Crystal Structure Theory and Applications
Vol.2 No.4(2013), Article ID:40595,7 pages DOI:10.4236/csta.2013.24020
Synthesis, Characterization, Crystal Studies of (E)-3-(3-(4-Fluorophenyl)-1-isopropyl-1H-indol-2-yl) Acrylaldehyde
Department of Chemistry, Karnatak University, Dharwad, India
Email: *jseetharam@yahoo.com
Copyright © 2013 Veerendra Kumar A. Kalalbandi, J. Seetharamappa. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received August 30, 2013; revised September 27, 2013; accepted October 25, 2013
Keywords: Single Crystal Structure; X-Ray Diffraction; Intermediate of Fluvastatin; Indoles
ABSTRACT
We have synthesized and developed single crystals of the title compound (E)-3-(3-(4-fluorophenyl)-1-isopropyl-1Hindol-2-yl) acrylaldehyde, which is a key intermediate of anti-cholesterol fluvastatin drug. It crystallized under orthorhombic system with a space group Pna21. The dihedral angle between the indole mean plane and 4-F-phenyl ring was observed to be 111.5 (3)˚ in the title molecule. Further, strong hydrogen bonds were not found in the crystal structure.
1. Introduction
Indole and its derivatives have been a topic of research interest and continue to be one of the dynamic areas of heterocyclic chemistry, particularly due to their natural occurrence and pharmacological activities [1]. A large number of indole derivatives are at the fore as pharmacologically active lead compounds for drug development [2]. Many drugs contain indole moiety, either as a basic prototype or as an attached group to invoke particular properties [3]. The incorporation of indole nucleus, a biologically accepted pharmacophore in medicinal compounds, has made it versatile heterocyclic possessing wide spectrum of biological activities [4,5] like anticancer, antimicrobial, anti-HIV, antitubercular, antiviral, antidepressant, cardiovascular activity, antihypertensive, etc., [6-9]. Furthermore, fluvastatin, which is a synthetic member of the statin class, contains indole moiety in its molecular makeup and the title molecule is its key intermediate.
In the course of synthesizing some fluvastatin derivatives, we have developed single crystals of key intermediate, (E)-3-(3-(4-fluorophenyl)-1-isopropyl-1H-indol-2- yl) acrylaldehyde and its molecular structure were determined. In the present paper, we report the synthesis, characterization by FTIR, 1H NMR and 13C NMR and its crystal properties.
2. Experimental
2.1. Synthesis of the Title Compound
The title compound (E)-3-(3-(4-fluorophenyl)-1-isopropyl-1H-indol-2-yl) acrylaldehyde was synthesized by following the reported method [10,11]. Friedel-Crafts condensation of fluorobenzene with chloroacetyl chloride in the presence of AlCl3 gave 4-Fluoro phenacyl chloride. Phenacyl chloride was then condensed with N-isopropylaniline in the presence of DMF to yield 1-(4-fluorophenyl)-2-(isopropyl(phenyl)amino) ethanone. Further, this was cyclized by means of ZnCl2 to yield 3-(4- fluorophenyl)-1-isopropyl-1H-indole. The condensation of 3-(4-fluorophenyl)-1-isopropyl-1H-indole with 3-(Nmethyl-N-phenylamino) acrolein in the presence of POCl3 in acetonitrile yielded the title compound. The synthetic route for the synthesis of title compound is portrayed in Scheme 1. The title compound was characterized by FT-IR, 1H NMR and 13C NMR spectra. Further, pale yellow colored crystals suitable for X-ray diffraction analysis were grown by slow evaporation of a dilute solution of title compound in chloroform (m. p.136˚C - 138˚C) at room temperature.
2.2. Physical and Spectral Measurements
Melting point was determined in open capillary tubes on
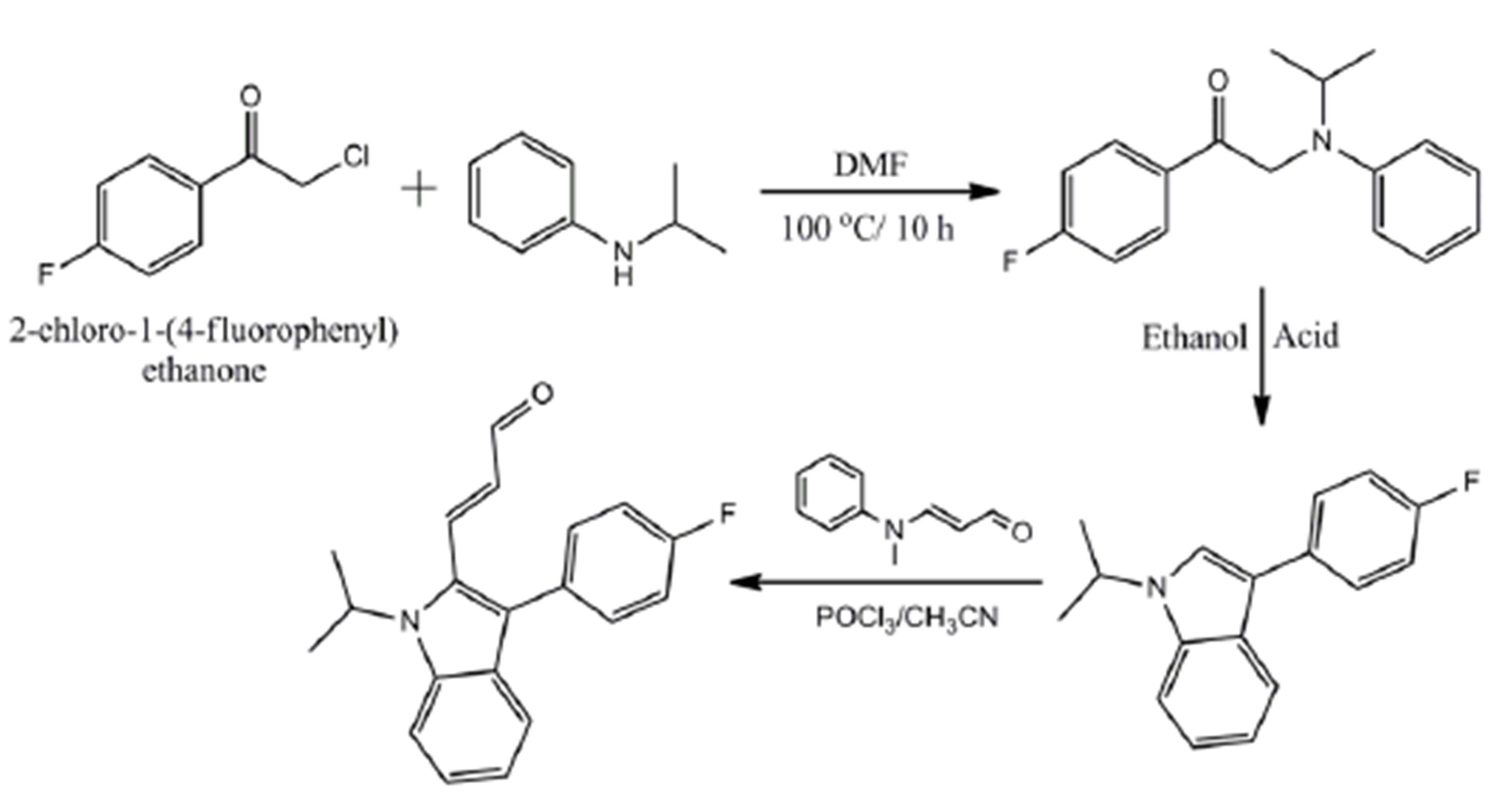
Scheme 1. Synthetic route for the synthesis of title compound.
a melting point apparatus of Concord Instruments (P) Ltd., Bangalore and is uncorrected. The IR absorption spectrum was recorded on a Shimadzu FT-IR-8400S Spectrophotometer using KBr pellets and is reported as wave numbers (ν cm−1). The 1H NMR spectrum was determined on a Bruker AV400 II at 400 MHz. The 13C NMR spectrum was recorded on Bruker DSX-300(S) AV-III 400(L) model at 100 MHz. All materials were purchased from commercial companies and are used directly. The solvents were dried by refluxing with appropriate drying agents and distilled before use.
2.3. Single-Crystal X-Ray Crystallography
Pale yellow colored crystals of the size of 0.30 × 0.20 × 0.20 mm was selected for data collection under a polarizing microscope and it was mounted on glass fiber for X-ray diffraction data collection. The high resolution Xray diffraction data sets were collected on a Bruker SMART APEX2 CCD Diffractometer using Mo Kα radiation (λ = 0.71073 Å) at ambient temperature. The crystal-to-detector distance was fixed at 40 mm. The diffraction data have been scaled for absorption effect by the multi-scanning method. The total exposure time was 0.98 h. The frames were integrated with the Bruker SAINT Software package using a narrow-frame algorithm. The structure was solved and refined using the Bruker SHELXTL [12] Software Package, using the space group Pna21, with Z = 4 for the formula unit, C20H18FNO. The ORTEP, packing and planes diagrams are generated using the Mercury 3.1.
3. Results and Discussion
3.1. Crystal Structure
The integration of the crystal data of the title compound (E)-3-(3-(4-fluorophenyl)-1-isopropyl-1H-indol-2-yl) acrylaldehyde using an orthorhombic unit cell yielded a total of 5558 reflections to a maximum θ angle of 23.92˚ (0.88 Å resolution), of which 2408 were independent (average redundancy 2.308, completeness = 99.5%, Rint =
2.25%, Rsig = 2.93%) and 2108 (87.54%) were greater than 2σ(F2). The final cell constants of a = 12.4637(4) Å, b = 9.9386(3) Å, c = 13.0272(3) Å, volume = 1613.70(8) Å3, are based upon the refinement of the XYZ-centroids of 1826 reflections above 20 σ(I) with 6.10˚ < 2θ < 47.84˚. Data were corrected for absorption effects using the multi-scan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.978. The crystal data and structure refinement parameters are given in Table 1.
The final anisotropic full-matrix least-squares refinement on F2 with 210 variables converged at R1 = 4.26%, for the observed data and wR2 = 8.48% for all data. The goodness-of-fit was 1.028. The largest peak in the final

Table 1. Crystal data and structure refinement.
difference electron density synthesis was 0.151 e. Å−3 and the largest hole was −0.105 e. Å−3 with an RMS deviation of 0.026 e. Å−3. On the basis of the final model, the calculated density was 1.265 mg/m3 and F (000), 648 e−.
3.2. Molecular Skeleton
The structure of the compound consisted of butenal and fluorobenzene fragments that connected to N-isopropylindole moiety. The ORTEP diagram of the title compound is given in Figure 1. The 4-F-phenyl ring C4-C3 form dihedral angle with the mean plane of the nine membered indole ring system was 111.5 (3)˚ (Figure 2). The sum of the bond angles around N1 [359.9 (3)˚] indicated sp2 hybridization [13]. The packing diagram (Figure 3) of the title molecule showed 4 molecules pack in a unit cell and no strong hydrogen bonds in the crystal structure were observed. However, weak inter molecular hydrogen bonds observed between C11H11···O1 by a distance of 2.692 Å are shown in Figure 4. All bond lengths and bond angles are in normal ranges and are given in Table 2. Atomic coordinates and equivalent isotropic displacement parameters are shown in Table 3. Anisotropic dis-placement parameters are shown in Table 4. Hydrogen coordinates and isotropic displacement parameters are shown in Table 5. Torsion angles are shown in Table 6.
3.3. Spectral Studies
3.3.1. FTIR Spectrum
The FT-IR spectrum of the title compound shows the strong characteristic absorption band at 1666.38 cm−1 due to aldehyde carbonyl stretching frequency. The shift in the band to lower wavenumber was due to the presence of α, β unsaturation. The band at 1610.45 cm−1 was observed due to C=C stretching.
3.3.2. 1H NMR Spectrum
The 1H NMR spectrum of the title compound showed two singlets at δ 1.64 and 1.66 ppm due to two methyl protons. The doublet at δ 9.61 ppm was attributed to aldehyde proton; it coupled with adjacent C8-CH protons with coupling constant of 7.6 Hz and the doublet at δ 7.95 ppm was accounted for vinylic proton with J value of 16 Hz. Further, C7 proton splits C8 proton signal into doublet (J = 16 Hz) which is split by C9 aldehyde proton into new doublets (J = 7.6 Hz) producing four-line spectrum (doublet of doublet) at δ 6.115 ppm. The signals appeared around δ 7.70 - 7.78 ppm were attributed to aromatic protons. 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.649 (s, 3H, C19-CH3), 1.666 (s, 3H, C20-CH3), 5.107 (m, 1H, C18-CH), 6.115 (dd, 1H, J = 16, 7.6 Hz, C8-CH), 7.959 (d, 1H, J = 16Hz, C7-CH), 9.619 (d, 1H, J = 7.6Hz, C9-CHO), 7.709 - 7.782 (m, 8H, Ar-H,).
3.3.3. 13C NMR Spectrum
The 13C NMR of the title compound exhibited a signal at δ 194.02 ppm for carbonyl carbon of the aldehyde group. The low intensity signal at δ 162.62 ppm was assigned to C1 carbon attached fluorine atom and the methyl carbons were observed at δ 21.37 ppm. The signal at δ 47.39 ppm was attributed to CH carbon of the isopropyl group. The peaks appeared between δ 112.60 - 160.20 ppm were assigned to aromatic and vinylic carbon atoms. 13C NMR (CDCl3, 100 MHz, δ ppm): 21.37 CH3, 47.39 C18-CH, 162.62 C1-CF, 194.02 C9-CHO, 112, 115, 119, 120, 124, 127, 130, 132, 136, 141, 160 Ar-C.
4. Conclusion
The title compound, a key starting material for the syn-
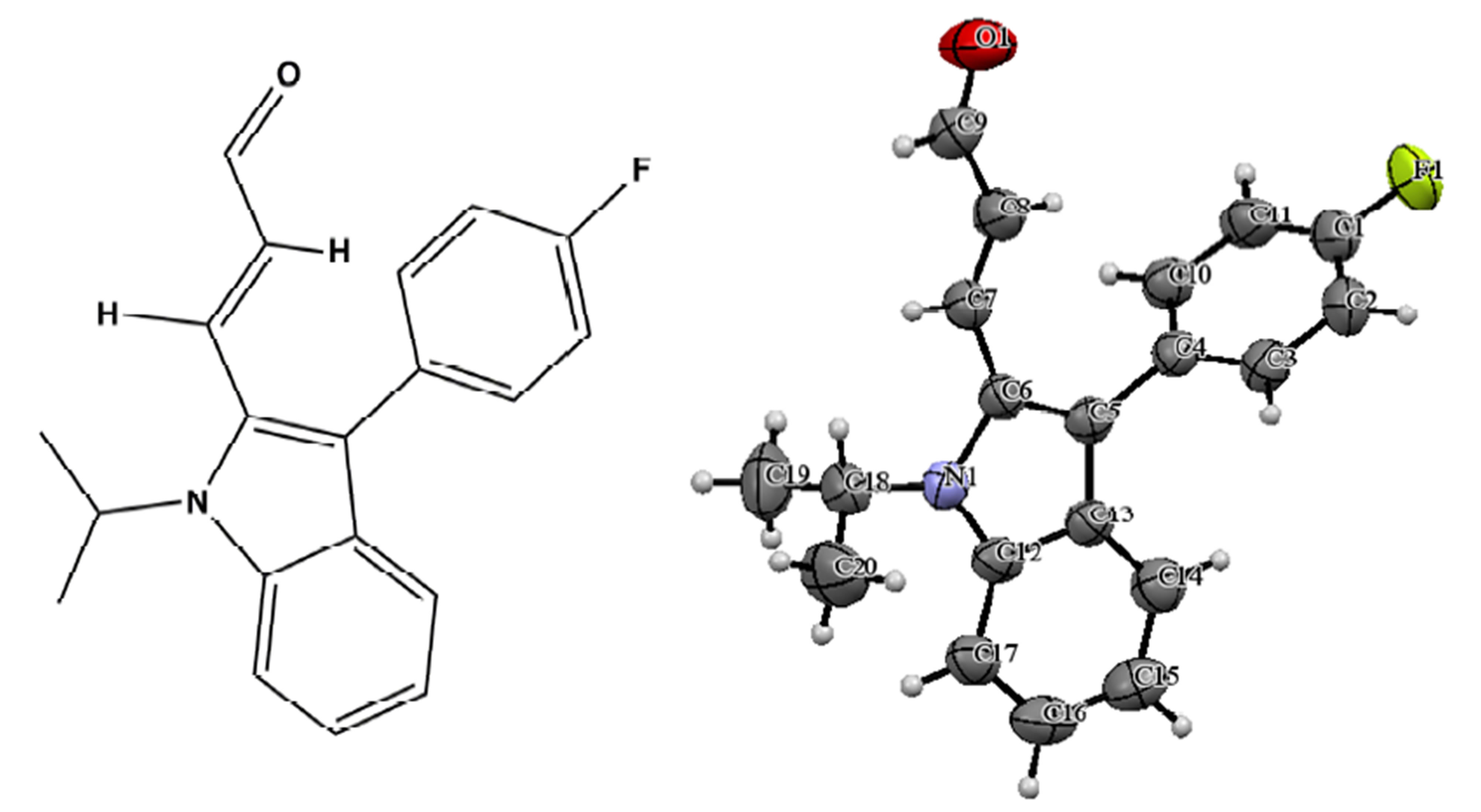
Figure 1. ORTEP diagram of the title molecule.
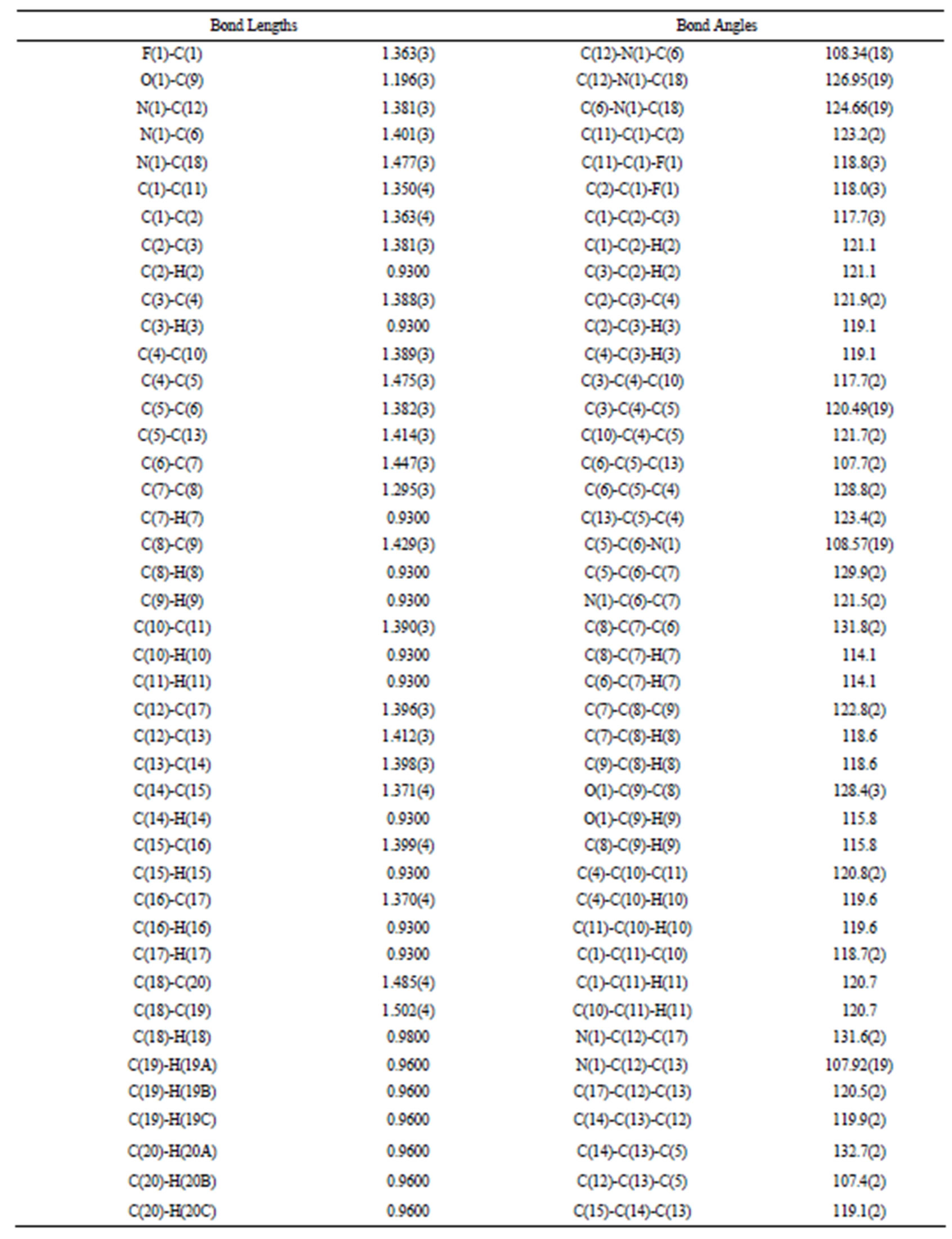
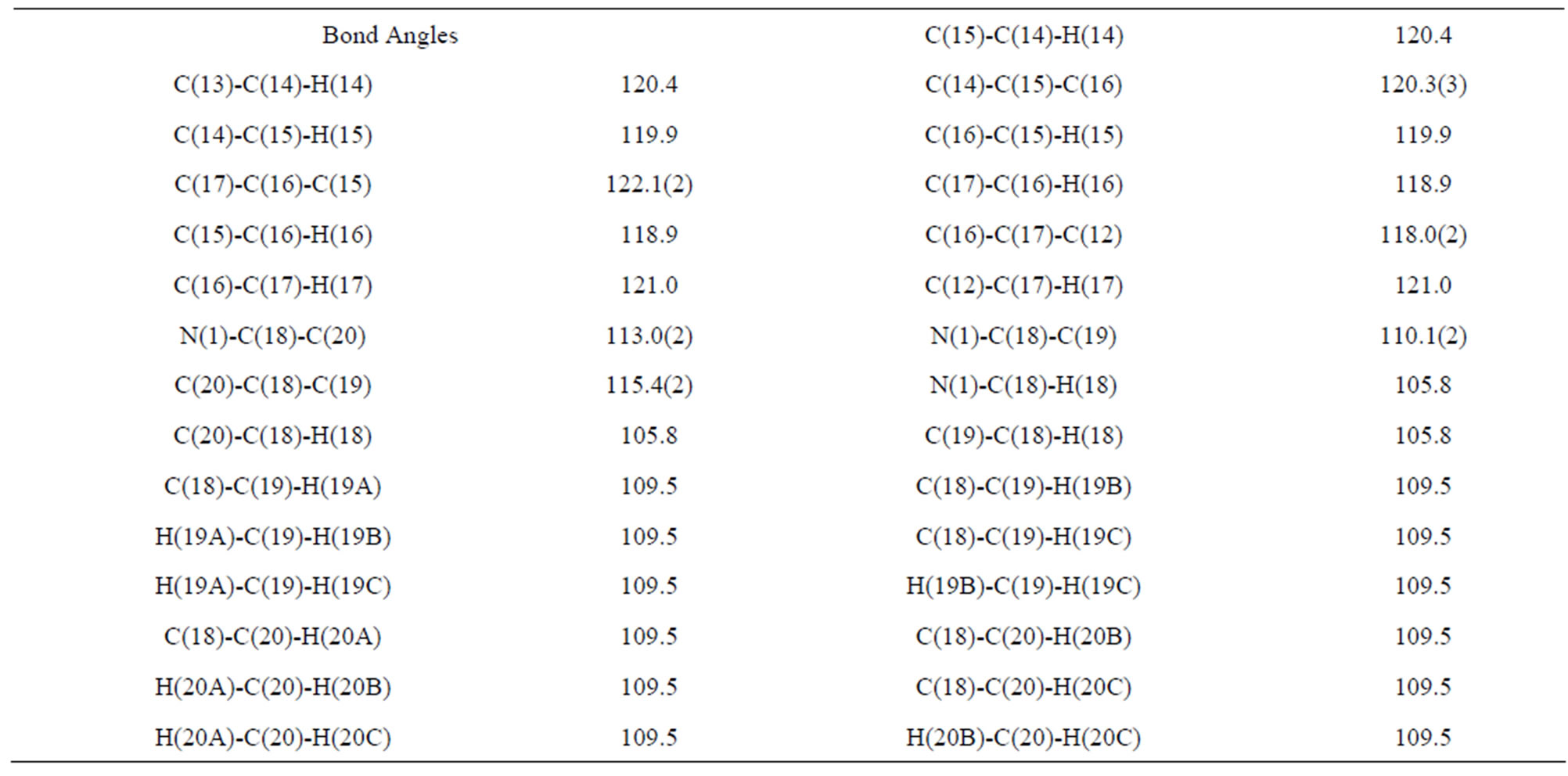
Table 2. Bond lengths [Å] and angles [˚].
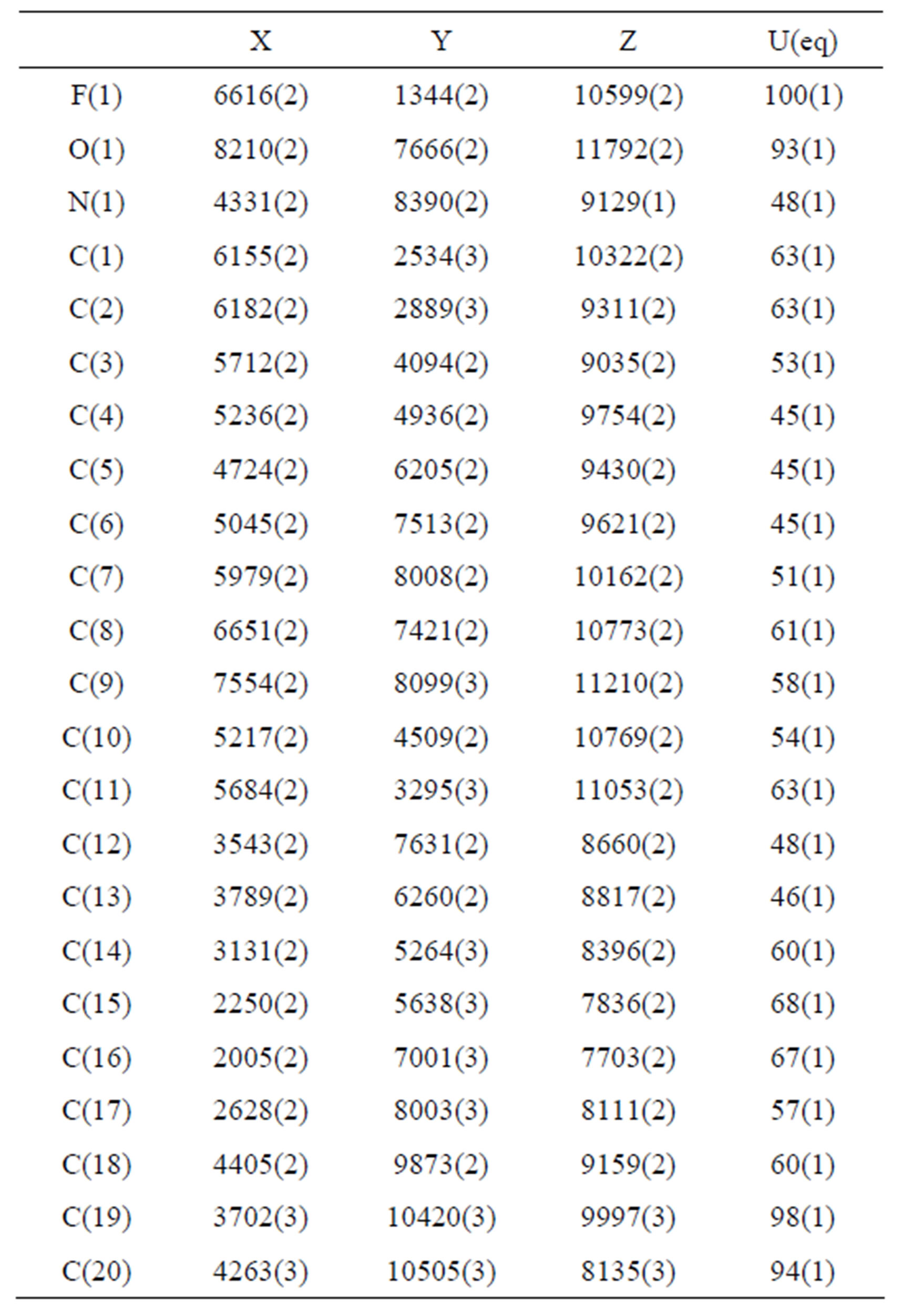
Table 3. Atomic coordinates (×104) and equivalent isotropic displacement parameters (Å2 × 103). U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.

Table 4. Anisotropic displacement parameters (Å × 103). The anisotropic displacement factor exponent takes the form: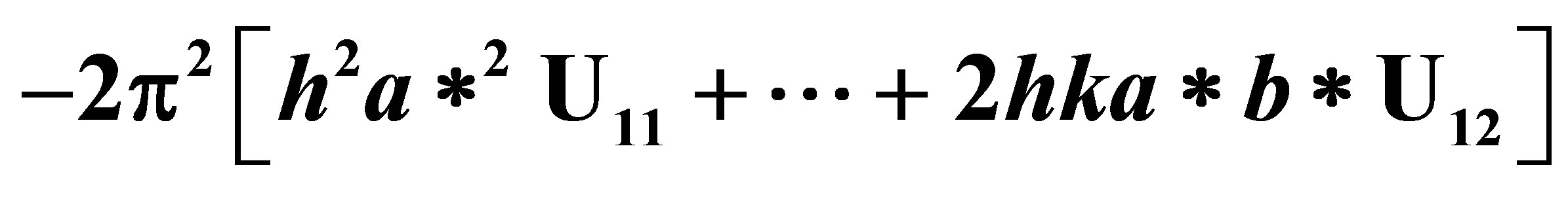 .
.
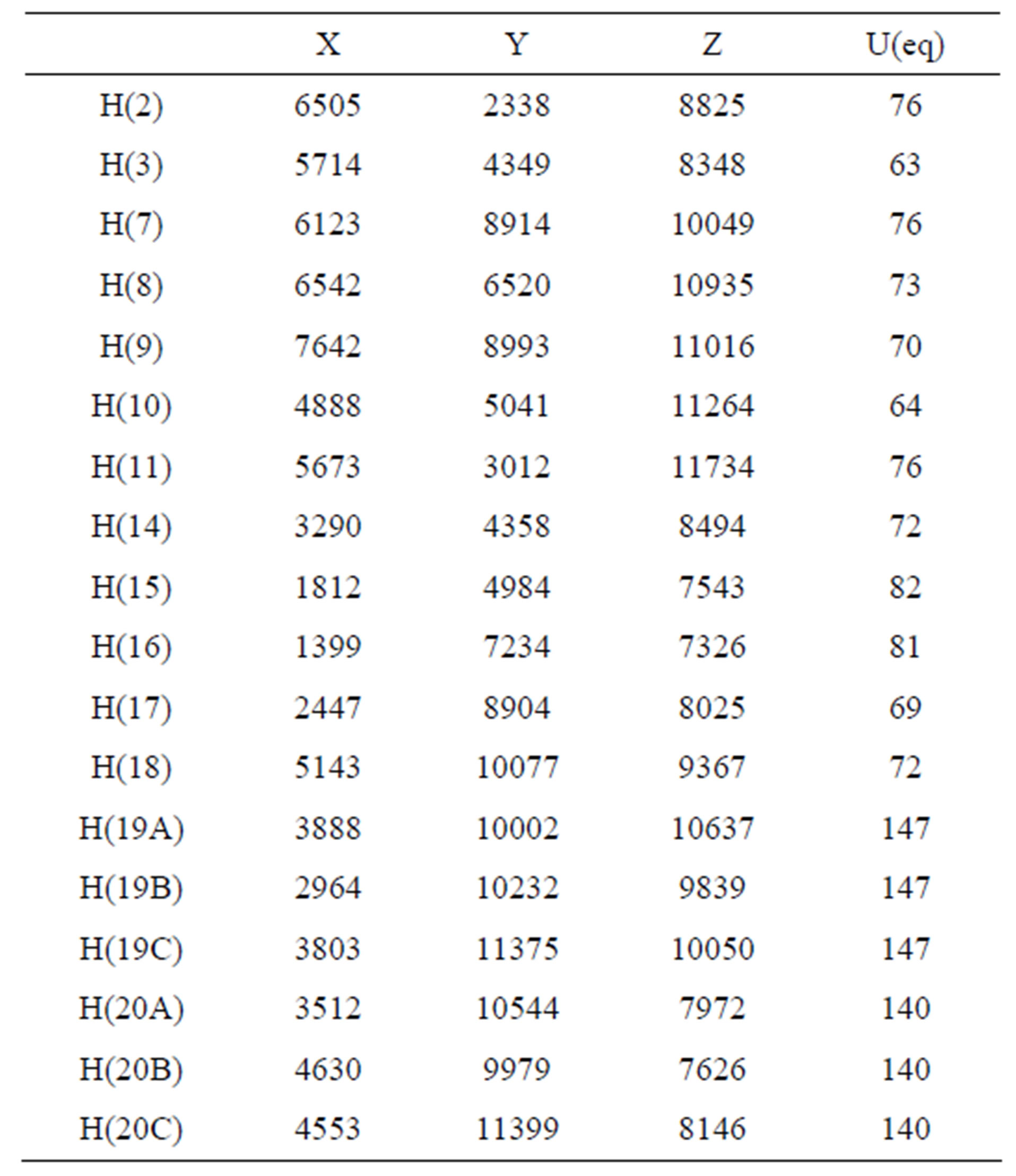
Table 5. Hydrogen coordinates (×104) and isotropic displacement parameters (Å2 × 103).
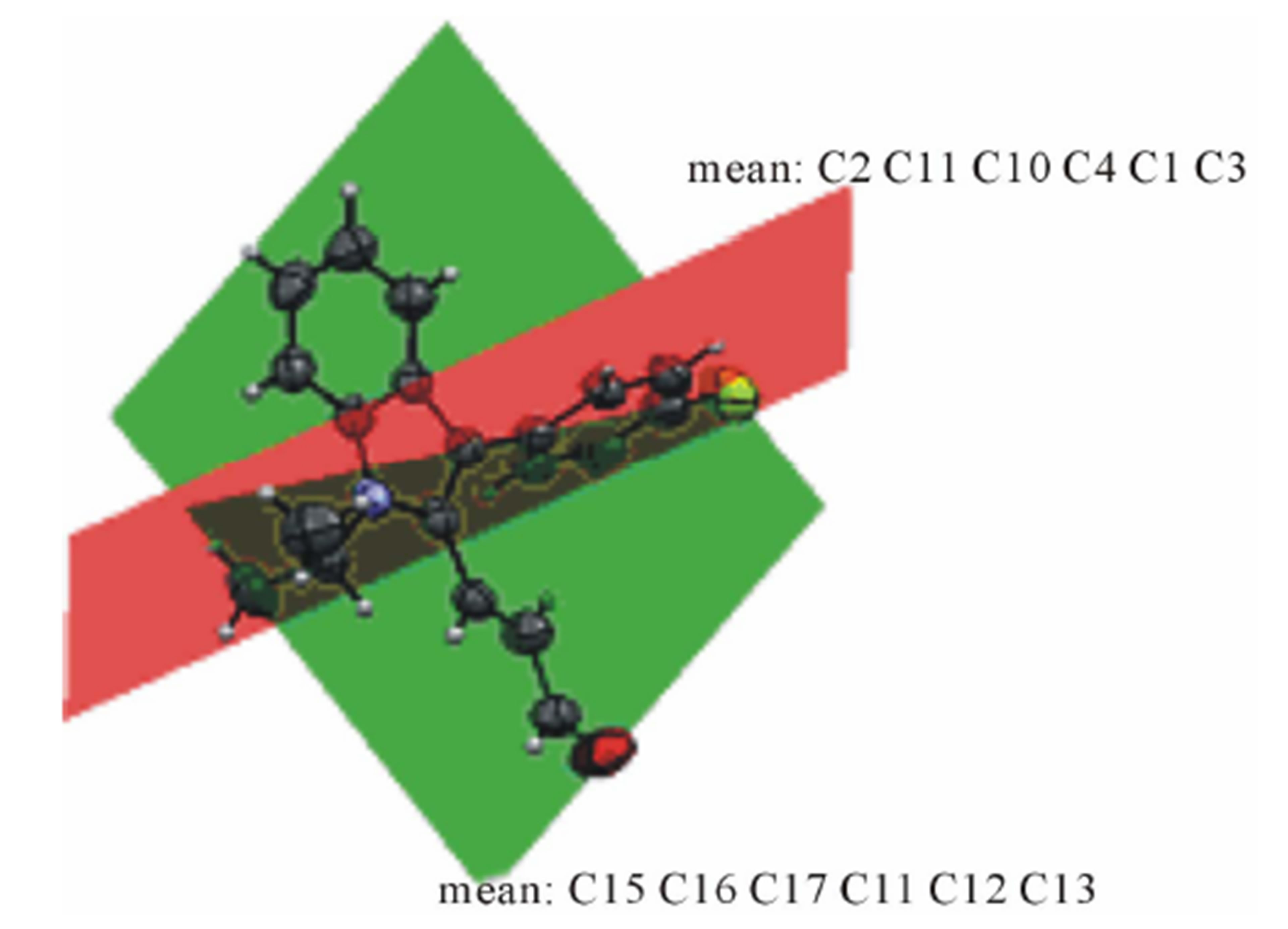
Figure 2. Dihedral angle formed between indole plane and benzene plane.
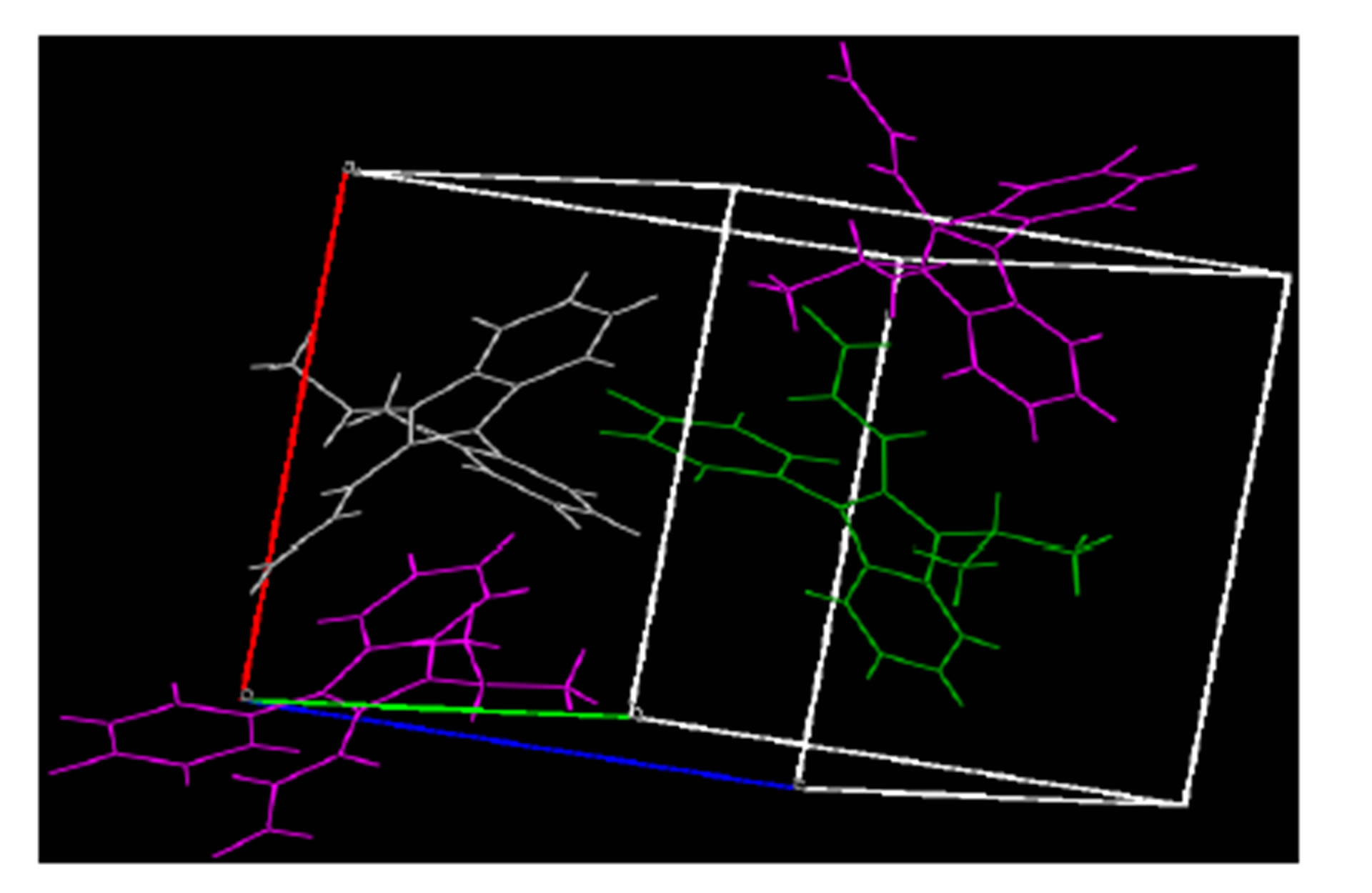
Figure 3. Packing diagram of title compound, ab Plane, colored by symmetry operation.
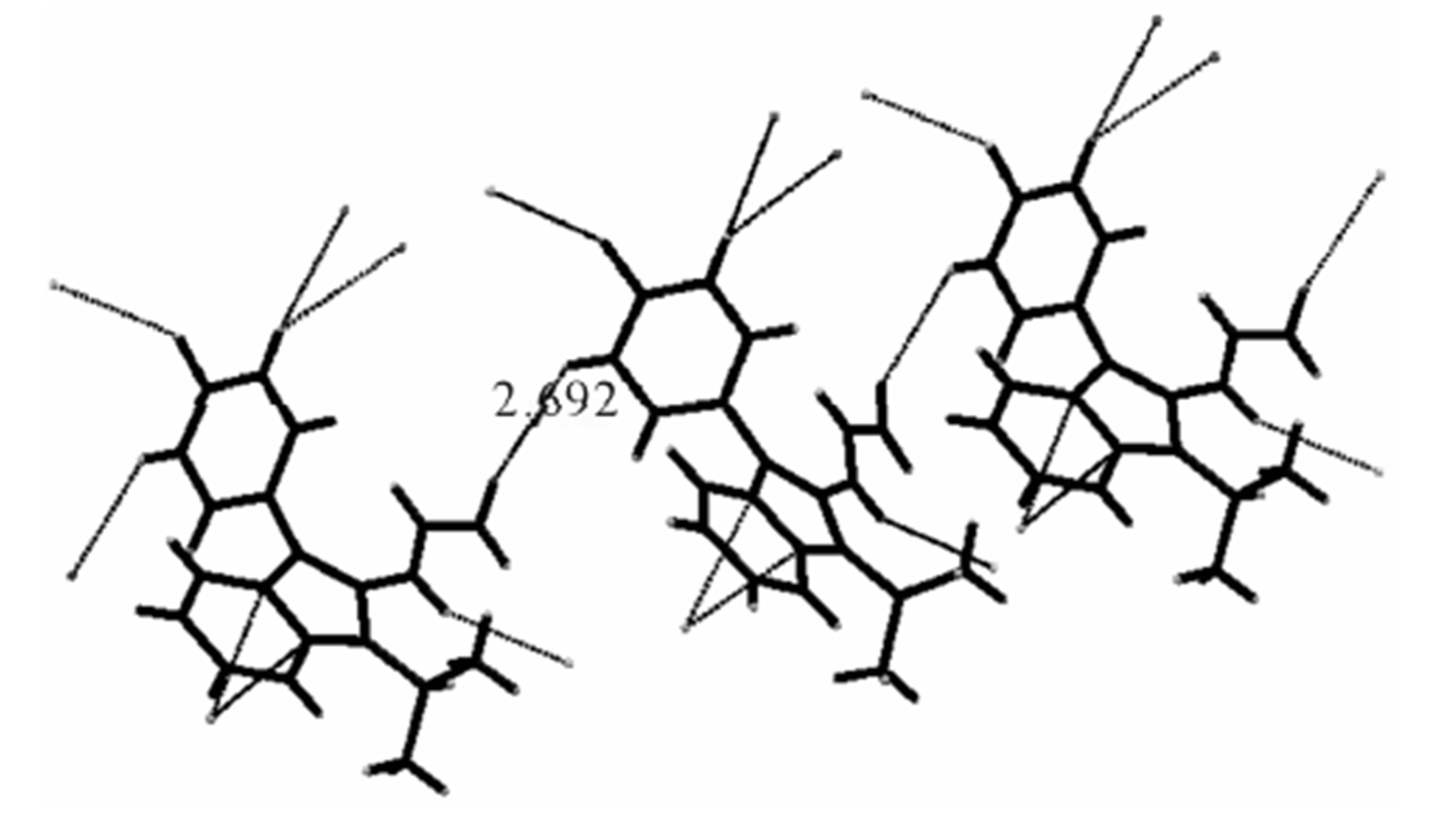
Figure 4. Diagram showing inter molecular weak Hydrogen bonding.
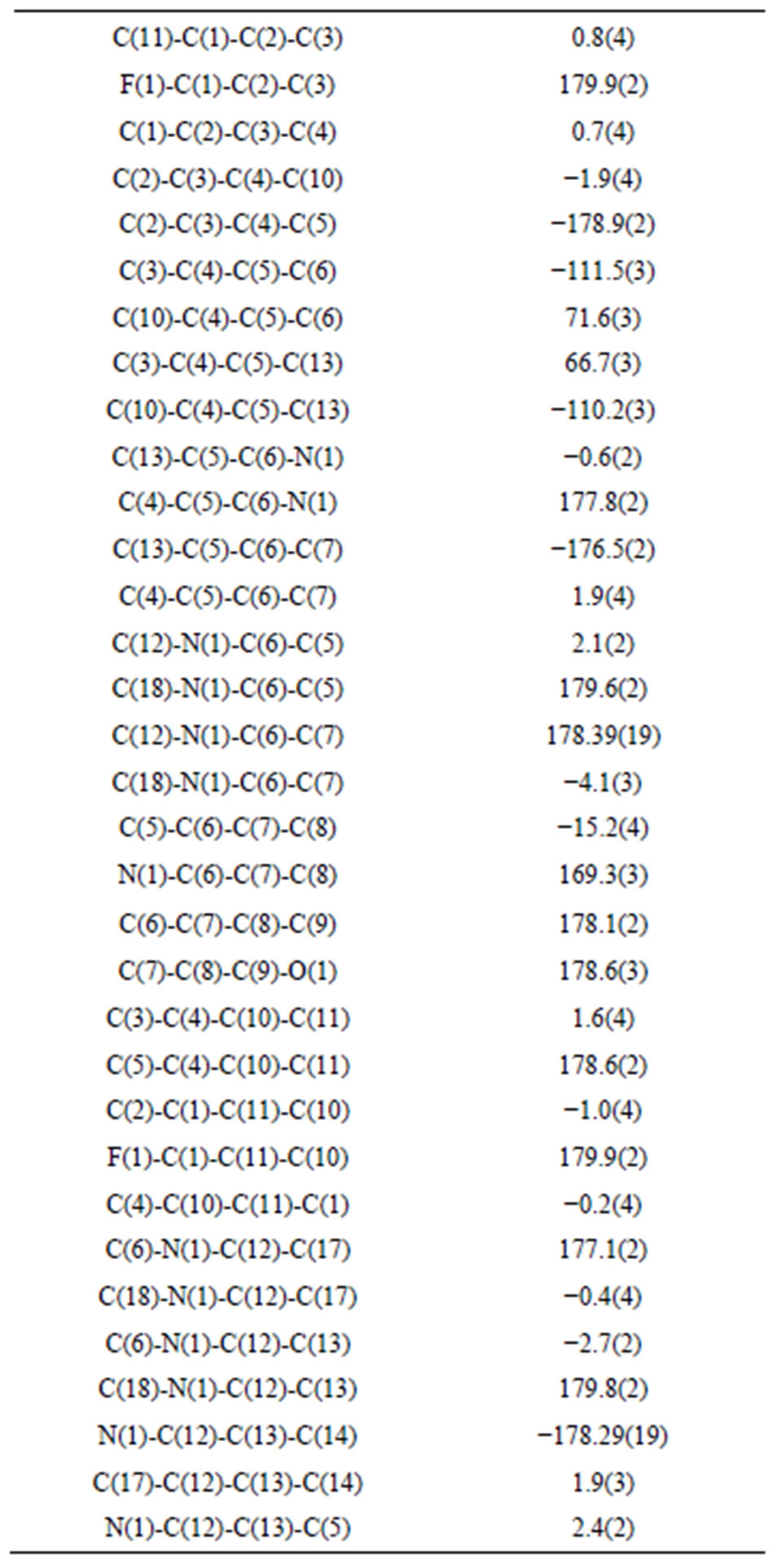

Table 6. Torsion angles [˚].
thesis of fluvastatin was synthesized, its spectral data were discussed and single crystals were grown. The crystal parameters were evaluated. Bond lengths and bond angles are in normal ranges. No classical hydrogen bonding was observed. The 4-F-phenyl ring C4-C3 form dihedral angle with the mean plane of the nine membered indole ring system was 111.5 (3)˚.
5. Acknowledgements
The authors acknowledge the University Scientific Instrumentation Centre (USIC), Karnatak University, Dharwad for SC-XRD data and NMR Research Centre, Indian Institute of Science (IISc), Bengaluru, India for carrying out the spectral analyses.
REFERENCES
- R. J. Sundberg, “Indoles,” Academic Press, New York, 1996.
- F. R. de sa alves, E. Z. Barreiro and C. A. M. Fraga, “From Nature to Drug Discovery: The Indole Scaffold as a Privileged Structure,” Mini-Reviews in Medicinal Chemistry, Vol. 9, No. 7, 2009, pp. 782-793. http://dx.doi.org/10.2174/138955709788452649
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, “Biomedical Importance of Indoles,” Molecules, Vol. 18, No. 6, 2013, pp. 6620-6662. http://dx.doi.org/10.3390/molecules18066620
- D. A. Horton, G. T. Bourne and M. L. Smythe, “The Combinatorial Synthesis of Bicyclic Privileged Structures or Privileged Structures,” Chemical Reviews, Vol. 103, No. 3, 2003, pp. 893-930. http://dx.doi.org/10.1021/cr020033s
- D. M. Wang, M. N. Sun and G. Liu, “Substituent Diversity-Directed Synthesis of Indole Derivatives,” Journal of Combonatorial Chemistry, Vol. 11, No. 4, pp. 556-575. http://dx.doi.org/10.1021/cc800198p
- V. Sharma, P. Kumar and D. Pathak, “Biological Importance of the Indole Nucleus in Recent Years: A Comprehensive Review,” Journal of Heterocyclic Chemistry, Vol. 47, No. 3, 2010, pp. 491-502. http://dx.doi.org/10.1002/jhet.349
- S. Rossiter, L. K. Folkes and P. Wardman, “Halogenated Indole-3-Acetic Acid as Oxidatively Activated Prodrugs with Potential for Targeted Cancer Therapy,” Bioorganic and Medicinal Chemistry Letters, Vol. 12, No. 2002, pp. 2523-2526. http://dx.doi.org/10.1016/S0960-894X(02)00505-X
- E. Abele, R. Abele, O. Dzenitis and E. Lukevics, “Indole and Isatin Oximes: Synthesis, Reactions and Biological Activity,” Chemistry of Heterocyclic Compounds, Vol. 39, No. 1, 2003, pp. 3-35. http://dx.doi.org/10.1023/A:1023008422464
- H. Panwar, R. S. Verma, V. K. Srivastava and A. Kumar, “Synthesis of Some Substituted Azetidinonyl and Thiazolidinonyl-1,3,4-thiadiazino[6,5-b]indoles as Prospective Antimicrobial Agents,” Indian Journal of Chemistry, Vol. 45B, No. 9, 2006, pp. 2099-2104.
- R. E. Walkup and J. Linder, “2-Formylation of 3-Arylindoles,” Tetrahedron Letters, Vol. 26, No. l8, 1985, pp. 2155-2158. http://dx.doi.org/10.1016/S0040-4039(00)98949-4
- G. T. Lee, J. C. Amedio Jr., R. Underwood, K. Prasad and O. Repic, “Vinylformylation Utilizing Propeniminium Salts,” Journal of Organic Chemistry, Vol. 57, No. 11, 1992, pp. 3250-3252. http://dx.doi.org/10.1021/jo00037a055
- G. M. Sheldrick, “A Short History of SHELX,” Acta Crystallographica Section A, Vol. A64, 2008, pp. 112- 122. http://dx.doi.org/10.1107/S0108767307043930
- R. Senthamizhselvi, G. Bhaskar, P. R. Sheshadri, P. T. Perumal and K. Illangovan, “3-(1,2-Di-p-tolylvinyl)-2- methyl-1H-indole,” Acta Crystallographica Section E, Vol. E68, 2012, p. o1815. http://dx.doi.org/10.1107/S1600536812021617
NOTES
*Corresponding author.