, while promotion gets stronger, suppression gets weaker with a strong feedback interaction between them (influences FS3 and FP3 in Figures 5 and 6). Since the knowledge regarding cellular mechanisms in premalignant lesions is very sparse, the assumption is made that different progressive and suppressive mechanisms in field tissue are (somewhat) similar to those activated in early malignant cells [35]. Suppression activities attempt to keep the tissue in homeostasis by counteracting promotion (see Figures 5 and 6).
Field tissue is subject to events that favor neoplastic transformation. The field tissue corresponds to atypical adenomatous hyperplasia (AAH), which is considered a precursor lesion for some types of lung adenocarcinoma [22,78-81]. Eventually, as smoking continues, the most advanced cells of the field phenotype change into an early adenocarcinoma. The point at which this occurs is also the boundary for the scope of this conceptual mechanism and the starting point for a discussion of adenocarcinoma biological phenomena.
4.2.1. Field Promotion Property
The generalization of various cellular processes that stimulate or maintain the persistent cellular stress state is a field property called promotion. In this context, the term describes the processes that drive the progression of field tissue to a more damaged state. Its essence is still very close to pre-field stress, but with critical additional features that make promotion an even more active driver towards malignancy. This capacity arises from the ge-
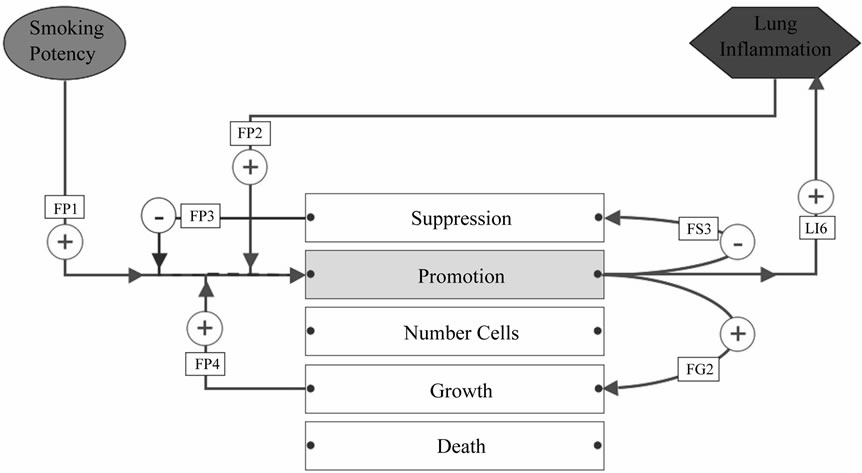
Figure 5. Influences on the field phenotype promotion property. Names of the influences (inside rectangular boxes) are cross-references to Table 1. Red lines indicate pro-tumorigenic influences; blue ones are anti-tumorigenic influences. FP1: Smoking induces gain-offunction mutations, amplifications, and over-expression of oncogenes as well as loss-offunction mutations, deletions and epigenetic silencing of tumor suppressor genes. FP2: Lung inflammation causes over-expression of DNA methyltransferase genes. FG2: Abnormally activated proto-oncogenes and silenced tumor suppressors increases proliferation. FP4: Increased proliferation predisposes to errors. FP3: DNA repair attempts to prevent mutations. FS3: Mutations and LOH inactivate tumor suppressors, methylation causes silencing. LI6: Mutations alter the expression of inflammatory mediator genes.
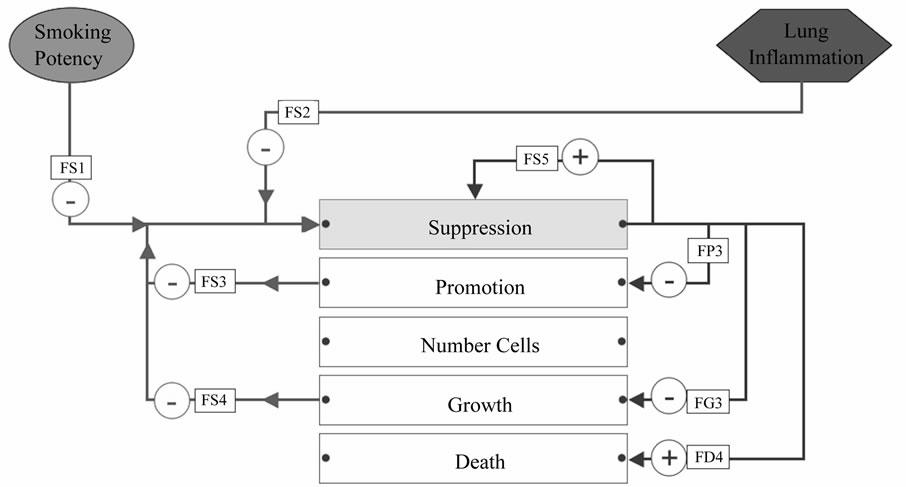
Figure 6. Influences on the field phenotype suppression property. Names of the influences (inside rectangular boxes) are cross-references to Table 1. Red lines indicate pro-tumorigenic influences; blue ones are anti-tumorigenic influences. FS1: Smoking creates DNA adducts that impair DNA repair. FS2: Inflammatory cytokines repress apoptotic pathways, by stimulating anti-apoptotic mechanisms and reduced expression of mismatch repair genes. FS3: Mutations and LOH inactivate tumor suppressors, methylation causes silencing. Loss of p53 function, over-expression of anti-apoptotic genes and down regulation of pro-apoptotic genes. FS4: Increased growth rate predisposes to more mutations in tumor suppressors. FS5: DNA repair attempts to correct errors in DNA to prevent mutations in tumor suppressors. FP3: DNA repair attempts to correct errors in DNA to prevent mutations in oncogenes. FD4: Apoptosis removes abnormal cells. FG3: Tumor suppressors, cell cycle control, and senescence interfere with proliferation.
nomic instabilities created by smoking and the chronic inflammation of the lung in heavy smokers. Tobacco carcinogens introduce bulky adducts to DNA, which may increase the chance of base mis-incorporation (influence FP1 in Figure 5). While there is already transient DNA damage in the pre-field cells, the chance for persevering base mis-incorporation is further augmented when the field proliferation rate is increased (influence FP4 in Figure 5) [35]. The gain-of-function mutations and amplifications targeted to “critical loci” lead to abnormal activation of oncogenes that greatly affect field cell behavior [34]. The activated inflammatory cells release cytokines, growth factors, reactive oxygen species and reactive nitrogen intermediates, which can also induce DNA damage and genomic instability (influence FP2 in Figure 5) [35]. To some extent, promotion is kept in check by suppression, which tries to repair the DNA, thus preventing further damage and mutagenesis (influence FP3 in Figure 5). However, with the constant influence of continued heavy smoking, this framework assumes that eventually promotion overcomes any suppressive influences and the net effect is for the field phenotypes to progress towards malignancy.
4.2.2. Field Suppression Property
Suppression, similar to the integrity property in prefield, is an aggregate property that generalizes the various processes of the field phenotype to repair damaged cells or to make the decision to activate pathways that dispose of cells that are beyond repair, thereby resisting the progression towards malignant transformation. Consequently, suppression includes all vital defensive and integrity-maintaining cellular processes as well as processes that actively fight against progression towards malignant transformation. A paradigm of suppression capabilities is the p53-induced cell cycle arrest and entry into senescence as well as apoptosis induction [35]. Suppression also hinders DNA adducts and mutagenesis-O(6)- methylguanine-DNA methyltransferase (MGMT) removes cytotoxic adducts from DNA (influence FS5 in Figure 6) [61]. In addition to the constant need for defensive activities against promotion (influence FS3 in Figure 6), the suppressive capacity of field tissue is further reduced by both continuing smoking influences (influence FS1 in Figure 6) and severe inflammation (influence in FS2 in Figure 6). Chronic inflammation leads to reduced expression of DNA mismatch repair genes and weakened enzymatic activity of their gene products. It also increases overall genomic instability which leads to point mutations and allelic loss of tumor suppressor genes (also “critical loci”), resulting in a weakening of suppression (influence FS2 in Figure 6) [82,83]. In addition to the DNA changes, tumor suppressor genes are silenced by DNA hypermethylation [84-86]. The abnormal methylation of the MGMT promoter is also linked to impaired repair [83]. Field tissue proliferates at an abnormally high rate compared to naïve tissue causing increased chances of base mis-incorporation due to insufficient time to remove the bulky DNA adducts during proliferation (influence FS4 in Figures 6 and 7). Smoker’s lungs have increased levels of DNA methyltransferase and increased promoter methylation, which could be caused by a direct smoking effect (influence FS1 in Figure 6) [84-86] or an effect mediated by chronic inflammation (influence FS2 in Figure 6) [87], or both.
4.2.3. Field Growth, Death and Cell Counts
In contrast to homeostatic pre-field tissue, field tissue undergoes excessive proliferation. A major contributing factor is the smoking-induced chronic inflammation environment that surrounds the field cells as they develop [32,87]. The chronically inflamed lung secretes cytokines that provide strong growth stimuli (influence FG1 in Figure 7) and the increasing genomic instability (due to both smoking and ongoing inflammation) further strengthens the already elevated oncogene signaling thereby further stimulating growth (influence FG2 in Figure 7) [32,87]. The processes that aim to balance the excessive growth include suppression, via p53 that controls the cell cycle and that can induce senescence (influence FG3 in Figure 7) as well as cytokines with anti-proliferative activities coming from the immune surveillance capability (influence FG4 in Figure 7) [35]. While these antigrowth mechanisms may still suffice in early field, homeostatic control is disturbed in the late field. The estimate is that late field growth rates reflect those observed in (atypical) adenomatous hyperplasia (AAH), the preneoplastic lesion commonly associated with lung adenocarcinoma [78-81]. AAH is characterized as a lesion/ nodule that is smaller than 5 mm in diameter [88], is of clonal origin [89,90] and consists of proliferating alveolar Type II cells that may present varying degrees of cellular atypia [91-93]. For early field, only those lesions that proliferate, but do not present appreciable atypia can be considered to stay within the conceptual framework’s scope. Based on various Ki-67 staining results on AAH lesions, we assume that there are 2 to 3 times more cells undergoing division in late as compared to early field, and this is still approximately 3 times less than in early adenocarcinoma [88,92,94-96]. Growth rate differences between late pre-field and early field are difficult to estimate. The only data available on histologically normal (non-cancerous) lung tissue are from the airway and it is evident that the growth rates are not comparable to that of the lung parenchyma. Ki-67 stained nuclei detected in a healthy smoker large airway epithelium are more numerous than in AAH of the lung parenchyma [88,94, 95,97,98].
The concept of the field death property includes both extrinsic and intrinsic processes. Natural killer (NK) cells of the immune surveillance system recognize and remove abnormal cells (influence FD3 in Figure 8) [99]. As in pre-field, necrosis and apoptosis are assumed to be caused by smoking itself (influence FD1 in Figure 8),
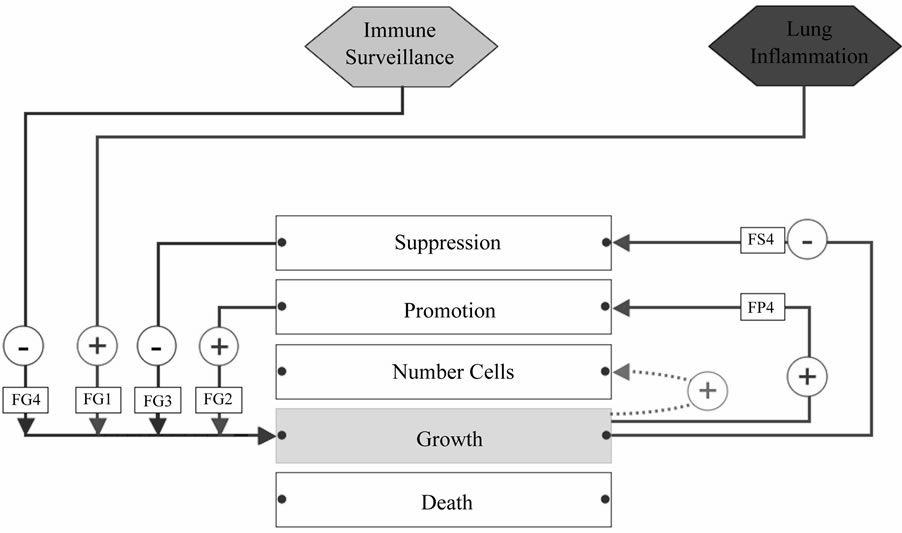
Figure 7. Influences on the field phenotype growth rate property. Names of the influences (inside rectangular boxes) are cross-references to Table 1. Red lines indicate pro-tumorigenic influences; blue ones are anti-tumorigenic influences. FG1: Chronic inflammation provides strong growth stimulus. FG2: Abnormal oncogene activation stimulates growth. FP4: Increased proliferation predisposes to errors. FG4: Cytokines with anti-proliferative activities inhibit growth. FG3: Tumor suppressors, cell cycle control, and senescence interfere with proliferation. FS4: Increased growth rate predisposes to more mutations in tumor suppressors.
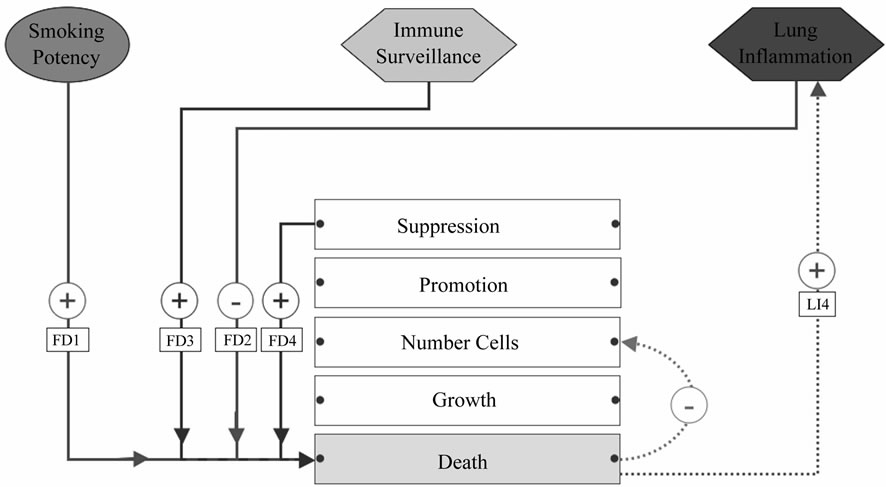
Figure 8. Influences on the field phenotype death rate property. Names of the influences (inside rectangular boxes) are cross-references to Table 1. Red lines indicate pro-tumorigenic influences; blue ones are anti-tumorigenic influences. FD1: Smoking cytotoxic effects kill cells. FD3: NK cells kill abnormal field cells. LI4: Necrotic cell death promotes inflammation. FD4: Apoptosis gets rid of abnormal cells. FD2: Inflammation derived pro-survival cytokines decreases the death rate.
but in field tissue, apoptosis is also induced as a protective mechanism within suppression to eliminate cells that are too damaged (influence FD4 in Figure 8) [98,100, 101]. However, even this influence is assumed compromised during advancement towards late field stages. Studies on AAH lesions have demonstrated that antiapoptotic proteins, such as bcl-2 and survivin, are more frequently detected in high versus low-grade lesions suggesting that the apoptotic processes are challenged [96,99,102,103]. At the current level of granularity of the description of the biological processes, different death processes are not separated, but treated as one aggregate property. Only smoking induced necrosis contributes to inflammation [27,32,98,104-106] (influence LI4 in Figure 8 and Table 1). The chronic inflammatory environment in field lesions inhibits cell death by means of pro-survival cytokines (influence FD2 in Figure 8) resulting in increasingly disturbed tissue homeostasis [32, 35,87].
To the best of our knowledge, direct measurements on death rates in precancerous lesions in the lung parenchyma are not available, and for the reasons mentioned above, large airway death measurements probably do not reflect the situation in the parenchyma. It is assumed that the increased growth rate, together with compromised apoptotic activity, results in net tissue growth, i.e. the death rate is smaller than the growth rate, as evidenced by the AAH lesions appearing in smoker lungs. Some indication of the size of the net growth rate in field tissue can be derived from lesion/nodule follow-on studies— the lesion doubling rates for AAH has been reported as 988 ± 470 days [107]. While AAH nicely represents the precancerous situation of lung adenocarcinoma, it does not necessarily mirror unique smoking dependent pathology as these lesions also precede non-smoker adenocarcinoma [108].
The field growth and death properties (see Table 2 at the end of the text) are influenced by smoking, other field properties, and lung biology. However, they also contribute influences of their own (see Figures 7 and 8) to the overall field progression.
5. CESSATION CHANGES AND SMOKING EXPOSED TISSUE
The cessation of cigarette smoking leads to elaborate and orchestrated changes in the lungs and the smokingtransformed tissues. When smoking abstinence commences there are indications of an acute recovery phase as suggested for example, by the rapid decrease in free lung inflammatory cells [109,110]. There is also evidence for a longer lasting recovery phase in terms of other inflammationand tissue-related parameters which may be dependent on the clearance of smoke toxins and metabolites. The clearance of smoke components and metabolites from the body is gradual [111] leading to a commensurate gradual decay in the smoking potency as tobacco products and metabolites clear. The lungs and tissues react more slowly leading to a “momentum of smoking” whereby inflammation may continue to increase for some time after the commencement of cessation. As cessation continues there is a concomitant improvement in lung health; lung inflammation stops worsening and immune surveillance stops degrading. A point of inflection is reached, after which time only im-


Table 2. Lung and tissue properties. All lung and tissue properties are indicated with a short description and references to the biology of the property.
provement occurs. After the point of inflection, the immune surveillance recovers (by “self-healing”; influence IS3 in Figure 3) leading to a progressive and sustained resolution of lung inflammation [9].
Cessation phenomenology is complex for a heavy, chronic smoker who has field lesions. The adenocarcinoma development risk appears to remain at a high level as in a current smoker [112], probably because self-perpetuating inflammation persists, attenuating and preventing efficient healing. Initially, as tobacco combustion products clear from the lungs, the field phenotype may continue its progression unabated for several years due to the “momentum” of smoking and the sustained impact from inflammation. However, after the inflection period, as sustained healing begins, field tissue may no longer continue to accumulate more genomic and epigenetic instabilities. This lack of additional accumulation of instabilities is reflected in a stabilization of the field promotion and suppression properties. With these two properties stagnant, the overall progression towards malignancy stops and there is no longer an increased risk of forming an adenocarcinoma.
As inflammation resolves with an accompanying decrease in growth stimulating and death inhibiting cytokines, there is a decreasing field growth stimulus. Simultaneously the immune surveillance capacity recovers [113]. The combined effect may now become a negative net growth rate resulting in a loss of cells in the field lesions. Increasingly, as abnormal cells are eliminated, the oncogene signaling and over-expression of antiapoptotic factors are diminished and this further reduces the field growth rate and speeds up cellular death rate. After a period of time-as the exact length of time appears to vary and is dependent on the level of pre-cessation smoking levels—the net growth turns negative. The field lesions start to shrink and may eventually disappear (see Figure 1 step 7). However, even if the number of field cells is diminishing, the model can still produce malignancy through continued progression of the remaining field cells. We believe that this balance between losing field cells and continuing field progression explains some of the observed epidemiological risk patterns in cessation. The smoking history (age, duration and level of smoking) prior to cessation probably contributes to this balance.
These assumptions are based on the epidemiological patterns of risk from former heavy smokers developing lung cancers. Thus the model’s timing patterns are expected to be similar. That risk gradually decreases while asymptotically approaching, but never touching the level for never-smokers, even after decades of abstinence. The dose-dependent drop in the relative risk of lung cancer mortality goes from 20 to two-fold after approximately 25 years of cessation [114-119]. For smokers where the lung tissue has only reached the pre-field state, a relatively speedy recovery of the lung tissue is expected. Even in heavy smokers, where both pre-field tissue and field lesions co-exist, we expect the pre-field tissues to regress if abstinence lasts long enough. Similar to the situation with field tissue, pre-field tissues may continue their independent progression during the period before the point of inflection, due to chronic lung inflammation. Once smoking ceases long enough and after the point where inflammation is not advancing any further (the inflection point), the pre-field stress slowly resolves and cell death is gradually reduced. Since the feedback from pre-field stress to pre-field integrity decreases (influence PFI3 in Figure 4), pre-field integrity can recover and eventually reach a capacity close to that of never-smokers. These reversions of the pre-field properties are equivalent to a reversion of pre-field progression (Figure 1 step 8) until a quasi-never smoker state is reached (Figure 1 step 10). Similar to the logic for the field situation, these assumptions are made to correspond to the gene expression profiles for smokers after 20 years [13].
However, since there are no detailed data available on smoking-related effects on the lung epithelium of exsmokers with more than 20 years abstinence, it is not known if lung tissue can ever return to a “true” naïve (never-smoker) state. With continuing abstinence, it is a plausible assumption that at some point, the lung again behaves similarly to naïve tissue, i.e. as a stable, “quasinormal” tissue which will not cause inflammation and will not change, progress, or transform further unless exposed to additional stimulus. The model concept of the smoke-exposed tissue phenotype (SET) is based on the observation that some smoking related effects on lung tissue may persist for many years after cessation since the overall human gene expression patterns become quasi-normal [13]. Gene expression profiling of epithelial cells from bronchoscopy reveals that long-time exsmokers (for up to 20 years) still harbor a smoking specific imprint in their expression profiles [9,13], and show genetic alterations such as loss of heterozygosity (LOH) [120]. Even though the SET is in tissue homeostasis and largely “normal”, these differences from a never smoker lung—“smoking scars”—act as a reminder of the serious tissue damage in former heavy smokers. The assumption of the formation and the timing conditions for SET tissue is dependent upon gene expression patterns. Since it is unclear exactly how SET tissue would behave if smoking were to resume, we have chosen not to address this issue as it is beyond the main questions of how naive tissue is transformed into an adenocarcinoma or how cessation may reverse some of the effects of smoking.
Accordingly, since the pre-field and field changes occur independently and simultaneously for up to several decades post-cessation, there could be a mixture of field and pre-field tissues existing for some time. If all the field tissue dies off, the remaining pre-field tissue may become SET tissue if abstinence lasts long enough. Since it is only formed after a very long term of progression, there is no need to account for its properties in the model.
6. DISCUSSION
Some modeling approaches on cigarette smoking and lung cancer already have existed, but they are largely epidemiological and thus based on disease endpoints such as tumor incidence or mortality (e.g. [121,122]). Our modeling approach is very different from these. First, our model focuses only on the early events during lung carcinogenesis; any tumor incidence findings are out of scope. Secondly, we put strong emphasis on the biological mechanisms rather than modeling the exposure parameters and the disease endpoint.
The field cancerization of the lung has been fairly well-described in the literature [24,123] as how histologically normal-appearing tissue adjacent to a site of neoplasia shares molecular abnormalities with the neoplasm. We have built our biological model based on this theory and extended it to cover already the smoking stressed lung tissue that existed well before the neoplastic lesion. The biological conceptual model is a collection of all the influences (Table 1) affecting the tissue phenotypes and their properties, and the inherent tissue and lung properties (Table 2). Oftentimes, when the biology is described and justified, the influences are indicated one at a time. So the higher level of connectivity and interactivity may be hidden. When these influences are visualized collectively as a directed graph (see Figure 9), the overall high degree of mutual interactivity emerges. From this visualization, the complexity of the entire biology can clearly be seen including the various feedback loops (also evident in more detail on Figures 3-8). The richness of the connectivity coupled with the tissue transformation rules (summarized in Figure 1) contributes to the intricacies of both the smoking and cessation phenomena and adenocarcinoma development. That combination of network and rules has very non-linear aspects:
• The pre-field phenotype and its properties have limitations on how far they can still progress or regress. That limitation is an inherent saturation (or boundary condition) effect.
• Similarly, field tissue and its properties cannot revert, which is a rule that affects the properties differently than those of pre-field, where progression of the tissue and its properties can revert during long term abstinence. In addition, field tissue will proliferate (during active smoking) or start to die off (during long term cessation) and that qualitative change in behavior reflects one aspect of the non-linear nature of the entire system.
• The creation of the first distinctly identified field lesions is a discontinuous event (from the requirement of having mutations occur).
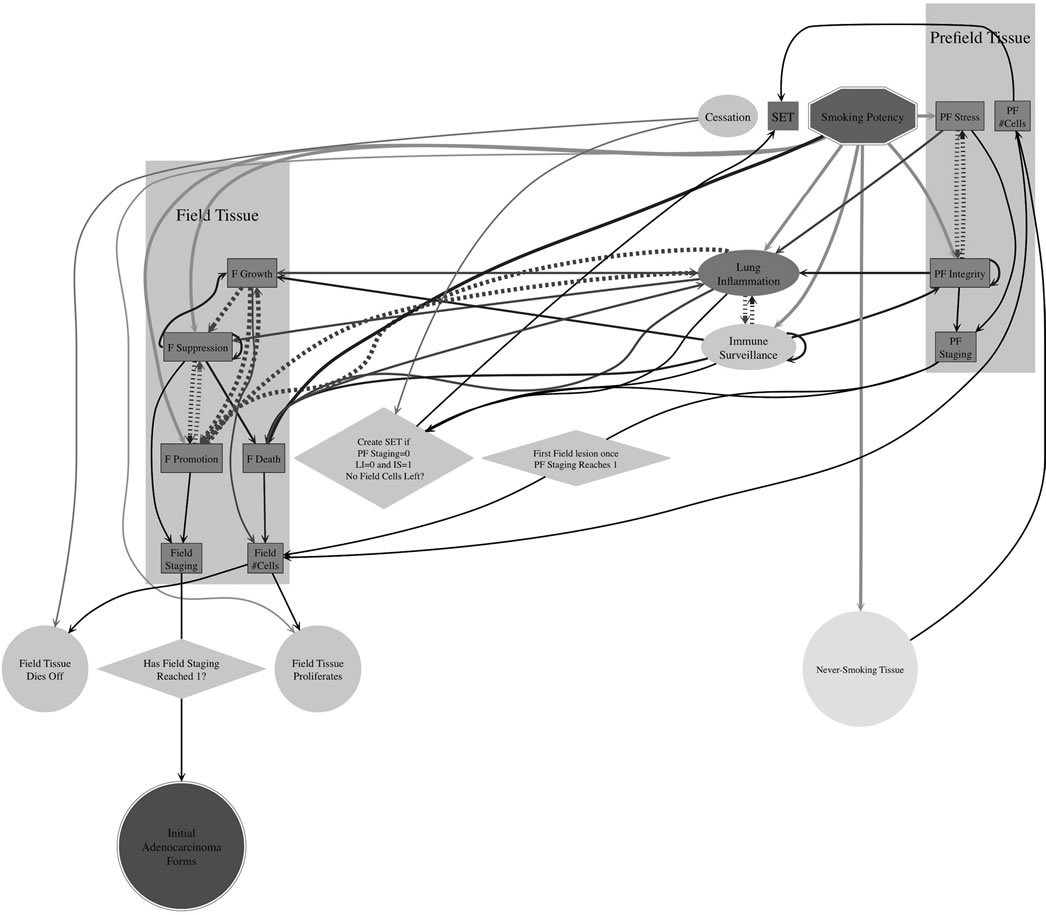
Figure 9. The entire conceptual framework as an influence network. When all the lung and tissue properties (Table 2) and influences (Table 1) are combined, the resulting model is a network—a directed graph—of the interactions (i.e. processes) that modify the behavior of every property in the model. This conceptual network illustrates the non-linearity and complexity of the underlying adenocarcinoma development biology and its representation in the model. This figure is not meant to be read in detail, but to provide a high-level overview of the biological concepts so as to highlight the complexity of the relationships in a visual manner that is not apparent from the text alone. The diagram was generated using the graph layout program Graphviz, http://www.graphviz.org.
• There are several feedback loops within the system as proand anti-progressive influences compete to drive the system away or towards adenocarcinoma development. Some are large cycles and indirect; some loops are direct:
- Lung inflammation and immune surveillance (Figure 3);
- The pre-field integrity and stress properties (Figure 4);
- The field suppression and promotion properties (Figures 5 and 6).
Should it be desired, the current conceptual framework, can be extended by adding more details, whether physiological, genomic or molecular. Such additions could include more properties or influences, new phenotypes, breaking influences (processes) into finer detail, etc. We fully expect this to happen as new experimental data become available or as there is increased understanding of adenocarcinoma development.
The framework is only as good as the current knowledge of smoking-dependent lung adenocarcinoma development allows. However, even though the knowledge is limited, the framework is built upon what is known and on reasonable assumptions to fill gaps. Nonetheless, the framework is an initial effort to build a comprehensive integrative framework that can explain a wide range of smoking related phenomena. There are many areas where it can be improved, for example including additional attributes such as aging of the lung, gender or ethnic differences. In general, we have tried to indicate areas where there are gaps in the biology or where educated guesses are necessary to fill gaps. All these are good candidates for new experiments to determine quantitative data for the processes (or reasonable surrogates) in the model, either to validate the model, determine faults or to indicate the need for a further hierarchical decomposition of any of the concepts. Such experiments would ideally record accurate cigarette smoking histories, the ages and demographics of the smokers, and measure as many simultaneous indicators of lung health and adenocarcinoma development. Some further salient experimental measurements that would be of particular use are:
• The histological changes and associated kinetics when a naive lung is initially exposed to cigarette smoke;
• The long term consequences of smoking abstinence— especially over 20 years;
• The growth and death rates of field tissue;
• Size of the Type II target tissues, in normal, smokingstressed and diseased lungs, and indexed by age, gender, weight and ethnic group.
Another area of exploitation of the conceptual framework would be to use it as the basis for a quantitative, predictive mathematical model, by translating the biological influences into a set of rate equations to quantitatively predict outcomes in realistic smoking scenarios.
One of the issues in building this biological framework and looking for the corresponding quantitative data has been that much of the quantitative clinical data have recorded cigarette smoking histories using an inaccurate quantitative measure—pack years. Ignoring the issue of the reliability of smoker’s memories, it would be nice if the dose, duration and age of smoking were broken apart when reported in data. A related issue is the frequent reporting of dose, regardless of the units, in bins, instead of actual values—e.g. light, heavy, etc. Such discriminations may be critical, especially when trying to distinguish behaviors in pre-field from field tissues.
Lung adenocarcinoma tumorigenesis is very complex and conventional approaches that depend upon population averages may not be the best way to understand the “timing” of the tumor development following prolonged cigarette smoking. Combining traditional epidemiological studies, new experiments motivated by the biological framework will provide a powerful new capability for understanding lung adenocarcinoma development risk associated with cigarette smoking. Integrative, system thinking is a unique way to deepen our understanding of the development of smoking-induced lung adenocarcinoma.
7. ACKNOWLEDGEMENTS
This work would not have been possible without the extensive help, insights, and contributions of Thomas Mueller, whom the authors most gratefully thank. The authors would also like to thank James Battey, Emilija Veljkovic, and Jutta Schüller from Philip Morris International R&D for their careful review and valuable comments on the manuscript.
![]()
![]()
REFERENCES
- Herbst, R.S., Heymach, J.V. and Lippman, S.M. (2008) Lung cancer. The New England Journal of Medicine, 359, 1367-1380.
- CDC (2004) The health consequences of smoking: Report of the surgeon general. US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health.
- Parkin, D.M., Bray, F., Ferlay, J., et al. (2005) Global cancer statistics, 2002. CA: A Cancer Journal for Clinicians, 55, 74-108. doi:10.3322/canjclin.55.2.74
- Burns, D.M., Anderson, C.M. and Gray, N. (2011) Do changes in cigarette design influence the rise in adenocarcinoma of the lung? Cancer Causes & Control, 22, 13- 22. doi:10.1007/s10552-010-9660-0
- Franceschi, S. and Bidoli, E. (1999) The epidemiology of lung cancer. Annals of Oncology, 10, S3-S6. doi:10.1093/annonc/10.suppl_5.S3
- Sun, S., Schiller, J.H. and Gazdar, A.F. (2007) Lung cancer in never smokers—A different disease. Nature Reviews Cancer, 7, 778-790.
- Lantuejoul, S., Salameire, D., Salon, C., et al. (2009) Pulmonarypreneoplasia—Sequential molecular carcinogenetic events. Histopathology, 54, 43-54. doi:10.1111/j.1365-2559.2008.03182.x
- Peto, R., Darby, S., Deo, H., et al. (2000) Smoking, smoking cessation, and lung cancer in the UK since 1950: Combination of national statistics with two case—Control studies. British Medical Journal, 321, 323-329. doi:10.1136/bmj.321.7257.323
- Beane, J., Sebastiani, P., Liu, G., et al. (2007) Reversible and permanent effects of tobacco smoke exposure on airway epithelial gene expression. Genome Biology, 8, R201. doi:10.1186/gb-2007-8-9-r201
- Gower, A.C., Steiling, K., Brothers, J.F., et al. (2011) Transcriptomic studies of the airway field of injury associated with smoking-related lung disease. Proceedings of the American Thoracic Society, 8, 173-179. doi:10.1513/pats.201011-066MS
- Izzotti, A., Bagnasco, M., Cartiglia, C., et al. (2005) Chemoprevention of genome, transcriptome, and proteome alterations induced by cigarette smoke in rat lung. European Journal of Cancer, 41, 1864-1874. doi:10.1016/j.ejca.2005.04.011
- Powell, C.A., Spira, A., Derti, A., et al. (2003) Gene expression in lung adenocarcinomas of smokers and nonsmokers. American Journal of Respiratory Cell and Molecular Biology, 29, 157-162. doi:10.1165/rcmb.2002-0183RC
- Spira, A., Beane, J., Shah, V., et al. (2004) Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proceedings of the National Academy of Sciences of the United States of America, 101, 10143-10148. doi:10.1073/pnas.0401422101
- Spira, A., Schembri, F., Beane, J., et al. (2004) Impact of cigarette smoke on the normal airway transcriptome. Chest, 125, 115S.
- Spira, A., Beane, J.E., Shah, V., et al. (2007) Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nature Medicine, 13, 361-366.
- Stav, D., Bar, I. and Sandbank, J. (2008) Gene expression subtraction of non-cancerous lung from smokers and non-smokers with adenocarcinoma, as a predictor for smokers developing lung cancer. Journal of Experimental & Clinical Cancer Research, 27, 45. doi:10.1186/1756-9966-27-45
- Gebel, S., Gerstmayer, B., Bosio, A., et al. (2004) Gene expression profiling in respiratory tissues from rats exposed to mainstream cigarette smoke. Carcinogenesis, 25, 169-178. doi:10.1093/carcin/bgg193
- Gebel, S., Gerstmayer, B., Kuhl, P., et al. (2006) The kinetics of transcriptomic changes induced by cigarette smoke in rat lungs reveals a specific program of defense, inflammation, and circadian clock gene expression. Toxicological Sciences, 93, 422-431. doi:10.1093/toxsci/kfl071
- Meng, Q.R., Gideon, K.M., Harbo, S.J., et al. (2006) Gene expression profiling in lung tissues from mice exposed to cigarette smoke, lipopolysaccharide, or smoke plus lipopolysaccharide by inhalation. Inhalation Toxicology, 18, 555-568.
- Schwartz, A.G., Prysak, G.M., Bock, C.H., et al. (2007) The molecular epidemiology of lung cancer. Carcinogenesis, 28, 507-518. doi:10.1093/carcin/bgl253
- Kerr, K.M. (2001) Pulmonary preinvasive neoplasia. Journal of Clinical Pathology, 54, 257-271. doi:10.1136/jcp.54.4.257
- Wistuba, I.I. and Gazdar, A.F. (2006) Lung cancer preneoplasia. Annual Review, 1, 331-348. doi:10.1146/annurev.pathol.1.110304.100103
- Borczuk, A.C. and Powell, C.A. (2007) Expression profiling and lung cancer development. Proceedings of the American Thoracic Society, 4, 127-132. doi:10.1513/pats.200607-143JG
- Steiling, K., Ryan, J., Brody, J.S. et al. (2008) The field of tissue injury in the lung and airway. Cancer Prevention Research, 1, 396-403. doi:10.1158/1940-6207.CAPR-08-0174
- Stampfli, M.R. and Anderson, G.P. (2009) How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nature Reviews Immunology, 9, 377-384.
- Wooten, J.B., Chouchane, S. and McGrath, T.E. (2011) Tobacco smoke constituents affecting oxidative stress. Cigarette Smoke and Oxidative Stress, 5-46.
- Aoshiba, K. and Nagai, A. (2003) Oxidative stress, cell death, and other damage to alveolar epithelial cells induced by cigarette smoke. Tobacco Induced Diseases, 1, 219-226.
- Seet, R.C., Lee, C.Y., Loke, W.M., et al. (2011) Biomarkers of oxidative damage in cigarette smokers: Which biomarkers might reflect acute versus chronic oxidative stress? Free Radical Biology and Medicine, 50, 1787- 1793. doi:10.1016/j.freeradbiomed.2011.03.019
- Chen, G.Y. and Nunez, G. (2010) Sterile inflammation: Sensing and reacting to damage. Nature Reviews Immunology, 10, 826-837.
- Mian, M.F., Lauzon, N.M., Stampfli, M.R., et al. (2008) Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. Journal of Leukocyte Biology, 83, 774-784. doi:10.1189/jlb.0707481
- Lu, L.M., Zavitz, C.C., Chen, B., et al. (2007) Cigarette smoke impairs NK cell-dependent tumor immune surveillance. The Journal of Immunology, 178, 936-943.
- Walser, T., Cui, X., Yanagawa, J., et al. (2008) Smoking and lung cancer: The role of inflammation. Proceedings of the American Thoracic Society, 5, 811-815. doi:10.1513/pats.200809-100TH
- Nathan, C. and Ding, A. (2010) Nonresolving inflammation. Cell, 140, 871-882. doi:10.1016/j.cell.2010.02.029
- Grivennikov, S.I. and Karin, M. (2010) Inflammation and oncogenesis: A vicious connection. Current Opinion in Genetics & Development, 20, 65-71. doi:10.1016/j.gde.2009.11.004
- Hanahan, D. and Weinberg, R.A. (2011) Hallmarks of cancer: The next generation. Cell, 144, 646-674. doi:10.1016/j.cell.2011.02.013
- Takahashi, H., Ogata, H., Nishigaki, R., et al. (2010) Tobacco smoke promotes lung tumorigenesis by triggering IKKbetaand JNK1-dependent inflammation. Cancer Cell, 17, 89-97. doi:10.1016/j.ccr.2009.12.008
- Botelho, F.M., Gaschler, G.J., Kianpour, S., et al. (2010) Innate immune processes are sufficient for driving cigarette smoke-induced inflammation in mice. American Journal of Respiratory Cell and Molecular Biology, 42, 394-403. doi:10.1165/rcmb.2008-0301OC
- Yang, I.A., Relan, V., Wright, C.M., et al. (2011) Common pathogenic mechanisms and pathways in the development of COPD and lung cancer. Expert Opinion on Therapeutic Targets, 15, 439-456.
- Ginsberg, R.J., Vokes, E.E and Raben, A. (1997) Cancer principles and practices of oncology. 5th Edition, Lippincott-Raven, Philadelphia.
- Mason, R.J., Dobbs, L.G., Greenleaf, R.D., et al. (1977) Alveolar type II cells. Federation Proceedings, 36, 2697- 2702.
- Ochs, M. (2006) A brief update on lung stereology. Journal of Microscopy, 222, 188-200. doi:10.1111/j.1365-2818.2006.01587.x
- Cavarra, E., Fardin, P., Fineschi, S., et al. (2009) Early response of gene clusters is associated with mouse lung resistance or sensitivity to cigarette smoke. American Journal of Physiology—Lung Cellular and Molecular Physiology, 296, L418-L429. doi:10.1152/ajplung.90382.2008
- Stevenson, C.S., Docx, C., Webster, R., et al. (2007) Comprehensive gene expression profiling of rat lung reveals distinct acute and chronic responses to cigarette smoke inhalation. American Journal of Physiology— Lung Cellular and Molecular Physiology, 293, L1183- L1193. doi:10.1152/ajplung.00105.2007
- Beane, J., Spira, A. and Lenburg, M.E. (2009) Clinical impact of high-throughput gene expression studies in lung cancer. Journal of Thoracic Oncology, 4, 109-118.
- Nguyen, T., Sherratt, P.J. and Pickett, C.B. (2003) Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annual Reviews of Pharmacology and Toxicology, 43, 233-260. doi:10.1146/annurev.pharmtox.43.100901.140229
- Benjamin, R.M. (2011) Exposure to tobacco smoke causes immediate damage: A report of the surgeon general. Public Health Reports, 126, 158-159.
- Wickenden, J.A., Clarke, M.C., Rossi, A.G., et al. (2003) Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosis. American Journal of Respiratory Cell and Molecular Biology, 29, 562- 570. doi:10.1165/rcmb.2002-0235OC
- Damico, R., Simms, T., Kim, B.S., et al. (2011) P53 mediates cigarette smoke-induced apoptosis of pulmonary endothelial cells: Inhibitory effects of macrophage migration inhibitor factor. American Journal of Respiratory Cell and Molecular Biology, 44, 323-332. doi:10.1165/rcmb.2009-0379OC
- Kasahara, Y., Tuder, R.M., Cool, C.D., et al. (2001) Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. American Journal of Respiratory and Critical Care, 163, 737-744. doi:10.1164/ajrccm.163.3.2002117
- Liu, H., Ma, L., Wu, J., et al. (2009) Apoptosis of alveolar wall cells in chronic obstructive pulmonary disease patients with pulmonary emphysema is involved in emphysematous changes. Journal of Huazhong University of Science and Technology [Medical Sciences], 29, 466-469. doi:10.1007/s11596-009-0415-7
- Yokohori, N., Aoshiba, K. and Nagai, A. (2004) Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest, 125, 626- 632.
- Hodge, S., Hodge, G., Scicchitano, R., et al. (2003) Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunology & Cell Biology, 81, 289-296. doi:10.1046/j.1440-1711.2003.t01-1-01170.x
- Hodge, S., Hodge, G., Ahern, J., et al. (2007) Smoking alters alveolar macrophage recognition and phagocytic ability: Implications in chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology, 37, 748-755. doi:10.1165/rcmb.2007-0025OC
- Kazeros, A., Harvey, B.G., Carolan, B.J., et al. (2008) Overexpression of apoptotic cell removal receptor MERTK in alveolar macrophages of cigarette smokers. American Journal of Respiratory Cell and Molecular Biology, 39, 747-757. doi:10.1165/rcmb.2007-0306OC
- Crosby, L.M. and Waters, C.M. (2010) Epithelial repair mechanisms in the lung. American Journal of Physiology—Lung Cellular and Molecular Physiology, 298, L715- L731. doi:10.1152/ajplung.00361.2009
- Gardner, A., Borthwick, L.A. and Fisher, A.J. (2010) Lung epithelial wound healing in health and disease. Expert Review of Respiratory Medicine, 4, 647-660.
- Rennard, S.I., Togo, S. and Holz, O. (2006) Cigarette smoke inhibits alveolar repair: A mechanism for the development of emphysema. Proceedings of the American Thoracic Society, 3, 703-708. doi:10.1513/pats.200605-121SF
- Hodge, S., Hodge, G., Scicchitano, R., et al. (2003) Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunology & Cell Biology, 81, 289-296. doi:10.1046/j.1440-1711.2003.t01-1-01170.x
- Hodge, S., Hodge, G., Ahern, J., et al. (2007) Smoking alters alveolar macrophage recognition and phagocytic ability: Implications in chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology, 37, 748-755. doi:10.1165/rcmb.2007-0025OC
- [61] Vandivier, R.W., Henson, P.M. and Douglas, I.S. (2006) Burying the dead: The impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest, 129, 1673-1682. doi:10.1378/chest.129.6.1673
- [62] Hang, B. (2010) Formation and repair of tobacco carcinogen-derived bulky DNA adducts. Journal of Nucleic Acids, 2010, Article ID: 709521. doi:10.4061/2010/709521
- [63] Van der Toorn, M., Slebos, D.J., de Bruin, H.G., et al. (2007) Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. American Journal of Physiology—Lung Cellular and Molecular Physiology, 292, L1211- L1218. doi:10.1152/ajplung.00291.2006
- [64] Medzhitov, R. (2008) Origin and physiological roles of inflammation. Nature, 454, 428-435.
- [65] Cantin, A.M. (2010) Cellular response to cigarette smoke and oxidants: Adapting to survive. Proceedings of the American Thoracic Society, 7, 368-375. doi:10.1513/pats.201001-014AW
- [66] Bellomi, M., Veronesi, G., Rampinelli, C., et al. (2007) Evolution of lung nodules < or =5 mm detected with low-dose CT in asymptomatic smokers. British Journal of Radiology, 80, 708-712.
- [67] Diederich, S., Wormanns, D., Semik, M., et al. (2002) Screening for early lung cancer with low-dose spiral CT: Prevalence in 817 asymptomatic smokers. Radiology, 222, 773-781. doi:10.1148/radiol.2223010490
- [68] Diederich, S., Thomas, M., Semik, M., et al. (2004) Screening for early lung cancer with low-dose spiral computed tomography: Results of annual follow-up examinations in asymptomatic smokers. European Radiology, 14, 691- 702. doi:10.1007/s00330-003-2200-5
- [69] Diederich, S., Hansen, J. and Wormanns, D. (2005) Resolving small pulmonary nodules: CT features. European Radiology, 15, 2064-2069. doi:10.1007/s00330-005-2836-4
- [70] Kodama, K., Higashiyama, M., Yokouchi, H., et al. (2002) Natural history of pure ground-glass opacity after longterm follow-up of more than 2 years. The Annals of Thoracic Surgery, 73, 386-392. doi:10.1016/S0003-4975(01)03410-5
- [71] Kuriyama, K., Seto, M., Kasugai, T., et al. (1999) Ground-glass opacity on thin-section CT: Value in differentiating subtypes of adenocarcinoma of the lung. American Journal of Roentgenology, 173, 465-469. doi:10.2214/ajr.173.2.10430155
- [72] Novello, S., Fava, C., Borasio, P., et al. (2005) Three-year findings of an early lung cancer detection feasibility study with low-dose spiral computed tomography in heavy smokers. Annals of Oncology, 16, 1662-1666. doi:10.1093/annonc/mdi314
- [73] Revel, M.P., Lefort, C., Bissery, A., et al. (2004) Pulmonary nodules: Preliminary experience with three-dimensional evaluation. Radiology, 231, 459-466. doi:10.1148/radiol.2312030241
- [74] Revel, M.P., Bissery, A., Bienvenu, M., et al. (2004) Are two-dimensional CT measurements of small noncalcified pulmonary nodules reliable? Radiology, 231, 453-458. doi:10.1148/radiol.2312030167
- [75] Revel, M.P., Merlin, A., Peyrard, S., et al. (2006) Software volumetric evaluation of doubling times for differrentiating benign versus malignant pulmonary nodules. American Journal of Roentgenology, 187, 135-142. doi:10.2214/AJR.05.1228
- [76] Nomori, H., Horio, H., Naruke, T., et al. (2001) A case of lung cancer with extensive pleural adhesion, which could be resected by a thoracoscopic middle lobectomy. Kyobu Geka, 54, 388-390.
- [77] Suzuki, K., Takahashi, K., Yoshida, J., et al. (1998) Synchronous double primary lung carcinomas associated with multiple atypical adenomatous hyperplasia. Lung Cancer, 19, 131-139. doi:10.1016/S0169-5002(97)00082-2
- [78] Weng, S., Tsuchiya, E., Satoh, Y., et al. (1990) Multiple atypical adenomatous hyperplasia of type II pneumonocytes and bronchiolo-alveolar carcinoma. Histopathology, 16, 101-103.
- [79] Chapman, A.D. and Kerr, K.M. (2000) The association between atypical adenomatous hyperplasia and primary lung cancer. British Journal of Cancer, 83, 632-636. doi:10.1054/bjoc.2000.1317
- [80] Kerr, K.M., Carey, F.A., King, G., et al. (1994) Atypical alveolar hyperplasia: Relationship with pulmonary adenocarcinoma, p53, and c-erbB-2 expression. The Journal of Pathology, 174, 249-256. doi:10.1002/path.1711740404
- [81] Mori, M., Tezuka, F., Chiba, R., et al. (1996) Atypical adenomatous hyperplasia and adenocarcinoma of the human lung: Their heterology in form and analogy in immunohistochemical characteristics. Cancer, 77, 665-674. doi:10.1002/(SICI)1097-0142(19960215)77:4<665::AID-CNCR12>3.0.CO;2-Z
- [82] Mori, M., Rao, S.K., Popper, H.H., et al. (2001) Atypical adenomatous hyperplasia of the lung: A probable forerunner in the development of adenocarcinoma of the lung. Modern Pathology, 14, 72-84. doi:10.1038/modpathol.3880259
- [83] Christmann, M., Verbeek, B., Roos, W.P., et al. (2011) O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: Enzyme activity, promoter methylation and immunohistochemistry. Biochimica et Biophysica Acta, 1816, 179-190.
- [84] Leng, S., Bernauer, A.M., Hong, C., et al. (2011) The A/G allele of rs16906252 predicts for MGMT methylation and is selectively silenced in premalignant lesions from smokers and in lung adenocarcinomas. Clinical Cancer Research, 17, 2014-2023. doi:10.1158/1078-0432.CCR-10-3026
- [85] Damiani, L.A., Yingling, C.M., Leng, S., et al. (2008) Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Research, 68, 9005-9014. doi:10.1158/0008-5472.CAN-08-1276
- [86] Lin, R.K., Hsieh, Y.S., Lin, P., et al. (2010) The tobaccospecific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. Journal of Clinical Investigation, 120, 521-532. doi:10.1172/JCI40706
- [87] Liu, F., Killian, J.K., Yang, M., et al. (2010) Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene, 29, 3650-3664.
- [88] Schetter, A.J., Heegaard, N.H. and Harris, C.C. (2010) Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis, 31, 37-49. doi:10.1093/carcin/bgp272
- [89] Kitamura, H., Kameda, Y., Nakamura, N., et al. (1995) Proliferative potential and p53 overexpression in precursor and early stage lesions of bronchioloalveolar lung carcinoma. American Journal of Pathology, 146, 876- 887.
- [90] Nakayama, H., Noguchi, M., Tsuchiya, R., et al. (1990) Clonal growth of atypical adenomatous hyperplasia of the lung: Cytofluorometric analysis of nuclear DNA content. Modern Pathology, 3, 314-320.
- [91] Niho, S., Yokose, T., Suzuki, K., et al. (1999) Monoclonality of atypical adenomatous hyperplasia of the lung. American Journal of Pathology, 154, 249-254.
- [92] Carey, F.A., Wallace, W.A., Fergusson, R.J., et al. (1992) Alveolar atypical hyperplasia in association with primary pulmonary adenocarcinoma: A clinicopathological study of 10 cases. Thorax, 47, 1041-1043. doi:10.1136/thx.47.12.1041
- [93] Koga, T., Hashimoto, S., Sugio, K., et al. (2002) Lung adenocarcinoma with bronchioloalveolar carcinoma component is frequently associated with foci of high-grade atypical adenomatous hyperplasia. American Journal of ClinicalPathology, 117, 464-470. doi:10.1309/CHXA-3MH0-B7FD-JGUL
- [94] Mori, M., Kaji, M., Tezuka, F., et al. (1998) Comparative ultrastructural study of atypical adenomatous hyperplasia and adenocarcinoma of the human lung. Ultrastructural Pathology, 22, 459-466. doi:10.3109/01913129809032282
- [95] Hayashi, H., Miyamoto, H., Ito, T., et al. (1997) Analysis of p21Waf1/Cip1 expression in normal, premalignant, and malignant cells during the development of human lung adenocarcinoma. American Journal of Pathology, 151, 461-470.
- [96] Kurasono, Y., Ito, T., Kameda, Y., et al. (1998) Expression of cyclin D1, retinoblastoma gene protein, and p16 MTS1 protein in atypical adenomatous hyperplasia and adenocarcinoma of the lung. An immunohistochemical analysis. Virchows Archiv, 432, 207-215. doi:10.1007/s004280050157
- [97] Yokose, T., Ito, Y. and Ochiai, A. (2000) High prevalence of atypical adenomatous hyperplasia of the lung in autopsy specimens from elderly patients with malignant neoplasms. Lung Cancer, 29, 125-130. doi:10.1016/S0169-5002(00)00101-X
- [98] Munoz-Antonia, T., Muro-Cacho, C., Sharma, S., et al. (2007) Expression of TGFbeta type-II receptor in association with markers of proliferation and apoptosis in premalignant lung lesions. Cancer, 110, 1527-1531. doi:10.1002/cncr.22937
- [99] Tan, D.F., Huberman, J.A., Hyland, A., et al. (2001) MCM2—A promising marker for premalignant lesions of the lung: A cohort study. BMC Cancer, 1, 6. doi:10.1186/1471-2407-1-6
- [100] Swann, J.B., Coquet, J.M., Smyth, M.J., et al. (2007) CD1-restricted T cells and tumor immunity. Current Topics in Microbiology and Immunology, 314, 293-323. doi:10.1007/978-3-540-69511-0_12
- [101] Evan, G. (1994) Why we live and why we die. Chemistry & Biology, 1, 137-141. doi:10.1016/1074-5521(94)90003-5
- [102] Lowe, S.W., Cepero, E. and Evan, G. (2004) Intrinsic tumour suppression. Nature, 432, 307-315. doi:10.1038/nature03098
- [103] Akyurek, N., Memis, L., Ekinci, O., et al. (2006) Survivin expression in pre-invasive lesions and non-small cell lung carcinoma. Virchows Archiv, 449, 164-170. doi:10.1007/s00428-006-0239-9
- [104] Nakanishi, K., Kawai, T., Kumaki, F., et al. (2003) Survivin expression in atypical adenomatous hyperplasia of the lung. American Journal of Clinical Pathology, 120, 712-719. doi:10.1309/GWTN2JTAN6K73YDE
- [105] Domagala-Kulawik, J. (2008) Effects of cigarette smoke on the lung and systemic immunity. Journal of Physiology and Pharmacology, 59, 19-34.
- [106] Kode, A., Yang, S.R. and Rahman, I. (2006) Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respiratory Research, 7, 132. doi:10.1186/1465-9921-7-132
- [107] Piccinini, A.M. and Midwood, K.S. (2010) DAMPening inflammation by modulating TLR signalling. Mediators of Inflammation, 2010, Article ID: 672395.
- [108] Takashima, S., Sone, S., Li, F., et al. (2003) Indeterminate solitary pulmonary nodules revealed at population-based CT screening of the lung: Using first followup diagnostic CT to differentiate benign and malignant lesions. American Journal of Roentgenology, 180, 1255- 1263. doi:10.2214/ajr.180.5.1801255
- [109] Sartori, G., Cavazza, A., Bertolini, F., et al. (2008) A subset of lung adenocarcinomas and atypical adenomatous hyperplasia-associated foci are genotypically related: An EGFR, HER2, and K-ras mutational analysis. American Journal of Clinical Pathology, 129, 202-210. doi:10.1309/THU13F3JRJVWLM30
- [110] Chaudhuri, R., Livingston, E., McMahon, A.D., et al. (2006) Effects of smoking cessation on lung function and airway inflammation in smokers with asthma. American Journal of Respiratory and Critical Care, 174, 127-133. doi:10.1164/rccm.200510-1589OC
- [111] Karimi, R., Tornling, G., Grunewald, J., et al. (2012) Cell recovery in bronchoalveolar lavage fluid in smokers is dependent on cumulative smoking history. PLoS One, 7, e34232. doi:10.1371/journal.pone.0034232
- [112] Hecht, S.S., Carmella, S.G., Chen, M., et al. (1999) Quantitation of urinary metabolites of a tobacco-specific lung carcinogen after smoking cessation. Cancer Research, 59, 590-596.
- [113] Benhamou, E., Benhamou, S., Auquier, A., et al. (1989) Changes in patterns of cigarette smoking and lung cancer risk: Results of a case-control study. British Journal of Cancer, 60, 601-604. doi:10.1038/bjc.1989.322
- [114] Willemse, B.W., ten Hacken, N.H., Rutgers, B., et al. (2005) Association of current smoking with airway inflammation in chronic obstructive pulmonary disease and asymptomatic smokers. Respiratory Research, 6, 38. doi:10.1186/1465-9921-6-38
- [115] Lee, P. N. (2000) Epidemiology of lung cancer. http://www.pnlee.co.uk/Reports.htm
- [116] Alberg, A.J. and Samet, J.M. (2003) Epidemiology of lung cancer. Chest, 123, 21S-49S.
- [117] Barbone, F., Bovenzi, M., Cavallieri, F., et al. (1997) Cigarette smoking and histologic type of lung cancer in men. Chest, 112, 1474-1479. doi:10.1378/chest.112.6.1474
- [118] Ebbert, J.O., Yang, P., Vachon, C.M., et al. (2003) Lung cancer risk reduction after smoking cessation: Observations from a prospective cohort of women. Journal of Clinical Oncology, 21, 921-926.
- [119] Matos, E., Vilensky, M., Boffetta, P., et al. (1998) Lung cancer and smoking: A case-control study in Buenos Aires, Argentina. Lung Cancer, 21, 155-163. doi:10.1016/S0169-5002(98)00055-5
- [120] Pohlabeln, H., Jockel, K.H. and Muller, K.M. (1997) The relation between various histological types of lung cancer and the number of years since cessation of smoking. Lung Cancer, 18, 223-229. doi:10.1016/S0169-5002(97)00067-6
- [121] Wistuba, I.I., Lam, S., Behrens, C., et al. (1997) Molecular damage in the bronchial epithelium of current and former smokers. Journal of the National Cancer Institute, 89, 1366-1373. doi:10.1093/jnci/89.18.1366
- [122] Lubin, J.H. and Caporaso, N.E. (2007) Cigarette smoking and lung cancer: Modeling total exposure and intensity. Cancer Epidemiology, Biomarkers & Prevention, 15, 517- 523. doi:10.1158/1055-9965.EPI-05-0863
- [123] Thurston, S.W., Liu, G., Miller, D.P., et al. (2005) Modeling lung cancer risk in case-control studies using a new dose metric of smoking. Cancer Epidemiology, Biomarkers & Prevention, 14, 2296-2302. doi:10.1158/1055-9965.EPI-04-0393
- [124] Greenberg, A.K., Yee, H. and Rom, W.N. (2002) Preneoplastic lesions of the lung. Respiratory Research, 3, 20. doi:10.1186/rr170
- [125] Tollerud, D.J., Clark, J.W., Brown, L.M., et al. (1989) Association of cigarette smoking with decreased numbers of circulating natural killer cells. American Review of Respiratory Disease, 139, 194-198. doi:10.1164/ajrccm/139.1.194
- [126] Minematsu, N., Blumental-Perry, A. and Shapiro, S.D. (2011) Cigarette smoke inhibits engulfment of apoptotic cells by macrophages through inhibition of actin rearrangement. American Journal of Respiratory Cell and Molecular Biology, 44, 474-482. doi:10.1165/rcmb.2009-0463OC
- [127] Guzik, K., Skret, J., Smagur, J., et al. (2011) Cigarette smoke-exposed neutrophils die unconventionally but are rapidly phagocytosed by macrophages. Cell Death & Disease, 2, e131.
- [128] Holt, P.G. and Keast, D. (2011) Environmentally induced changes in immunological function: Acute and chronic effects of inhalation of tobacco smoke and other atmospheric contaminants in man and experimental animals. Bacteriological Reviews, 41, 205-216.
- [129] Sopori, M. (2002) Effects of cigarette smoke on the immune system. Nature Reviews Immunology, 2, 372-377.
- [130] Robbins, C.S., Dawe, D.E., Goncharova, S.I., et al. (2004) Cigarette smoke decreases pulmonary dendritic cells and impacts antiviral immune responsiveness. American Journal of Respiratory Cell and Molecular Biology, 30, 202- 211. doi:10.1165/rcmb.2003-0259OC
- [131] Chan, C.W. and Housseau, F. (2007) The “kiss of death” by dendritic cells to cancer cells. Cell Death & Differentiation, 15, 58-69. doi:10.1038/sj.cdd.4402235
- [132] Wissinger, E., Goulding, J. and Hussell, T. (2009) Immune homeostasis in the respiratory tract and its impact on heterologous infection. Seminars in Immunology, 21, 147-155. doi:10.1016/j.smim.2009.01.005
- [133] Grivennikov, S.I., Greten, F.R. and Karin, M. (2010) Immunity, inflammation, and cancer. Cell, 140, 883-899. doi:10.1016/j.cell.2010.01.025
- [134] Gebel, S., Diehl, S., Pype, J., et al. (2010) The transcriptome of Nrf2-/-mice provides evidence for impaired cell cycle progression in the development of cigarette smokeinduced emphysematous changes. Toxicological Sciences, 115, 238-252. doi:10.1093/toxsci/kfq039
- [135] Losa, D., Chanson, M. and Crespin, S. (2011) Connexins as therapeutic targets in lung disease. Expert Opinion on Therapeutic Targets, 15, 989-1002. doi:10.1517/14728222.2011.584875
- [136] Rangasamy, T., Cho, C.Y., Thimmulappa, R.K., et al. (2004) Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. Journal of Clinical Investigation, 114, 1248-1259.
- [137] Ogawa, Y., Duru, E.A. and Ameredes, B.T. (2008) Role of IL-10 in the resolution of airway inflammation. Current Molecular Medicine, 8, 437-445. doi:10.2174/156652408785160907
- [138] Ji, H., Houghton, A.M., Mariani, T.J., et al. (2005) K-ras activation generates an inflammatory response in lung tumors. Oncogene, 25, 2105-2112. doi:10.1038/sj.onc.1209237
- [139] Iliopoulos, D., Hirsch, H.A. and Struhl, K. (2009) An epigenetic switch involving NF-kB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell, 139, 693-706. doi:10.1016/j.cell.2009.10.014
- [140] Thorley, A.J. and Tetley, T.D. (2007) Pulmonary epithetlium, cigarette smoke, and chronic obstructive pulmonary disease. International Journal of Chronic Obstructive Pulmonary Disease, 2, 409-428.
- [141] Povey, A.C., O’Donnell, P., Barber, P., et al. (2006) Smoking is associated with a decrease of O6-alkylguanine-DNA alkyltransferase activity in bronchial epithelial cells. International Journal of Cancer, 119, 463-466. doi:10.1002/ijc.21790
- [142] Medzhitov, R. and Janeway Jr, C.A. (2002) Decoding the patterns of self and nonself by the innate immune system. Science, 296, 298-300. doi:10.1126/science.1068883
- [143] Hanahan, D. and Weinberg, R.A. (2000) The hallmarks of cancer. Cell, 100, 57-70. doi:10.1016/S0092-8674(00)81683-9
- [144] De Visser, K.E., Eichten, A. and Coussens, L.S.M. (2006) Paradoxical roles of the immune system during cancer development. Nature Reviews. Cancer, 6, 24-37.
- [145] Halliwell, B. (2011) Free radicals and antioxidants—Quo vadis? Trends in Pharmacological Sciences, 32, 125-130. doi:10.1016/j.tips.2010.12.002
- [146] Iliopoulos, D., Hirsch, H.A. and Struhl, K. (2009) An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell, 139, 693-706. doi:10.1016/j.cell.2009.10.014
- [147] Iliopoulos, D., Jaeger, S.A., Hirsch, H.A., et al. (2010) STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Molecular Cell, 39, 493-506. doi:10.1016/j.molcel.2010.07.023
- [148] Reuter, S., Gupta, S.C., Chaturvedi, M.M., et al. (2010) Oxidative stress, inflammation, and cancer: How are they linked? Free Radical Biology and Medicine, 49, 1603- 1616. doi:10.1016/j.freeradbiomed.2010.09.006
- [149] Schreiber, R.D., Old, L.J. and Smyth, M.J. (2011) Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science, 331, 1565-1570. doi:10.1126/science.1203486
- [150] Ochs, M., Nyengaard, J.R., Jung, A., et al. (2004) The number of alveoli in the human lung. American Journal of Respiratory and Critical Care, 169, 120-124. doi:10.1164/rccm.200308-1107OC
- [151] Dini, L. (2010) Phagocytosis of dying cells: Influence of smoking and static magnetic fields. Apoptosis, 15, 1147- 1164. doi:10.1007/s10495-010-0490-z
- [152] Rom, W.N., Hay, J.G., Lee, T.C., et al. (2000) Molecular and genetic aspects of lung cancer. American Journal of Respiratory and Critical Care, 161, 1355-1367. doi:10.1164/ajrccm.161.4.9908012
- [153] Wistuba, I.I. (2007) Genetics of preneoplasia: Lessons from lung cancer. Current Molecular Medicine, 7, 3-14. doi:10.2174/156652407779940468