Advances in Lung Cancer
Vol.2 No.1(2013), Article ID:28861,10 pages DOI:10.4236/alc.2013.21002
CD8 T cell response in a phase I study of therapeutic vaccination of advanced NSCLC with allogeneic tumor cells secreting endoplasmic reticulum-chaperone gp96-Ig-peptide complexes*
![]()
1Department of Medicine, Division of Hematology/Oncology, University of Miami, Miller School of Medicine, Miami, USA
2Sylvester Comprehensive Cancer Center, Biostatistics Core Facility, University of Miami, Miller School of Medicine, Miami, USA
3Department of Microbiology and Immunology, University of Miami, Miller School of Medicine, Miami, USA;
#Corresponding Author: epodack@miami.edu
Received 21 December 2012; revised 22 January 2013; accepted 8 February 2013
Keywords: Heat Shock Protein; Chaperone Gp-96; Non-Small Cell Lung Cancer; Gene Transfer Immunotherapy; Allogeneic Vaccine; Cytotoxic CD8 T Cells
ABSTRACT
Antigen containing, allogeneic cells secreting the genetically modified protein and peptide-chaperone gp96-Ig cross, prime and expand antigen specific CD8 T cells with therapeutic antitumor activity in mice. In a first in man phase I study, we now report the results of therapeutic vaccination of non-small cell lung cancer (NSCLC) patients with an established, allogeneic nonsmall cell lung adenocarcinoma cell line secreting gp96-Ig. Advanced NSCLC-patients stage IIIB or IV of any histological subtype were enrolled and treated with up to 36 vaccinations over the course of 18 weeks. Primary endpoint was safety, secondary endpoints tumor response and overall survival. Measurement of tumor antigen specific CD8 CTL responses is precluded by the lack of known NSCLC associated antigens. Therefore, we measured patient CD8 T cell-IFN-γ responses to allo-antigens of the vaccine cells as surrogate for tumor antigen specific CD8 CTL. In 7 of 18 treated patients tumor growth was stabilized, however none of the 18 patients had an objective tumor response by RECIST criteria. Of 15 patients evaluable for immune response, 11 responded to vaccination with more than twofold increase in CD8-IFN-γ frequency above baseline. These patients had a median survival time of 16.5 months. Four patients who had no CD8 response above base line had survival times from 2.1 to 6.7 months. Our data are consistent with the concept that generation of CD8 CTL by therapeutic vaccination may delay tumor growth and progression and mediate prolonged survival even in the absence of objective tumor responses. Further studies will be required to test this concept and promising result.
1. INTRODUCTION
Tumor rejection by the immune system requires the generation and clonal expansion of tumor antigen specific, major histocompatibility complex I (MHC I) restricted, cytotoxic CD8+ T cells (CTL). Activation, differentiation and expansion of tumor antigen specific CD8+ CTL is dependent on tumor antigen presentation by MHC I of activated dendritic cells (DC) to the T cell receptor (TCR) of cognate CD8 T cells. MHC I presentation of tumor antigens by activated DC entails antigen transfer from tumor cells to DC in a process known as “antigen cross-presentation”. The molecular mechanisms of antigen cross-presentation are under investigation. We have tested previously the use of B7-costimulation for this purpose in NSCLC with promising results [1] . One efficient pathway for tumor antigen cross presentation to and activation of DC, first described by Srivastava’s group, is via protein/peptide-chaperones [2-8] . In this first in human study, we here describe the use of genetically modified allogeneic tumor cells secreting protein/ peptide-chaperone gp96-Ig as tumor vaccine for the treatment of advanced non-small cell lung cancer.
Protein chaperones are ubiquitous and essential components in all cells preventing protein aggregation during de novo protein synthesis and folding and during protein degradation. When released from dying cells, chaperones are danger associate molecular patterns (DAMPs) that activate and are taken up by DC and potently cross present chaperone-associated client peptides via MHC I to antigen specific CD8 T cells. The biological function of endoplasmic reticulum (ER) chaperone gp96, also known as heat shock protein gp96, is unique in that it has the dual function of chaperoning 1) newly synthesized proteins and 2) proteasome generated peptides translocated into the ER via the transporter for antigen presentation (TAP). The latter are subsequently selected for MHC class I loading and presentation to CD8+ T cells [9] . In its function as DAMP, cell released gp96 functions as adjuvant for activating dendritic cells (DC) and macrophages [10] by binding to toll-like receptor (TLR) 2 and TLR4. By binding to CD91and triggering endocytosis, gp96 also imports its chaperoned client peptides into DC [11] where they are cross-presented by MHC I. Antigenic peptides mediate antigen specific CD8 T cell activation, clonal expansion and generation of antigen specific, MHC I restricted, CD8+ CTL. Importantly, all antigenic epitopes present in the gp96-associated clientpeptide pool released from a dying cell are cross presented and generate a polyepitope specific CD8 CTL response. Therefore gp96 derived from tumor cells generates CD8 CTL against all tumor associated antigens without the requirement of knowing the nature of the antigen. Antigen cross presentation by gp96 is fully active across MHC barriers allowing the use of allogeneic tumor cells as source of tumor antigen [9,12] . Self antigens chaperoned by gp96 are not antigenic since they are normally and continuously presented by MHC I and the peripheral TCR repertoire has been shaped in the thymus to delete self reactive CD8 T cells.
Srivastava and his collaborators were the first to recognize that the MHC I antigen cross presenting activity of gp96 had excellent potential for the use as cancer vaccine to generate antigen specific CD8 CTL in cancer patients. A vaccine trial in melanoma patients vaccinated with gp96 purified from autologous melanoma specimens showed safety and no statistical difference to the control group treated with dacarbazine, temozolomide, interleukin-2, or complete resection [13] .
Based on the published results cited, we reasoned that tumor cells secreting gp96 will be immunogenic and will be rejected by CD8 CTL. We generated the fusion protein gp96-Ig by deleting the C-terminal ER-retention sequence of gp96 and replacing it with the hinge, CH2 and CH3 domain of the heavy chain of IgG1 to generate gp96-Ig. Transfecting the gp96-Ig cDNA into murine tumor cells, we demonstrated that gp96-Ig secreting tumor cells upon transplantation into mice are rejected by CD8 CTL responses that generate protective memory whereas parental vector-transfected tumor cells expand and kill the recipient [14] . Figure 1 illustrates the vaccine principle in cartoon.
Gp96-Ig mediated antigen cross presentation and priming of CD8+ CTL is extraordinary sensitive and powerful. Femto-molar (10−15) amounts of gp96-Ig-chaperoned peptides are sufficient to induce a full antigen specific CD8 CTL response in mice in vivo [15,16] . Multiple antigens expressed by the gp96-Ig secreting cells will activate corresponding antigen specific CD8+ CTL responses as shown for multiple simian immunodeficiency virus antigens (SIV) [17] .
Can allogeneic cells be used as source of tumor antigens for gp96-Ig vaccination? Are tumor antigens, here NSCLC specific tumor antigens, shared between patients with different MHC? It is well documented that melanoma associated antigens are identical regardless of the MHC type of the patient [18-20] . Although non-small cell lung cancer (NSCLC) specific tumor associated antigens have not been identified, we considered it likely that NSCLC associated antigens are also shared between NSCLC patients with different MHC types. This hypothesis is supported by numerous gene expression analyses that have been conducted to find prognostic markers and that show similar patterns of gene up-regulation and down-regulation [21] . In support, we found antigen sharing in allogeneic small cell lung cancer (SCLC) in an analysis of over 40 HLA A1 or A2 restricted CD8 CTL clones that killed two allogeneic HLA A1 or HLA A2 positive SCLC lines [22] .
Tumors induce immune suppression which interferes with CTL responses. We have found in preclinical mod-

Figure 1. Principle of antigen-cross presentation via MHC I of DC to clonally expand antigen specific CD8 CTL by secretion of gp96-lg peptide complexes by allogeneic NSCLC vaccine cells AD100. The allogeneic irradiated cell line AD100 injected as vaccine secretes gp96-lg which chaperones tumor associated antigenic peptides (TAA) and self peptides. Gp96-lg activates DC via TLR2, TLR4 and CD91, and is endocytosed. Chaperoned peptides are cross presented by MHC I and cross prime TAA-specific CD8 CTL. DC also secrete IL-12 and recruit NK cells creating a Th1 environment.
els that frequent vaccination with gp96-Ig secreting tumor cells can overcome immune suppression induced by established tumors and restore CD8 CTL activation [23,24] . The method of frequent (up to twice weekly) vaccination is evaluated in patients for the first time in this study.
The preclinical data in mice and macaques provide a solid basis for considering the gp96-Ig technology for therapeutic vaccination of cancer patients. We selected non-small cell lung cancer (NSCLC) as our primary target because NSCLC, unlike melanoma, is non-immunogenic. Generation of a CD8 CTL response to NSCLC associated antigens therefore will find the tumor unfit to cope with CTL attack rendering NSCLC potentially susceptible to rejection by vaccine immunotherapy.
The annual mortality of non-small cell lung cancer (NSCLC) in the United States exceeds 156,940 patients and an estimated 221,130 new cases of lung cancer are expected in 2011 according to the American Cancer Society [25]. Results of treatment with chemotherapy for NSCLC are far from optimal and 5-year survival only increased from 14.2% to 18.0% from 1975 to 2005 (NCI, SEER Statistics Review 1975-2006). Older (>5 years) Phase III trials have typically demonstrated responses to therapy in less than 30% of patients, with median survival less than one year [26-28] . New drugs have been reported for NSCLC patients, but these regimens still result in complete responses in <10% of patients with little effect on survival [29-33] . There is a great need of other therapeutic options for our NSCLC patients and active immunotherapy is one of them, however there is no vaccine approved for NSCLC.
In this phase I study we evaluate the secreted gp96-Ig vaccine strategy for NSCLC. The NSCLC cell line AD100 was transfected with HLA A1 and human gp96- Ig and after irradiation used to vaccinate advanced NSCLC patients of any HLA type. Following results from preclinical studies [23] we increased the frequency of vaccination from biweekly to weekly and twice weekly for a total of 9, 18 or 36 intracutaneous vaccinations. The data show safety and a trend toward clinical benefit associated with increased IFN-γ screting CD8+ CTL levels.
2. PATIENTS, MATERIAL AND METHODS
2.1. Patient Selection
Inclusion criteria were: patients with histologically confirmed NSCLC stage IIIB, stage IV, or recurrent disease; at least one site of bi-dimensionally measurable disease; treated brain metastasis must be stable by CT scan or MRI for at least 8 weeks; patients must have received and failed at least two lines of therapy; age ≥ 18 years; ECOG performance status 0-2; life expectancy ≥ 3 months; signed informed consent. Laboratory parameters included: hemoglobin levels ≥ 10 g/dl; absolute neutrophil count ≥ 1500; platelets ≥ 100 k; creatinine clearance ≥ 50 ml/min; and <2.5× upper institution limit for normal, liver function tests (total and direct bilirubin, aspartate transaminase, alanine transaminase, and alkaline phosphatase). Exclusion criteria: active or symptomatic cardiac disease; pregnant or lactating women; known HIV infection; uncontrolled brain or spinal cord metastases; active infections; concomitant steroid or other immunosuppressive therapy; other active malignancies present within the past three years, except for basal and/ or squamous cell carcinoma(s) or in situ cervical cancer; meningeal carcinomatosis; current chemotherapy or radiation therapy, or other anti-tumor therapy during the last four weeks; immune deficiency syndromes including the following: rheumatoid arthritis, systemic lupus erythematousus, Sjogren’s disease, sarcoidosis, vasculitis, polymyositis, and glomerulonephritis. Also patients needed to have acceptable lung function defined as: FEV1 > 30% of the predicted value, or DLCO > 30% of the predicted value, or pCO2˚ < 45˚ mmHg.
2.2. Investigational Agent: Allogeneic NSCLC/AD100-gp96-Ig Vaccine
The use of allogeneic NSCLC tumor cells as source of tumor antigens chaperoned by secreted gp96-Ig and cross presented via MHC I to patient CD8 T cells is based on the hypothesis that allogeneic NSCLC tumors from different patients have a repertoire of identical tumor antigens similar to allogeneic melanomas and allogeneic small cell lung cancers [22,34,35] . Recent gene array analyses of NSCLC tumors are consistent with this hypothesis [36] . We stably transfected an established NSCLC adenocarcinoma cell line (AD100) with a plasmid encoding HLA-A1 and gp96-Ig and generated the AD100-A1-gp96-Ig live cell-based vaccine that continuously secretes gp96-Ig as measured by ELISA. Secreted gp96-Ig is expected to activate the innate immune response and stimulate patient dendritic cells (DC) to cross present gp96-Ig chaperoned, tumor associated antigens to CD8 T cells and generate polyepitope specific CD8 CTL to NSCLC in treated patients. The AD100- gp96-Ig vaccine cells were expanded under cGMP conditions, tested for gp96-Ig secretion as immunologically active vaccine agent, for HLA A1 expression for cell identity, for sterility and freedom of adventitious agents, irradiated at 12,000 rad, frozen in aliquots with 10% DMSO and stored at −135˚C until use.
2.3. Treatment Plan
Based on the results in the B7 vaccination study in which the CD8 T cell immune response was not affected by partial HLA matching of vaccine and patients ADDIN EN.CITE ADDIN EN.CITE.DATA [1] , the AD100-A1-gp96-Ig vaccine was given to patients irrespective of their HLA type. If treatment was completed a total maximum of 4.5 ´ 108 AD100-A1-gp96-Ig vaccine cells were administered intradermally in three 6-week courses over an 18-week period in 9, 18, 36 doses (Table 1). Intradermal injections on succeeding vaccination days were rotated to different limbs in a clockwise manner. On each visit clinical evaluation of the status of disease and adverse events was conducted.
Blood samples for immunological evaluation were obtained before the initial vaccination and on the last day of each course. Patients with either objective responses or stable disease (SD) and an acceptable toxicity profile (autoimmune < grade 2, and grade ≤ 3 for other body systems) were treated with a second and third course.
2.4. Trial Design
The trial was designed as a single institution Phase I, proof of principle study with safety as the primary endpoint and secondary objectives tumor response and survival. After accrual of 7 patients to DS-1 (9 vaccinations every other week) between July 2007 and October 2008, a protocol amendment took effect adding patient cohorts to receive 18 weekly (DS-2) or 36 twice weekly (DS-3) vaccinations. Assignment to DS-1, DS-2 and DS-3 then proceeded in an alternating manner with a planned total of 12 patients per cohort. The study was not powered for statistical comparison of DS cohorts but 12 patients per cohort was expected to provide the basis for recommending at least one vaccination schedule for further testing. Although the study was funded by foundation support and monitored by an independent data and safety monitoring committee, enrollment proceeded slowly due to institutional conflict of interest concerns and several audits that did however not reveal any safety concern. Despite an appropriate conflict of interest management plan and apparent safety, the study unfortunately had to be closed for completely study-unrelated institutional reasons in April 2011 at which time 11 patients had been enrolled and treated in DS-1, 4 in DS-2 and 3 in DS-3.
2.5. Adverse Events (AE)
All AEs were classified by type and severity according
(CTCAE v 3.0). No dose modification was made for any vaccine-related AEs if encountered. Vaccination was to the Common Terminology Criteria for Adverse Events discontinued for any patient experiencing a grade 2 or higher autoimmune AE or grade 3 or higher non-autoimmune AE that was possibly vaccine-related.
2.6. Clinical Response
Clinical response and progression of disease were evaluated according to criteria proposed by the Response Evaluation Criteria in Solid Tumors (RECIST) Committee. The study was approved by FDA (IND10940) and OBA, by the University of Miami IRB and IBC, and monitored by an external Safety Monitoring Committee operated by the DSM Services Division of the Western Institutional Review Board (WIRB) as part of the conflict of interest management plan. The clinical principal investigator had no financial conflict.
2.7. Immune Response
Since tumor associated tumor antigens have not been identified in NSCLC, tumor antigen specific CD8 CTL frequencies and activity cannot be determined. As surrogate assay we used the allo-antigen specific CD8-IFN response to vaccination as indicator of CD8 reactivity. We have documented previously the potency of gp96-Ig induced antigen specific cross-priming of polyepitope specific CD8 T cells in murine models and in non-human primates in the presence of allogeneic responses [15- 17,37] . Therefore we expect that patients immunized with AD100-A1-gp96-Ig will also generate NSCLCassociated antigen-specific CD8 CTL if allo-reactive IFN-γ screting CD8 cells are generated in vaccinated patients.
Patient CD8 T cells were purified from PBMC by negative selection. 2 × 104 CD8 T cells were restimulated in vitro in triplicate with 104 irradiated vaccine cells for 40 hours in ELIspot plates for IFN-γ-detection. Stimulators for specificity controls were K562 and melanoma tumor cells. In addition unstimulated CD8 cells served as negative controls. Data are reported as number IFN-secreting cells per 2 × 104 CD8 T cells and patients were characterized as immune responders (IR+) if they had more than a twofold increase from baseline.
Table 1. Dose-schedules (frequency of vaccinations).
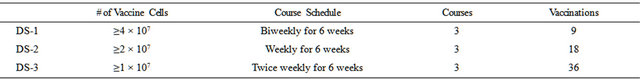
Three cohorts of 12 patients each were therapeutically vaccinated in 3 courses of 6 weeks. The total number of vaccine cells injected was the same in all three cohorts, if treatment was completed. The frequency of vaccination increased from injections every 2nd week in cohort 1 to weekly injections in cohort 2 and twice weekly injections in cohort 3.
2.8. Statistical Considerations
Patient characteristics, treatment received, and immune parameters (plotted in relation to treatment course)
were summarized with descriptive statistics. Progression-free (PFS) and overall survival (OS) were estimated by the Kaplan Meier method with 95% confidence intervals (CIs) based on the log-log transform method and Greenwood’s variance [38]. PFS was defined as the elapsed time from treatment start to documented evidence of disease progression or death from any cause, whichever was earlier, or to date of last progression-free assessment (censored observations). OS was the elapsed time from treatment start to death from any cause or to last contact (censored observations). Exploratory analyses included examination of prognostic factors for PFS and OS using the log rank test and Cox regression as well as plots and descriptive statistics to assess clinical outcome in relation to DS cohort and immune response (IR).
3. RESULTS
3.1. Expectations for Phase I Studies of Vaccine Immunotherapy of Cancer
The immune system performs immune surveillance to detect transformed cells and eliminates them in an immune response that requires Perforin-1 and IFN-γ before they form cancers [39,40] . The fact that cancers do arise indicates that the tumor has evaded immune surveillance. Once tumors are established the immunological balance is completely changed. There is now an excess of tumor associated antigens that can anergize antigen specific T cells and the tumor often mediates active immune suppression favoring tumor survival. Therapeutic vaccine immunotherapy is applied in this setting of tumor induced immune suppression and antigen excess, raising the following questions: 1) Is it possible and what is the best method and vaccination schedule to generate tumor specific CD8 T cell IFN-responses in heavily pretreated patients in the presence of established immune-suppressive cancers? 2) Are CD8 T cell IFN-γ responses to vaccination-therapy a good surrogate to assess vaccine responsiveness and predict improved clinical outcome?
In this phase I study we can only provide early data on safety, on overall survival and on the induction of CD8- IFN-γ responses. Premature closure of the study precludes statements about vaccine scheduling due to limited enrollment in DS-2 and DS-3. Although we observe CD8 T cell IFN-γ responses to therapeutic vaccination in a group of patients who also have longer survival than patients without a CD8 IFN-γ response, we cannot determine in a phase I study whether these two observations are causally related and whether CD8 responses are good surrogates for clinical response.
3.2. Clinical Response
Nineteen patients were consented. One patient was never treated due to early disease progression and clinical deterioration. Eleven patients were treated in DS-1, four in DS-2, and three in DS-3. Two patients, both in DS-3, completed the planned three-course treatment as they achieved and maintained disease stabilization through three courses of vaccination. Two patients in DS-1 and 3 in DS-2 completed two courses and eight patients (all in DS-1) completed one course. The remaining three patients (one in each cohort) progressed or died during the first treatment course. The median age of 18 evaluable patients was 67 years (range 38 - 86); the majority were female (11 patients). Most of the patients were White non-Hispanic (11 patients) and a third were White Hispanic (6 patients). Demographic characteristics for study patients are summarized in Table 2.
There were no treatment-related serious adverse events (SAE) or immune related events (IRE) with the vaccine or the vaccinations. Most of the AEs were grade 1 or 2. They included: erythema and skin induration that were transitory and usually resolved in 1 - 2 weeks. A summary of the most common treatment-related AEs is given in Table 3.
There were no complete or partial clinical responses. Seven of 18 patients (39%; 95% CI: 17.3% - 64.3%) achieved disease stabilization after the first course of vaccinetions (6 weeks) and eleven patients had disease progression. The seven patients with initial disease stabilization continued to a second (5 patients) and third (2 patients) course of vaccination and all but one of them exhibited an immune response. Five of these patients died at a median time of 16.5 months (range 6.7 to 20.0) and two were alive with 12.2 and 21.0 months follow up. Overall a total of 15 patients have died and three surviving patients have been followed for 12.2, 21.0, and 38.8 months. The Kaplan-Meier estimate of median survival was 8.1 months (95% CI: 6.7% - 18.2%), and the 1-, 2-, and 3-year rates were 44.4% (95% CI: 21.6% - 65.1%), 19.0% (95% CI: 4.8% - 40.3%), and 9.5% (95% CI: 0.8% - 32.1%), respectively). Overall survival is shown in Figure 2. Exploratory analysis of the prognostic effect of baseline factors found no associations with progression free survival but indicated longer OS for patients of Hispanic ethnicity (p = 0.020) as well as for those who were fully active per ECOG assessment (p = 0.043).
3.3. Dose Schedule
Although the study was not powered for comparison by DS, we observed longer progression-free times for most of the patients vaccinated weekly or twice weekly than for those vaccinated every other week (Figure 3). An effect of vaccination frequency on OS is not evident
Table 2. Baseline characteristics of 18 evaluable patients.

Distribution of age, gender, ethnicity, frequency of prior treatments, performance status, tumor-histology and disease stage among enrolled and treated patients.
Table 3. Most common treatment-related AEs.

Adverse events, type, frequency and grade related to vaccine-treatment.

Figure 2. Overall survival of all 18 patients with advanced NSCLC.
but immune response positive (IR+) patients lived longer most of the patients vaccinated weekly or twice weekly than IR− patients (Figure 3).
3.4. Immune Response
We were able to measure CD8 IFN-responses to in vitro restimulation with vaccine cells in 15 patients at baseline and following at least one course of vaccination; for 6 patients data were also available after the second treatment course and 2 patients were assessed after 3 courses. Controls were stimulated with melanoma cells or with K562 or left unstimulated. As summarized in Table 4 and illustrated in Figure 4, CD8 responses were highly variable and the distribution was skewed to the right. A more than twofold increase in the frequency of
Table 4. CD8 CTL Response by IFN-g ELI-Spot.
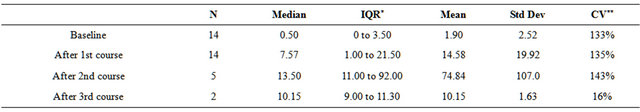
*IQR: Inner quartile range, 25th to 75th percentile; **CV: coefficient of variation = standard deviation as a percent of mean; Table 4. Patient CD8 T cells purified by negative selection from PBMC were restimulated in vitro with vaccine cells and IFN-g secretion determined in ELI-spot plates. Controls were unstimulated or stimulated with unrelated tumor cells as described in material and methods.
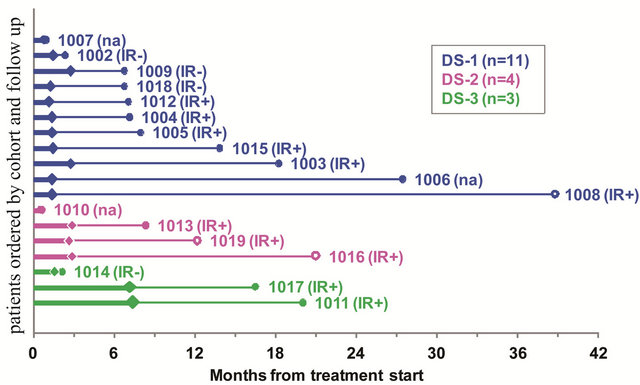
Figure 3. Time to progression (thick line) and additional follow up (thin line) by dose-schedule cohort. Patients are shown within cohort in order of increasing follow up (shortest at top). Filled diamonds indicate disease progression; open diamonds indicate stable disease at last assessment. Filled circles indicate death; open circles last follow up of surviving patients. IR+: more than twofold increase in CD8 from baseline. IR−: no CD8 immune response. na: not assessed for immune response.
IFN-γ secreting CD8 cells during all courses of vaccination was observed in 11 patients giving an overall immune response rate of 73.3% (95% CI: 44.9% to 92.2%), In spite of the absence of an objective tumor response the 11 patients who developed a CD8 CTL response during the first, second or third course of vaccination had an estimated median survival of 16.5 (95% CI: 7.1% - 20.0%) months (see Figure 3, patients marked IR+). In contrast, 4 patients (1002, 1009, 1014, 1018 in Figure 4) had minimal change or decreased frequency of IFN-γ secreting CD8 cells after 6 weeks of vaccination, indicating that their immune system was unable to mount an allogeneic CD8 response and probably also unable mediate CD8 responses to gp96TAAIg mediated antigen cross presentation; survival of these 4 patients was only 2.1, 2.3, 6.7 and 6.7 months (see Figure 3, marked IR−).
4. DISCUSSION
In this first in man vaccine trial injecting live, irradiated, allogeneic tumor cells secreting gp96-Ig we found the treatment safe with no vaccine related serious adverse events and only expected local adverse events at

Figure 4. CD8 IFN-γ response. Samples from 15 patients collected for immune response at base line and after at least one course of vaccination were available for analysis of the CD8 IFN-γ response. 20,000 purified patient CD8 T cells were stimulated with vaccine cells for 40h in ELI-spot plates and the frequency of IFN-γ secreting cells determined. + indicates first increase. Solid indicate immune response (IR+), dashed lines no response (IR−).
the injection site. Although the study is limited in lacking a control arm and in having been closed prematurely by the institution for reasons entirely unrelated to the study, it offers extremely interesting insights into the effects of therapeutic vaccine immunotherapy.
The mechanisms of immunotherapy are very different to those of chemotherapy. A single vaccination by antigen cross presentation will induce an antigen specific CD8 CTL response which, however, in the environment of tumor induced immune suppression and antigen excess will be very modest ADDIN EN.CITE ADDIN EN.CITE.DATA [23,24] and transient and undetectable within about one week [41,42] . Frequent vaccination before the original response is suppressed gradually builds up a more substantial response that in preclinical models has been able to completely reject tumors in a percentage of test subjects ADDIN EN.CITE ADDIN EN.CITE.DATA [23,24] . Building up this anti-tumor response in cancer patients may take 6 to 12 weeks with repeated vaccinations. Only when the CD8 CTL response, measured here as IFN-γ secretion, is sufficiently built up by vaccination, can it be expected to affect tumor growth. The anti tumor effect may be detected by slower and delayed tumor growth associated with prolonged survival, as is frequently seen in experimental animals ADDIN EN.CITE ADDIN EN.CITE.DATA [23,24] . Or tumors may be stabilized and arrested in growth or even be rejected. Unlike in chemotherapy, the therapeutic effects of anti tumor vaccine immunotherapy are expected to be delayed but may continue even after cessation of vaccination. Tumors may not completely stop to grow but may grow significantly slower resulting in longer survival. The determination of progression free survival (PFS) and of responses according to RECIST criteria therefore is of limited value for estimating anti tumor activity of vaccine immunotherapy.
We find in this study that none of the 18 patients had an objective tumor response by RECIST criteria, such as partial or complete remission. Nonetheless, 11 of the 18 lacking objective clinical response developed an immune response evidenced by increased frequency of vaccinespecific IFN-γ CD8 T cells. For these patients, the estimated median survival was 16.5 months (95% CI: 7.1% - 20.0%), and three of them were alive at last follow up of 12.2, 21.0 and 38.8 months. Patients who had no CD8 response had shorter survival times. While this select group of patients necessarily excludes those with early progression and death, and the small number of patients makes it difficult to assess the influence of baseline characteristics, their outcome suggests that further study of gp96 vaccination is warranted. We consider the data as suggestive evidence for beneficial effects of CD8 CTL responses in NSCLC patients.
The gp96-Ig secreting vaccine cells are allogeneic and are expected to generate allo-antigen specific responses concurrent with tumor-associated antigen-specific cross presentation by gp96-Ig and priming of MHC I restricted antigen specific CD8 CTL. Since we do not know the identity of putative NSCLC-associated antigens, we can only measure the allo-response and assume that cross presentation of tumor associated antigens will occur to a corresponding extent, similar to data in preclinical animal models in mice and macaques [16,17,24] . Preclinical studies have also shown that concurrent allogeneic responses do not interfere with or inhibit antigen cross presentation by gp96-Ig [9,16,17] . If no allo-CD8 response is observed, it is likely that the CD8 cells are also unable to respond to antigen cross presentation, probably as consequence to prior treatment regimens and due to overwhelming tumor induced immune suppression.
Due to the unfortunate, premature institutional closure of the trial, which was completely unrelated to this study, we could not complete enrollment in the two arms with increased frequency of vaccination of 18 times weekly or 36 times twice weekly. Nonetheless, 4 of the 5 immune response-evaluable patients in these two groups had increased CD8 responses and are included in the group of 11 patients with prolonged overall survival. The data are supportive for our preclinical findings that increased frequency of vaccination can surmount tumor induced suppression and increase CD8 CTL responses with clinical benefit.
Our method of preparing vaccines from established, allogeneic tumor cell lines by transfection with gp96-Ig, such as the NSCLC cell line AD100 in this study, provides a relatively simple and inexpensive way to conduct tumor-vaccine immunotherapy. Off the shelf allogeneic vaccines are of great advantage as therapeutic option. Vaccine cell-secreted gp96-Ig-tumor-peptide-chaperone complexes are danger signals for DC and generate patient autologous MHC I restricted CTL responses against tumor associated antigens. Normal self-peptides do not induce immune responses. Allogeneic cells have been shown to contain shared tumor antigens in melanoma and other tumors, thus it is reasonable to expect that NSCLC tumors share antigens. Indeed, recent microarray data indicate substantial identity of gene expression profiles in NSCLC tumors ADDIN EN.CITE ADDIN EN.CITE.DATA [36] .
In this single institution study of 18 patients, the tumor cell-based gp96-Ig secreting AD100-gp96-Ig vaccine was found to have an acceptable safety profile. Because there was a significant disease control rate in this heavily pretreated population, as well as a substantial CD8 CTL response in this Phase I trial, there is a strong basis for developing Phase II trials incorporating new combinations and strategies for a novel approach to the treatment of advanced NSCLC patients and to test the strategy in other tumors.
5. CONCLUSION
Vaccination of patients with a novel, first-in-man, allogeneic gp96-Ig transfected NSCLC-adenocarcinoma vaccine, in the setting of minimal toxicity and strong immunologic responses has proven to be a viable therapeutic intervention and will be important to explore further in this population of advanced NSCLC patients.
5.1. Authors’ Contributions
Conception and trial design: E. R. Podack, G. R. Walker, L. E. Raez. Development of vaccine and methodology: E. R. Podack. Acquisition of data: L. E. Raez, P. Baldie, E. Fisher, J. E. Gomez, K. Tolba, E. S. Santos, Analysis and Interpretation of data: L. E. Raez, G. R. Walker, E. Fisher, E. R. Podack. Writing and review of manuscript: G. R. Walker, L. E. Raez, E. R. Podack. Study supervision: E. R. Podack, L. E. Raez.
6. ACKNOWLEDGEMENTS
Supported by a grant from the Alliance of Cancer Gene Therapy (ACGT), New York and by NIH grant PO1 CA109094 to ERP; by the James & Esther King Award by Florida Biomedical Research Institute, Florida Department of Health to LER.
REFERENCES
- Raez, L.E., Cassileth, P.A., Schlesselman, J.J., Sridhar, K., Padmanabhan, S., Fisher, E.Z., Baldie, P.A., Podack and E.R. (2004) Allogeneic vaccination with a B7.1 HLA-A gene-modified adenocarcinoma cell line in patients with advanced non-small-cell lung cancer. Journal of Clinical Oncology, Official Journal of the American Society of Clinical Oncology, 22, 2800-2807. doi:10.1200/JCO.2004.10.197
- Srivastava, P.K., DeLeo, A.B. and Old, L.J. (1986) Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proceedings of the National Academy of Sciences of the United States of America, 83, 3407-3411. doi:10.1073/pnas.83.10.3407
- Udono, H., Levey, D.L. and Srivastava, P.K. (1994) Cellular requirements for tumor-specific immunity elicited by heat shock proteins: Tumor rejection antigen gp96 primes CD8+ T cells in vivo. Proceedings of the National Academy of Sciences of the United States of America, 91, 3077-3081. doi:10.1073/pnas.91.8.3077
- Udono, H. and Srivastava, P.K. (1993) Heat shock protein 70-associated peptides elicit specific cancer immunity. The Journal of Experimental Medicine, 178, 1391-1396. doi:10.1084/jem.178.4.1391
- Udono, H. and Srivastava, P.K. (1994) Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. Journal of Immunology, 152, 5398-5403.
- Binder, R.J. and Srivastava, P.K. (2005) Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nature Immunology, 6, 593-599. doi:10.1038/ni1201
- Yang, Y., Liu, B., Dai, J., Srivastava, P.K., Zammit, D.J., Lefrancois, L. and Li, Z. (2007) Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity, 26, 215-226. doi:10.1016/j.immuni.2006.12.005
- Li, Z. and Srivastava, P.K. (1993) Tumor rejection antigen grp96/ grp94 is an ATPase: Implications for protein folding and antigen presentation. The EMBO Journal, 12, 3143-3151.
- Arnold, D., Faath, S., Rammensee, H. and Schild, H. (1995) Cross-priming of minor histocompatibility antigen-specific cytotoxic T cells upon immunization with the heat shock protein gp96. The Journal of Experimental Medicine, 182, 885-889. doi:10.1084/jem.182.3.885
- Vabulas, R.M., Braedel, S., Hilf, N., Singh-Jasuja, H., Herter, S., Ahmad-Nejad, P., Kirschning, C.J., Da Costa, C., Rammensee, H.G., Wagner, H., et al. (2002) The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. The Journal of Biological Chemistry, 277, 20847- 20853. doi:10.1074/jbc.M200425200
- Binder, R.J., Han, D.K. and Srivastava, P.K. (2000) CD91: A receptor for heat shock protein gp96. Nature Immunology, 1, 151-155. doi:10.1038/77835
- Matzinger, P. and Bevan, M.J. (1977) Induction of H-2- restricted cytotoxic T cells: In vivo induction has the appearance of being unrestricted. Cellular Immunology, 33, 92-100. doi:10.1016/0008-8749(77)90137-X
- Testori, A., Richards, J., Whitman, E., Mann, G.B., Lutzky J, Camacho, L., Parmiani, G., Tosti, G., Kirkwood, J.M., Hoos, A., et al. (2008) Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma: The C-100-21 Study Group. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 26, 955-962. doi:10.1200/JCO.2007.11.9941
- Yamazaki, K., Nguyen, T. and Podack, E.R. (1999) Cutting edge: Tumor secreted heat shock-fusion protein elicits CD8 cells for rejection. Journal of Immunology, 163, 5178-5182.
- Oizumi, S., Strbo, N., Pahwa, S., Deyev, V. and Podack, E.R. (2007) Molecular and cellular requirements for enhanced antigen cross-presentation to CD8 cytotoxic T lymphocytes. Journal of Immunology, 179, 2310-2317.
- Strbo, N., Oizumi, S., Sotosek-Tokmadzic, V. and Podack, E.R. (2003) Perforin is required for innate and adaptive immunity induced by heat shock protein gp96. Immunity, 18, 381-390. doi:10.1016/S1074-7613(03)00056-6
- Strbo, N., Vaccari, M., Pahwa, S., Kolber, M.A., Fisher, E., Gonzalez, L., Doster, M.N., Hryniewicz, A., Felber, B.K., Pavlakis, G.N., et al. (2011) Gp96 SIV Ig immunization induces potent polyepitope specific, multifunctional memory responses in rectal and vaginal mucosa. Vaccine, 29, 2619-2625. doi:10.1016/j.vaccine.2011.01.044
- Sang, M., Wang, L., Ding, C., Zhou, X., Wang, B., Wang, L., Lian, Y. and Shan, B. (2011) Melanoma-associated antigen genes—An update. Cancer Letters, 302, 85-90. doi:10.1016/j.canlet.2010.10.021
- Gajewski, T.F. and Fallarino, F. (1995) Rational development of tumour antigen-specific immunization in melanoma. Therapeutic Immunology, 2, 211-225.
- Bystryn, J.C. (1995) Clinical activity of a polyvalent melanoma antigen vaccine. Recent Results in Cancer Research Fortschritte der Krebsforschung Progres dans les Recherches sur le Cancer, 139, 337-348. doi:10.1007/978-3-642-78771-3_26
- Glazer, C.A., Smith, I.M., Ochs, M.F., Begum, S., Westra, W., Chang, S.S., Sun, W., Bhan, S., Khan, Z., Ahrendt, S., et al. (2009) Integrative discovery of epigenetically derepressed cancer testis antigens in NSCLC. PloS One, 4, e8189. doi:10.1371/journal.pone.0008189
- Yamazaki, K., Spruill, G., Rhoderick, J., Spielman, J., Savaraj, N. and Podack, E.R. (1999) Small cell lung carcinomas express shared and private tumor antigens presented by HLA-A1 or HLA-A2. Cancer Research, 59, 4642-4650.
- Oizumi, S., Deyev, V., Yamazaki, K., Schreiber, T., Strbo, N., Rosenblatt, J. and Podack, E.R. (2008) Surmounting tumor-induced immune suppression by frequent vaccinetion or immunization in the absence of B cells. Journal of Immunotherapy, 31, 394-401. doi:10.1097/CJI.0b013e31816bc74d
- Schreiber, T.H., Deyev, V.V., Rosenblatt, J.D. and Podack, E.R. (2009) Tumor-induced suppression of CTL expansion and subjugation by gp96-Ig vaccination. Cancer Research, 69, 2026-2033. doi:10.1158/0008-5472.CAN-08-3706
- http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-029771.pdf
- Bonomi, P.D., Finkelstein, D.M., Ruckdeschel, J.C., Blum, R.H., Green, M.D., Mason, B., Hahn, R., Tormey, D.C., Harris, J., Comis, R. et al. (1989) Combination chemotherapy versus single agents followed by combination chemotherapy in stage IV non-small-cell lung cancer: A study of the Eastern Cooperative Oncology Group. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 7, 1602-1613.
- Schiller, J.H., Harrington, D., Belani, C.P., Langer, C., Sandler, A., Krook, J., Zhu, J. and Johnson, D.H. (2002) Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. The New England Journal of Medicine, 346, 92-98. doi:10.1056/NEJMoa011954
- Weick, J.K., Crowley, J., Natale, R.B., Hom, B.L., Rivkin, S., Coltman Jr., C.A., Taylor, S.A. and Livingston, R.B. (1991) A randomized trial of five cisplatin-containing treatments in patients with metastatic non-small-cell lung cancer: A Southwest Oncology Group study. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 9, 1157-1162.
- Miller, V.A.O.C.P., Soh, C. and Kabbinavar, F. A. (2009) randomized, double-blind, placebo-controlled, phase IIIb trial (ATLAS) comparing bevacizumab (B) therapy with or without erlotinib (E) after completion of chemotherapy with B for first-line treatment of locally advanced, recurrent, or metastatic non-small cell lung cancer (NSCLC). Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 27, LBA8002.
- Reck, M., von Pawel, J., Zatloukal, P., Ramlau, R., Gorbounova, V., Hirsh, V., Leighl, N., Mezger, J., Archer, V., Moore, N., et al. (2009) Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as firstline therapy for nonsquamous non-small-cell lung cancer: AVAil. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 27, 1227- 1234. doi:10.1200/JCO.2007.14.5466
- Sandler, A., Gray, R., Perry, M.C., Brahmer, J., Schiller, J.H., Dowlati, A., Lilenbaum, R. and Johnson, D.H. (2006) Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. The New England Journal of Medicine, 355, 2542-2550. doi:10.1056/NEJMoa061884
- Scagliotti, G., Novello, S., von Pawel, J., Reck, M., Pereira, J.R., Thomas, M., Abrao Miziara, J.E., Balint, B., De Marinis, F., Keller, A., et al. (2010) Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non-small-cell lung cancer. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 28, 1835-1842. doi:10.1200/JCO.2009.26.1321
- Treat, J.S.G., Peng, G., Monberg, M.J., Obasaju, C.K. and Socinski, M.A. (2011) Comparison of pemetrexed plus cisplatin with other first-line doublets in advanced nonsmall cell lung cancer (NSCLC): A combined analysis of three phase 3 trials. Lung Cancer. http://www.ncbi.nlm.nih.gov/pubmed/22115704.
- De Smet, C., Lurquin, C., De Plaen, E., Brasseur, F., Zarour H, De Backer, O., Coulie, P.G. and Boon, T. (1997) Genes coding for melanoma antigens recognised by cytolytic T lymphocytes. Eye (Lond), 11, 243-248. doi:10.1038/eye.1997.59
- Riley, J.P., Rosenberg, S.A. and Parkhurst, M.R. (2001) Identification of a New Shared HLA-A2.1 Restricted Epitope From the Melanoma Antigen Tyrosinase. Journal of Immunotherapy (1991), 24, 212-220.
- Singhal, S., Miller, D., Ramalingam, S. and Sun, S.Y. (2008) Gene expression profiling of non-small cell lung cancer. Lung Cancer, 60, 313-324. doi:10.1016/j.lungcan.2008.03.007
- Strbo, N., Pahwa, S., Kolber, M.A., Gonzalez, L., Fisher, E. and Podack, E.R. (2010) Cell-secreted Gp96-Ig-peptide complexes induce lamina propria and intraepithelial CD8+ cytotoxic T lymphocytes in the intestinal mucosa. Mucosal Immunology, 3, 182-192. doi:10.1038/mi.2009.127
- Collett, D. (2003) In Modeling Survival Data in Medical Research, 2nd Edition, CRC Press, New York.
- Schreiber, R.D., Old, L.J. and Smyth, M.J. (2011) Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science, 331, 1565-1570. doi:10.1126/science.1203486
- Koebel, C.M., Vermi, W., Swann, J.B., Zerafa, N., Rodig, S.J., Old, L.J., Smyth, M.J. and Schreiber, R.D. (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature, 450, 903-907. doi:10.1038/nature06309
- North, R.J. and Bursuker, I. (1984) T cell-mediated suppression of the concomitant antitumor immune response as an example of transplantation tolerance. Transplantation Proceedings, 16, 463-469.
- North, R.J. and DiGiacomo, A. (1986) Generation and Suppression of the Immune Resoponse to Immunogenic Tumors. In: Steinman, R.M. and North, R.J., Eds., Mechanisms of Host Resistance to Infectious Agents, Tumors, and Allografts, The Rockefeller University Press, New York, 387-396.
NOTES
*Disclosure of potential conflicts of interest: Dr. E. R. Podack and the University of Miami have financial interest and hold equity in a commercial enterprise developing this vaccine technology.