Modern Research in Catalysis
Vol.3 No.3(2014), Article ID:47850,21 pages
DOI:10.4236/mrc.2014.33010
The Nature of the True Catalyst in Transfer Hydrogenation with Alcohol Donors Using (arene)2Ru2Cl4(II)/TsDPEN Precursor
Judith Toubiana, Liraz Medina, Yoel Sasson*
Casali Center of Applied Chemistry, Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, Israel
Email: *ysasson@huji.ac.il
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 3 May 2014; revised 3 June 2014; accepted 15 June 2014
ABSTRACT
The widespread precatalyst (prepared in-situ or ex-situ) (arene) RuTsDPEN advocated for highly effectual asymmetric transfer hydrogenation (ATH) reactions with 2-propanol as hydrogen donor at ambient conditions, is proven to be unstable under the strong reducing conditions prevailing in the reaction mixtures (blend of alcohol and a base such as KOH). We assert that the true catalysts are the ruthenium metal nanoclusters formed swiftly under the reducing conditions of these systems. The TsDPEN ligand plays a critical role in the generation and formatting of the active catalyst including wreaking chiral properties to the so formed catalytic nanoparticles. Kinetic measurements, NMR, UV-visible spectroscopy, circular dichroism (CD) and TEM analyses corroborate this argument.
Keywords:Catalytic transfer Hydrogenation (CTH), Asymmetric Transfer Hydrogenation (ATH), Ruthenium Nanoparticles, Ruthenium Complexes, Nanocatalyst, Chiral Nanoparticles

1. Introduction
Homogeneous catalytic transfer hydrogenation (CTH), predominantly of C = O bonds, has developed through the last decades into a viable synthetic protocol which in numerous instances may replace standard hydrogenation techniques [1] -[3] . The nature and the performance of the catalysts (mostly homogeneous) used in CTH have improved dramatically since the original report, back in 1964, by Henbest and coworkers [4] . State of the art thenium (II) NNN complexes, recently disclosed by Yu et al. [5] , transforms ketones into alcohols using 2-propanol as a hydrogen donor, with astounding TOF of 720,000 h−1 at 820 and 55,800 h−1 at 280 achieving >99% yields within minutes. With chiral ligands, these catalysts also exhibit high enantioselectivity with ee of up to 99% [6] [7] .
A key milestone in the development of the contemporary transfer hydrogenation catalysis has been the contribution by Noyori and Ikariya et al. who in 1995 [8] [9] introduced the diamino ligand TsDPEN (along with amino alcohols) that when combined with a Ru(II)-arene precursor, generated in situ a catalyst that was proven active in the highly efficient asymmetric transfer hydrogenation (ATH) of ketones at room temperature (Scheme 1) [10] -[12] .
This process and its equivalent, where formic acid is used as hydrogen donor, [10] were implemented in numerous synthetic procedures and were widely successfully applied on a commercial scale [13] -[17] .
The contribution of Noyori et al. was not confined merely to the discovery and exploration of this particular catalytic system. He also authorized the archetype of CTH mechanisms with two novel paradigms in homogeneous catalysis by metal complexes. These are: a) The concept of “Outer Sphere” mechanism where the substrates do not directly coordinate to the metal center but act in response to interactions with the ligands only [18] and b) the idea of “Metal-Ligand bifunctional catalysis” where one of the ligands (such as primary or secondary amine) functions as a basic site that interact with the donor molecule via hydrogen bonding thus facilitating a proton transfer between the donor and the acceptor [19] -[21] . This novel mechanism was also proposed for the direct catalytic hydrogenation of ketones under hydrogen pressure [22] . Several experimental and theoretical studies supported the concerted hydrogen transfer process via the above mechanism [23] [24] . The concept of metal-ligand bifunctional catalysis was further developed by Handgraaf [25] and by Baratta [26] also for the alternative “inner sphere” mechanism where a metal alkoxide complex is a key intermediate.
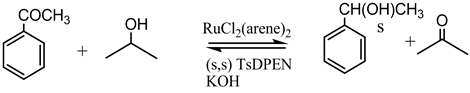
Noyori’s outer sphere bifunctional mechanism was corroborated experimentally through isolation and identification of the presumed intermediates [27] . Starting with dichlororuthenium (p-cymene) (II) (complex 4) and TsDPEN as an auxiliary ligand, the following three complexes (1 - 3) were synthesized and fully characterized (Figure 1).
The 18e complex 1 was considered as the catalyst precursor while 2 and 3 were advocated as reactive intermediates playing a major role in the ATH catalytic cycle [17] .
The structure and chirality of the three complexes 1 - 3 were confirmed via single crystal X-ray analysis and by 1H NMR. Another important tool used for the identification of these species is electrospray ionization combined with a mass spectrometer (ESI-MS) [28] -[30] . A state of the art development in this field has been the ambient ionization method desorption electrospray ionization (DESI) coupled with high resolution MS [31] . This method was shown to provide a straightforward approach for intercepting reactive species in real time without prior sample preparation. It was indeed demonstrated that DESI can intercept CTH intermediates in solution on the millisecond time scale [32] .
Numerous theoretical studies were also carried out to substantiate [33] [34] or to challenge [35] the concerted metal-ligand bifunctional CTH and ATH mechanisms.
While practicing the standard Noyori’s protocol in transfer hydrogenation of simple ketones such as acetophenone, using 2-propanol as a hydrogen donor (Scheme 1), and in replicating the preparation and characterization of the intermediates 1 - 3, we were intrigued by several puzzling observations as follows:
1) The Ru(II)TsDPEN catalyst is not stable under the reaction conditions and rapidly loses activity. Only one reaction batch is typically viable [36] [37] .
2) Through the ATH process the color of the reaction solution is changing with time indicating a continuous alteration in the state of the catalyst (the substrates and products are obviously colorless). In other words, this catalytic system is not operating in steady state as should be expected in an archetypal catalytic process.
3) The preparation of the putative catalytic intermediates 1 - 3, was not carried out under authentic CTH conditions (namely in the simultaneous presence of 2-propanol and KOH). We realized that upon exposure of 1 - 3 to KOH dissolved in 2-propanol at ambient temperature, and these intermediates swiftly react and transform to other species.
Upon inspection we came to the conclusion that the catalytic mechanism, originally proposed by Noyori and later adopted by numerous authors, cannot be correct simply since the catalyst precursor 4, the TsDPEN ligand and the intermediates 1 - 3 are all unstable under the strong reducing conditions of the CTH reaction where a mixture of 2-propanol and KOH is applied.
We believe that the true catalyst in Scheme 1 is ruthenium nanoclusters swiftly formed and uniquely shaped in the presence of the TsDPEN ligand under the reaction conditions. This assertion is corroborated by kinetic
Scheme 1. A typical ATH reaction with in-situ generated catalyst.
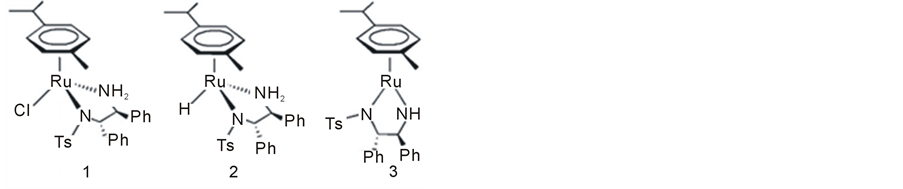

Figure 1. The starting arene complex (4) and the three Ru-TsDPEN intermediates (1 - 3) proposed by Noyori for the ATH reaction.
measurements, NMR, UV-visible spectroscopy, circular dichroism (CD) and TEM analyses presented in this paper.
2. Experimental Section
2.1. General Considerations
All operations were carried out under nitrogen atmosphere using standard Schlenk techniques. Solvents were distilled from the appropriate drying agents and degassed before use.
Complexes 1 - 3 were prepared according to literature protocols [27] [38] .
[RuCl2(p-cymene)]2, RuCl(p-cymene)[(R,R)-TsDPEN] , ethanol amine and (1R,2R)-(−)-N-p-Tosyl-1,2-diphenylethylenediamine (TsDPEN) were purchased from Aldrich.
2.2. Catalysts Characterization
For TEM analysis 3.5 microliter of 0.25 mM of the catalytic solution (with or without addition of 0.01 mmol of KOH) was deposited and dried onto a carbon coated 400 mesh copper TEM grid. The samples were analyzed with 200 kV FEG scanning-transmission electron microscope (STEM) Tecnai F20 G2 (FEI Company, USA). Imaging of the samples was done in bright field (BF) TEM mode and STEM mode with high angular annular dark-field STEM detector (HAADF). Elemental composition was analyzed with energy dispersion X-Ray spectroscopy (EDS) in STEM mode.
Transmission Electron Microscopy (TEM): Performed with the high resolution TEM Tecnai F20 G2. The microscope combines ultra-high resolution performance (point resolution 0.24 nm, line resolution 0.1 nm, limit of information 0.15 nm, HR STEM resolution < 0.2 nm) with extended analytical abilities and equipped with energy dispersive X-ray spectroscopy EDS detector.
For UV-visible and CD analyses, a sample of 3ml of the solution was poured into glass cuvette and directly analysed.
UV-Vis spectra were measured with a UV-Vis (Varian EL-03097225) spectrophotometer using isopropanol as the reference.
Circular dichroism (CD) spectra were recorded on a JASCO J-810 Spectrophotometer (JASCO, Japan) using the supplied Spectra-Manager software.
For H-NMR and diffusion NMR analysis, after the solution was evaporated and dried under vaacum at room temperature, a small sample is dissolved in CDCl3 in order to fill the NMR tube by 4 - 5 cm.
H-NMR spectra were recorded on a 500 MHz Burker Avance II instrument. Diffusion rates are measured by COSY-NMR.
2.3. Procedure for the Catalytic Transfer Hydrogenation of Ketones
Degassed propan-2-ol (40 ml) was added to a mixture of [RuCl2(p-cymene)]2 (6.124 mg, 0.01 mmol) and (1R, 2R)-(−)-N-p-Tosyl-1, 2-diphenylethylenediamine (14.66 mg, 0.04 mmol). The mixture was heated to 82˚C for 30 min under nitrogen (pretreatment). After cooling to 30˚C, acetophenone (0.48 g, 4.0 mmol) was added followed by of 1 ml of 0.1 M KOH solution in 2-propanol (0.01 mmol). The mixture was maintained at 30˚C for 24 hours. Samples were taken from the mixture at certain intervals and the conversion was determined by GC analyses.
2.4. Catalytic Activity Measurements
Samples taken from the reaction mixtures were analyzed by GC and HPLC.
For GC we used a Thermo Focus Gas chromatograph (Thermo scientific) equipped with a 95%-dimethyl-polysiloxane-5%-diphenyl packed column (GC Cap. Column 30 m × 0.25 mm × 0.25 µm) (Zebron™ ZB-5) and an FID detector. Helium was used as a carrier gas at a pressure of 50 kPa. Retention time of acetophenone: 5.30 and of 1-phenylethanol: 5.19 minutes.
3. Results and Discussion
3.1. Kinetics
Scheme 1 was tested under the original protocol defined in the prior art [8] [9] . The kinetic profile of the reaction is shown in Figure S1 in appendix. The catalyst performed exactly as reported (TOF of 69 min−1 after 30 min). The reaction stopped after 15 hours at 92% conversion. Using a chiral HPLC column [39] we verified the high ee (97%) of the product (R) 1-phenylethanol. However, upon addition of a batch of fresh substrate to the mixture after 15 hours, no further reaction was observed. Catalytic activity could be resumed only after the addition of a new batch of a catalyst (4 + TsDPEN + KOH). The lifetime of the catalyst in this system is thus limited to 12 - 15 hours at ambient temperature (Figure S1 in Appendix).
Remarkably, when elemental mercury (100:1 molar ratio Hg:Ru) was added to the above reaction mixture the reaction rate dropped by 52% to TOF of 33 min−1. This is an indication that the active catalyst in this reaction is possibly not a homogeneous metal complex.
The color changes observed at different stages of this run are shown in Figure 2(b). We noted that the reaction solution remained clear without any precipitation formed even after 15 hours although it turned into dark black accompanied with a total loss of activity. The color changes clearly suggest that the catalyst is not in a steady state through the process and its nature is varying with time.
As originally reported, in the absence of the TsDPEN ligand, precursor 4 exhibited a very poor catalytic activity. Thus, with 4 used as a sole catalyst, after the standard pretreatment and addition of KOH and substrate, conversion of barely 1% was measured in Scheme 1 after 1 hour and 8% after 15 hours (at 30˚C). Suspension of black particles appeared after 2.5 hours and precipitation was clearly observed in the mixture after 5 hours (Figure 2(a)). The catalyst was totally inactive after the first batch. Interestingly, the performance of 4 as a single catalyst in transfer hydrogenation was remarkably improved at reflux temperature (82˚C). Nonetheless the addition of TsDPEN under these conditions had no effect on the catalysis and no enantioselectivity [39] was observed. We also noticed that the lifetime of catalyst 4 in Scheme 1 at 82˚C is the same (approximately 12 hours) in the presence or in the absence of TsDPEN. As described above this is also the lifetime of the advocated catalyst at ambient temperature.
3.2. The Role of the Asserted Intermediates 1 - 3
The “metal-ligand bifunctional” and the “outsphere” mechanisms proposed by Noyori were allegedly authenticated by physical isolation and characterization of the intermediate complexes 1 - 3 and by the stoichiometric reversible reaction of 2 with acetone [27] Nonetheless, none of these proclaimed intermediates was actually prepared under the genuine CTH or ATH reaction conditions, namely in the simultaneous presence of the alcohol donor (2-propanol) and the base (KOH).
We have replicated the original three steps synthetic protocol and recorded the UV-visible spectra of the three species 1 - 3. To a solution of 4 in methylene chloride (Figure 3(a)) 1 equivalent of TsDPEN and of solid KOH were added at ambient temperature to generate the orange solution of 1 (Figure 3(b)). Then, 1 in methylene chloride solution was converted to the deep purple complex 3 by contacting it at room temperature with an aqueous KOH solution (Figure 3(c)). Finally the methylene chloride solution of 3 was washed with water to

Figure 2. Appearance and color of Scheme 1 mixtures as function of time (after pretreatment). (a) Complex 4 + KOH; (b) Complex 4 + TsDPEN + KOH.

Figure 3. Appearance and color of solutions of complexes 4 (a), 1 (b), 3 (c), 2 (d) and 2 + KOH (e).
remove the KOH, the solvent was evaporated under vacuum, and upon addition of 2-propanol in the absence of a base, 3 was transformed to dark brown ruthenium hydride complex 2 (Figure 3(d)).
The UV-visible spectra of complexes 1 - 3 and the appearance of their solutions are shown in Figure 3 and in Figure 4 respectively. The original complex 4 in CH2Cl2 displays three absorbance peaks at 271, 346 and 459 nm (Figure 3(a), Figure 4 red line). After addition of TsDPEN and KOH in methylene chloride the peak at 271 nm disappeared, the peak at 459 nm moved to 452 nm and the peak at 346 nm did not change (Figure 3(b), Figure 4 green line).
Washing the above solution with aqueous KOH generated the purple complex 3 (Figure 3(c)) that displayed two new absorption peaks at 305 and 550 nm (Figure 4 blue line). After evaporation of the methylene chloride and addition of 2-propanol, the brown complex 2 (Figure 3(d)) was formed with absorption peaks at 356 and 459 nm (Figure 4 brown line). After exposing the as-prepared complex 2 (in 2-propanol) to the authentic conditions of the ATH/CTH reaction, by adding two equivalents of KOH to the solution, an instant color change from brown to black was observed (Figure 3(e)). The UV-Visible spectrum of the resulting solution showed a flat absorption line with no peaks (Figure 4 purple line).
In view of these observations we can safely argue that none of the intermediate complexes 1 - 3 is present in the actual ATH/CTH reaction mixture and evidently no other soluble catalytic intermediate is present in the system. We may conclude that the molecular mechanism proposed by Noyori and coworkers cannot be utterly accurate.
UV-visible absorption spectrophotometry is considered as a practical method for monitoring the evolution of metallic species in the course of the preparation of colloidal metal nanoclusters [40] -[43] . In view of the Mie theory [44] that allows the reckoning of the spectrum of a ruthenium (and other metallic) colloids, we may contend that Ru(0) nanoclusters were evidently formed in the above solution upon the addition of KOH.
Complex 1 can be prepared and purified in an alternative method according to an Organic Syntheses procedure [38] . This procedure utilizes conditions that are different from the standard ATH/CTH reaction settings e.g. the base used is triethylamine and not KOH. Complex 1 prepared via this procedure behaved similarly to what is described above, showing the same flat UV-visible absorption spectra, when exposed to 2-propanol in the presence of KOH.
3.3. UV-Vis and TEM Analyses
We have examined the UV-Visible spectra of a solution of complex 4 in 2-propanol without added ligands (heated to 82˚C for 30 minutes followed by cooling and addition of 2 equivalents of KOH at 30˚C) as a function of time.

Figure 4. UV-Vis spectra of complexes 4, 1, 3, 2 and 2 + KOH + 2-propanol.
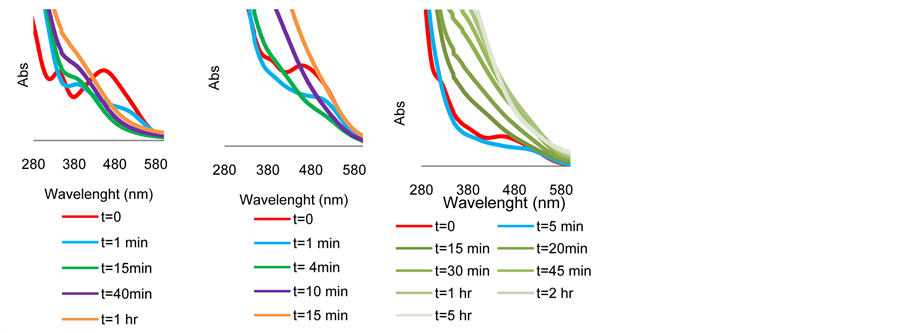
The spectrum is shown in Figure 5(a). It is evident that the original solution of 4 in 2-propanol exhibits two peaks with λ max at 341 and 453 nm (red line). 1 minute after the addition of KOH there is a clear shift of these peaks to 392 and 540 nm respectively (blue line). Traces of the two peaks remain after 15 and 40 minutes but both completely disappear after one hour where a flat absorption curve is observed (brown line). Ru(0) nanoclusters were seemingly quantitatively formed in the above solution after one hour. This was also corroborated by TEM analysis of the sample where particles of the size 3 - 4 nm are clearly observed as shown in Figure 6(a).
Remarkably, when TsDPEN was added to the above experiment, the original absorbance peaks disappeared much faster and after merely 4 minutes, a flat absorption line was apparent. This is shown in Figure 5(b). The same observation was made when a ketonic substrate is present [45] (Figure S3 in Appendix). TEM and STEM analyses of these mixtures clearly show that no particles are observed when 4 and TsDPEN are blended in 2-propanol at 82˚C (Figure 6(b)) but upon cooling and addition of KOH, particles of the size of 1 nm are blurrily detected (Figures 6(c) and Figure 6(d)). Immediate disappearance of the UV-Vis peaks is detected when the solution is maintained 82˚C after addition of KOH. No change in the particles size is observed (see TEM Figure S4 in Appendix). When we tracked the conduct of the UV-Visible spectra of the 4 + TsDPEN + KOH mixture beyond the 4 minutes required for the total disappearance of the original peaks, we observed an interesting dynamic behavior as a function of time as shown in Figure 5(c). During the process the flat absorption line (attributed to scattering absorbance of the nanoclusters solution between 300 - 600 nm) monotonically moved upwards for a period of 2 hours and then stopped. It is known that the shape, size and surface composition of nanoclusters in solution influence the nature of the absorption spectra in the UV-Visible range [46] -[48] .
Although we cannot realize any quantitative information from this observed phenomenon, we may safely assume that nanoparticles are evidently formed in the ATH reaction mixture after 6 - 10 minutes. These particles are gradually varying in nature for the next 2 hours. As noted previously, the absence of any peak in the UV-Vis region may suggest that NPs are the only catalytic species present in solution during the reaction. These species lose their catalytic activity after approximately 10 - 15 hours. No solid precipitation and no change in the particle size accompany this loss of catalytic activity.
It is therefore evident that the TsDPEN ligand advocated for the ATH reactions actually strongly enhance the transformation of complex 4 via reduction into ruthenium nanoclusters. At the same time the clusters formed in the presence of this ligand are far more catalytically active in comparison with the nanoparticles formed from complex 4 in the absence of the TsDPEN. It seems that with TsDPEN the in-situ formed active nanoclusters are particularly stable in solution as no precipitation is formed even after prolonged time (weeks). Nonetheless, the catalytic activity in transfer hydrogenation is lost within 10 - 15 hours at 30˚C. Our attempts to isolate these soluble nanoclusters failed. Our experiments at 82˚C confirm that TsDPEN fails to stabilize the ruthenium clusters at this temperature. Swift reaction is observed but black precipitate is observed at the end of the reaction.
We also realized that the pretreatment previously proposed (boiling 4 + TsDPEN in 2-propanol for 30 minutes) prior to the addition of KOH and the substrate, is actually redundant. The same kinetic results and general behavior were observed when 4, TsDPEN, KOH, 2-propanol and acetophenone were directly mixed at ambient temperature under inert atmosphere without any pretreatment.
 (a) (b) (c)
(a) (b) (c)
Figure 5. UV-Visible spectral change of intermediates at 30˚C in 2-propanol as function of time (a) Complex 4 + KOH up to 1 hr. (b) Complex 4 + TsDPEN + KOH up to 15 min. (c) Complex 4 + TsDPEN + KOH up to 5 hrs.
 (a)
(a) (b)
(b) (c)
(c) (d)
(d)
Figure 6. (a) TEM image of nanoparticles formed at 30˚C from 4 in 2-propanol and KOH (b) TEM image of 4 + TsDPEN in 2-propanol at 82˚C without KOH; (c) TEM image of the same after addition of KOH at 30˚C; (d) STEM image of the same.
3.4. Circular Dichroism (CD) Analyses
We measured the CD spectrum of (R,R) TsDPEN in 2-propanol. This is shown in Figure 7(a). Upon addition of one equivalent of 4 and boiling the solution at 82˚C for 30 minutes, we obtained the CD spectrum presented in Figure 7(b). The latter spectrum exhibits two cotton effect peaks at 235 and 245 nm that in general looks quite similar to the CD spectrum of the free TsDPEN ligand. However upon cooling of the above solution and addition of 5 equivalents of KOH the CD spectrum observed is totally different. This is shown in Figure 7(c) where new peaks at lower wavelength (220 - 235 nm) are apparent.
This suggests the creation of a new type of chirality. The latter can feasibly be attributed to the generation of chiral nanoparticles. Interestingly, upon heating the above solution to 82˚C the chirality is lost (see Figure S5 in Appendix). This explains the lack of enantioselectivity when the ATH reaction is carried out at 82˚C.
3.5. Diffusion NMR Spectroscopy
Diffusion ordered spectroscopy (DOSY) [49] -[51] obtained through pulsed field gradient (PFG) NMR experiments [52] [53] has become a multi-purpose tool in the chemical laboratory due to its capacity to gain insight into the behaviour of solution systems through the measurement of diffusion coefficients (D) and their correlation to molecular properties such as size, weight, shape etc. or to the nature of interaction between different molecules [54] [55] .
DOSY techniques were previously applied to study the dynamics of formation of nanoparticles, exhibiting lower diffusion rate than the starting molecules [56] [57] . Although the metallic nanoparticles themselves cannot be detected by NMR, measurements of diffusion coefficients of species adsorbed on the particle’s surface, allow
 (a)
(a) (b)
(b) (c)
(c)
Figure 7. CD spectra of (a) TsDPEN in 2-propanol (b) after addition of 4 and boiling (c) upon addition of KOH at 30˚C.
to perform the test and draw certain critical and definite conclusions concerning the formation and behaviour of metallic nanoclusters. In the present case these adsorbed species probably comprise adsorbed cymene and TsDPEN molecules or fragments thereof and possibly, metallic hydrides. The self-diffusion constant is measured in m2 s−1 and for metallic nanoparticles it is expected to be smaller, by several orders of magnitude, than that of typical organic or organometallic molecules.
We initially determined the self-diffusion coefficients of complex 4 and of TsDPEN in chloroform-d at 25˚C. We measured D = 8.13 × 10 − 10 m2 s−1 for complex 4 and 9.55 × 10 − 10 m2 s−1 for TsDPEN. Using the Stokes-Einstein equation we estimated the molecular diameter of complex 4 at 9.9A and of TsDPEN at 8.4 A which is in accordance with molecular models (note the viscosity of chloroform-d: 0.542 mPa s).
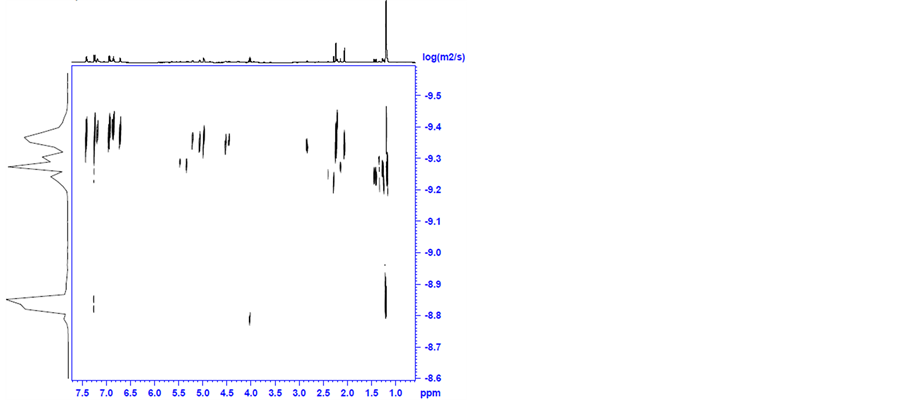
Then, 4 and TsDPEN (2 equivalents) mixture in 2-propanol, was heated for 30 minutes to 82˚C under nitrogen (the standard pretreatment conditions) and was analysed by diffusion 1H NMR spectroscopy. The NMR spectrum of the mixture is shown in Figure S6 in appendix. The DOSY spectra are shown in Figure 8(a) and Figure 8(b).
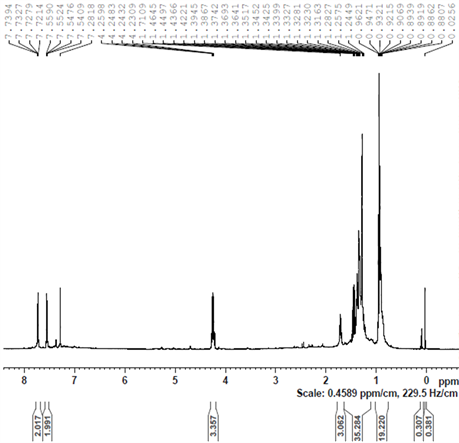
It is apparent that the original peaks of the coordinated cymene in complex 4 at δ = 5.0 (for aromatic H); 2.9 (for CH(CH3)2); 2.1 (for CH3) and 1.2 (for CH(CH3)2) ppm and of the TsDPEN ligand at δ = 7.3 − 7.0 (for aromatic H); 4.0 (for CHN); 2.3 (for CH3 in p-Ts) ppm are all recognized with a small shift to higher field indicating formation of a complex. This is also supported by a new hydride peak clearly observed at −9.0 ppm. The latter was measured to have a diffusion coefficient of 5 × 10 − 10 m2 s−1. Traces of fragments at δ = 4 and 1.4 ppm with diffusion coefficient of 1.5 × 10 − 9 m2 s−1 are also observable.
Using the Stokes-Einstein equation we estimated the van der Waals diameter of the main product to be 1.60 nm and of the smaller fragment as 5.3 A. The hydride is evidently a constituent of the larger molecule.
It is clear that a new complex is indeed formed between 4 and TsDPEN even prior to the addition of KOH. Based on the diameter measured we assume that this complex maintains the dimeric nature of 4 and that at least one of the original two chlorine bridges is not broken. This intermediate is clearly not one of the complexes 1 - 3. Examining the 1H NMR spectra of pure complex 1 revealed that it is different in nature from the adduct formed between 4 and TsDPEN in 2-propanol at 82˚C. As noted in view of the TEM analyses no nanoclusters are formed at this stage.
A dramatic change in the NMR spectrum is observed when KOH is added to the above solution at 30˚C (after it was heated in 2-propanol to 82˚C under nitrogen for 30 minutes). This is shown in Figure 9(b). It is clear that the original characteristic peaks of the coordinated cymene in complex 4 and of the added TsDPEN ligand have almost entirely disappeared in this spectrum. Instead, new peaks at 1.7, 1.3 and 0.9 ppm appeared which can be attributed to unidentified organic fragments adsorbed on the metallic ruthenium cluster. The latter were measured to have a diffusion coefficient of 7.9 × 10 − 10 m2 s−1 which corresponds to a van der Waals diameter of 1.0 nm (Figure 9(a)).
We assume that these are the catalytically active ruthenium (0) nanoclusters. Since the catalysis is enantioselective we believe that the organic fragments adsorbed to the nanoparticles surface maintain some of the chirality introduced by the original TsDPEN ligand.
Remarkably we did not detect in Figure 9(b) any peaks with δ < 0 that could be assigned to an intermediate hydride species such as complex 2. We may assume that at the point, where the ATH reaction actually starts, the original TsDPEN and p-cymene ligands associated with the ruthenium core are evidently transformed into different species. We may infer that under the ATH reaction conditions, with the formation of catalytically active ruthenium nanoclusters, the p-cymene and the TsDPEN ligands disintegrate to form fragments that remain
 (a)
(a) (b)
(b)
Figure 8. DOSY spectra of the complex formed by mixing 4 and TsDPEN in 2-propanol at 82˚C.
 (a)
(a)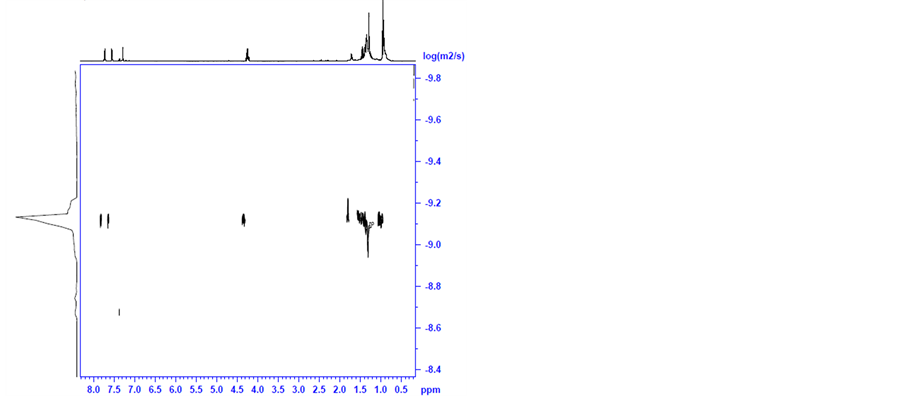 (b)
(b)
Figure 9. DOSY (a) and H-NMR (b) spectra of the ruthenium species, prepared by addition of KOH to 4 + TsDPEN at 30˚C (after drying, in chloroform-d).
partially adsorbed to the catalyst surface. This reduction-crumbling process, where TsDPEN has a pivotal role, is vital for the formation of the catalytically active chiral nanoclusters.
Upon heating the above solution to 82˚C total destruction of ligand fragments was detected, explaining the loss of the chirality (see Figure S7 in Appendix).
3.6. The Critical Role of the TsDPEN Ligand
As was shown above, the added TsDPEN ligand empowers and facilitates the reduction of complex 4, in the presence of 2-propanol and KOH, into catalytic nanoclusters and stabilizes the latter against precipitation as long as the temperature is maintained below 30˚C (black suspension was observed at 82˚C after several hours). The catalytic activity is nonetheless totally lost after approximately 12 - 15 hours at 30˚C.
Aiming to determine whether one should deem TsDPEN as a ligand or as a non-stoichiometric additive, we studied the kinetic behaviour of Scheme 1 with different 4: TsDPEN ratios under the standard reaction conditions. Results are summarized in Table1
In view of the data presented in Table 1 it is clear that the 4: TsDPEN molar ratio advocated by Noyori et al. (1:1 or 1:2, entries 2 and 3) results in effective catalysis. Nonetheless, entry 4 shows that a ratio of 1:0.5 is not less effectual. If the active catalyst is indeed a stoichiometric complex formed between 4 and TsDPEN we would expect that the experiments in entry 5 and in entry 4 would give similar results and that experiment 6 would show much inferior performance.
This is evidently not the case and it seems that TsDPEN should conceivably be considered as a critical additive for the generation of the true active catalyst and not as a stoichiometric ligand. On the other hand we noted that with excess of TsDPEN the reaction is critically retarded (entry 1).
3.7. The Crucial Function of pH in CTH Reactions
The significance of the base as a cocatalyst in CTH/ATH reactions is well established. It was argued that when 4 is used as a precursor, at least two equivalents of a base are required to convert complex 4 into complex 3. Nonetheless, in the cited procedures up to 1:5 Ru:KOH molar ratio was typically used. At the same time several authors claimed for CTH reactions “without base” [58] [59] . We have now confirmed that when 4 and TsDPEN are used in Scheme 1, a base is indeed essential for both the active catalyst in-situ formation and for promoting the CTH reaction. However, for the former function stoichiometric amount (relative to Ru) of base is needed while a trace dose is sufficient for the latter task.
Consequently, we noted that if at least one equivalent of KOH is provided, the critical factor is not the Ru:base molar ratio, but rather the pH of the reaction mixture. As long as the medium is acidic no CTH reaction is observed. We found that the normal pH of a solution of complex 4 + TsDPEN in 2-propanol is 3.6. Upon addition of 0.5 equivalent of KOH the pH rises to 5.4 - 5.8 and with 1.0 equivalent it increases to 6.3. Evidently this is not sufficient for the CTH reaction to initiate.
In a series of experiments we have examined the conversion in Scheme 1 with catalyst 4 + TsDPEN under standard conditions but with different Ru:KOH ratios and diverse KOH concentrations (controlled by different volumes of 2-propanol used). Results are summarized in Table2
Entries 1 - 5 (where no reaction is observed) clearly imply that at least one equivalent of a base is critical for the reaction to start. This is however not sufficient as long as the pH is below 7 (entry 5). In fact, even with Ru:KOH of 1:2 there is only inconsiderable reaction when the pH is 6.4 (entry 8). Once basic pH is attained the process advances smoothly. The color of the reaction mixtures is another indicator to the status of the catalyst. As long as the clear orange-brown solution is observed no reaction is apparent. At higher pHs (>8) the solution turns dark black indicating that productive reaction is taking place.
There is evidently no advantage in using larger excess of KOH. Ru:KOH ratio of 1:5 resulted in the highest conversion (95%). Additional base was not beneficial and actually lower conversion was obtained (entries 12 - 13).
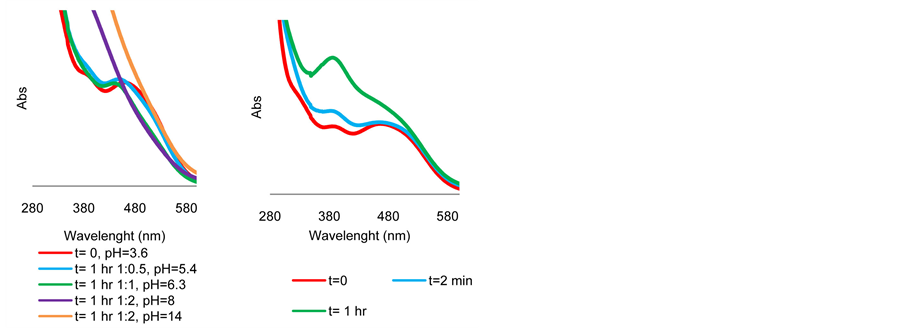
The UV-visible spectra of the samples listed in Table 2 (after 1 hour reaction time) plainly demonstrate that a critical Ru:KOH ratio and a minimal pH is essential for the formation of the catalytic nanoparticles and for initiation of the CTH process. In Figure S8(a) in Appendix the spectra of the solution in entries 1, 2, 5, 9, 10 in Table 2 are shown as function of time. In Figure S8(b) in appendix the UV-Visible spectrum of entry 8 is shown as function of time. In the latter experiment the pH is below 7.0 (6.4) and the absorbance peaks are shift

Table 1 . The effect of Ru:TsDPEN ratio on the kinetics of Scheme 1.
Table 2. Conversion and appearance of the reaction mixtures in Scheme 1 catalyzed by in situ prepared catalyst 4 + TsDPEN + KOH as function of initial Ru:KOH molar ratio, KOH concentration and pH.
ing as function of time from 458 nm to 386 nm but they do not disappear even after one hour.
In view of these findings we may suggest that the ruthenium complexes with bridging halides are resilient to reduction by 2-propanol in the absence of a base. These dimeric complexes are reduced by alcohols only in the presence of KOH or similar strong base which is consumed in the process.
Consequently one equivalent of e.g. KOH is required per equivalent of ruthenium to accomplish the reduction of Ru(II) to Ru(0) nanoparticles. Interestingly, monomeric ruthenium complexes (such as 1 - 3) do not require a base for the latter reduction step and pure 2-propanol is sufficient for this task. However, the ATH/CTH reaction that follows necessitates the presence of traces of base just to secure pH > 7. If this requirement is met, an ATH/CTH reaction “without base” will swiftly take place.
3.8. The Significance of the Pretreatment
We examined the consequence of the pretreatment originally advocated for Scheme 1 (20 - 30 minutes reflux of 4 with the amino ligand in 2-propanol prior to the addition of KOH and the substrate). For that aim Scheme 1 was performed without any pretreatment. Complex 4 was mixed with the standard amounts of 2-propanol, TsDPEN, KOH and acetophenone at room temperature. The CTH process started immediately and conversion of 90% was measured after 15 hours. This is the very same result that was attained when conducting the suggested pretreatment prior to the addition of KOH and the substrate. The recommended pretreatment is evidently not necessary for the formation of the active catalytic species and for the CTH reaction to take place. This was also substantiated by spectroscopy as described below.
The UV-Visible spectra of the ternary mixture of 4, TsDPEN and KOH in 2-propanol at room temperature as function of time is shown in Figure S9 in appendix. It is clear that the original homogeneous complex decomposes and disappears in less than 15 minutes, identical to Figure 5(b).
Conversely, when 4 was mixed with KOH in 2-propanol at 30˚C for one hour prior to the addition of TsDPEN and the acetophenone substrate, a very slow CTH reaction was observed with less than 12% conversion after 8 hours and with total deactivation of the catalyst.
Consequently it is obvious that the ruthenium nanoclusters formed in the absence of TsDPEN are different in nature in comparison with the clusters shaped when the amino ligand is present. The former species are significantly less active and considerably less robust catalysts in the CTH/ATH reaction.
Surprisingly, however, when 4 in 2-propanol was mixed with TsDPEN and KOH at 30˚C for 24 hours, and only then the acetophenone substrate was added, the reaction started normally and reached 90% conversion (and catalyst deactivation) at the regular time (15 hours). This remarkable observation clearly suggests that the active nanocluster catalyst is stable in the absence of the substrate and that acetophenone evidently has a critical role in the deactivation process that follows.
3.9. Rationale of the Catalyst Deactivation
In our previous study [60] on RuCl2(PPh3)3 precatalyst in CTH reactions the cause for the catalyst deactivation was evidently observed by the naked eye: agglomeration and precipitation of metallic nanoclusters took place concurrently with the loss of catalytic activity. In the latter example the base was found to be the main drive for the agglomeration and deactivation of the nanocatalyst. The case of the present catalyst (4 + TsDPEN + KOH) is visibly different. No solid precipitation is observed at 30˚C and the solution remained clear and stable (though eventually catalytically inactive) for several weeks. Pretreatment of complex 4 with TsDPEN and KOH during 24 hours at 30˚C did not alter the normal behaviour of Scheme 1 after the addition of acetophenone. The TsDPEN ligand distinctly prevents the agglomeration and precipitation of the in-situ formed catalytic nanoparticles but did not avert the loss of catalytic activity after a certain time. Addition of more TsDPEN in the course of the reaction did not avert the catalyst deactivation. Reducing the amount and the concentration of the KOH used also did not extend the lifespan of the catalyst (Table 2) Attempts to enforce agglomeration by adding flocculants such as aluminium sulphate (15 equivalents relative to the catalyst) did not affect the catalysis of Scheme 1.
The reason for the deactivation of this catalytic system is probably not related to agglomeration. In fact the deactivation is most likely not caused by change in size of the catalytic nanoparticles. TEM analyses of the catalyst 4 + TsDPEN + KOH at different stages of Scheme 1 suggest that once a size of 2 - 3 nm is attained, there is no further growth in the size of the nanoclusters. This is also supported by the NMR and the UV-Visible measurements.
In order to determine whether the catalyst deactivation is a result of a product or a substrate poisoning we treated 4 + TsDPEN with KOH in 2-propanol under the standard conditions with excess of the product 1-phenylethanol for 10 hours prior to addition of the substrate. This pretreatment did not affect the normal behaviour of Scheme 1. Same was realized after pretreatment with acetone. Pretreatment with excess of substrate (acetophenone) also did not disturb the performance of Scheme 1.
At this stage we cannot point at the specific reason for the catalyst deactivation. However since the deactivation is taking place only when all the reaction components are present namely complex 4, the TsDPEN ligand, KOH, 2-propanol and the substrate (acetophenone) we presume that a certain time dependent surface modification process of the catalytic nanoclusters is taking place in presence of the above ingredients which render the catalyst inactive within 10 - 15 hours. Interestingly we have also found that this deactivation process is essentially independent of temperature. When Scheme 1 was conducted under reflux (82˚C) a much faster reaction was monitored (100% conversion after 95 minutes) but the catalyst deactivation rate remained essentially the same. Consequently 4 catalytic cycles could be carried out at 82˚C before the catalyst stopped working after 12 hours.
3.10. Origins of Chirality
The hypothesis that ruthenium nanoparticle is the true catalyst in these CTH/ATH reactions calls for rationalization of an intriguing issue: what is the origin of the exceptionally high enantioselectivity typically observed in the presence of the chiral TsDPEN ligand.
Reviewing previous studies where chirally modified nanoclusters were deliberately prepared we realized that normally only moderate enantioselectivities were achieved. Ruthenium colloidal systems stabilized by chiral ligands were found to be active in the hydrogenation of arene derivatives [61] -[64] . Aplication of preprepared ruthenium NPs stabilized by chiral aminoalcohols and with oxazoline derived ligands in AH and ATH reactions were reported by Chaudert and coworkers [65] . Average ees were observed. Han et al. [66] have shown that (R)-BINAP ligands imprint chirally active sites on rhodium nanoparticles. These chirally stabilized rhodium NPs exhibited fairly high regioselectivity and chiral induction ability for the asymmetric hydroformylation of olefins (59% ee) [67] . Chirally modified Pd nanoparticles were used in asymmetric allylic alkylation again with only mediocre success [68] . Conversely, very high ees were reported by Jiang et al. who used cinchona and phosphine modified iridium catalyst for the enantioselective hydrogenation of aromatic ketones [69] .
Although we have presented only circumstantial evidences for the chirality issue (which is not the main aim of this study) we assert that the in-situ formed ruthenium nanoclusters in the presence of TsDPEN are chiral in nature and are thus accountable for inducing the asymmetric transfer hydrogenation. Chiral nanoscale materials have attracted intensive interest in recent years [70] -[76] . Several models were proposed to account for the optical activity found in chiral ligand-protected metallic nanoclusters (CLPMC) [77] [78] . One theory assumes a chiral metallic core, [79] second offers an achiral core but with chiral ligands adsorbed on the surface resulting in dissymmetric field effects [80] . A third model suggests a chiral footprint caused by a local deformation due to the ligand-cluster interactions [81] . Nonetheless, to date no plausible explanation of the physicochemical origin of this phenomenon was provided. This was attributed to the lack of precise structural information on CLPMCs and the difficulty in quantifying their chiroptical response. However, in a recent circular dichroism study on chiral gold nanoparticles it was concluded that the chirality results from mixing of ligand orbitals with those of the surface gold atoms of the nanoparticles [82] . We believe that a similar phenomenon takes place in the in-situ formation of chiral ruthenium nanoparticles stabilized with TsDPEN ligand. Concurrent with the reduction of the precursor 4, the TsDPEN ligand evidently functions simultaneously as a template, a structure directing agent and also as a stabilizing surface ligand leading to the formation of chiral ruthenium nanoclusters which are responsible for the asymmetric catalysis in these ATH reactions.
4. Conclusion
We have presented several independent compelling evidences suggesting that the true catalysts in CTH/ATH reactions in presence of Ru (II) arene complexes augmented with TsDPEN ligand in basic conditions with 2-propanol as a hydrogen donor are ruthenium nanoclusters formed in-situ via reduction in the reaction mixture. The diamine ligand actually facilitates the reduction process and moulds the characteristics of the catalytic nanoparticles. In the presence of the optically active TsDPEN ligand, chiral ligand-protected metallic nanoclusters (CLPMC) are apparently formed which are responsible for the high enantioselectivity observed. The three previously proposed intermediates complexes 1 - 3 are not playing any role in the CTH process where the catalyst is prepared in situ. The lifespan of the nanocluster catalysts is limited (typically 10 - 15 hours). We could not yet enlighten the specific reason for the catalyst deactivation, but we noted that it does not result from growth and agglomeration of the particles, nor is it caused by specific poisoning by any of the reaction components. The deactivation is attributed to some modifications of the nanoparticles that are apparently taking place only when all the reaction ingredients, including the hydrogen acceptor, are present.
Acknowledgements
We thank G. Tzafriri for technical assistance, Dr. I. Popov for performing the TEM analyses and Dr. R. Hoffman for the NMR analyses.
References
- Brieger, G. and Nestrick, T. (1974) Catalytic Transfer Hydrogenation. Chemical Reviews, 74, 567-580. http://dx.doi.org/10.1021/cr60291a003
- Johnstone, R.A.W., Wilby, A.H. and Entwistle, I.D. (1985) Heterogeneous Catalytic Transfer Hydrogenation and Its Relation to Other Methods for Reduction of Organic Compounds. Chemical Reviews, 85, 129-170. http://dx.doi.org/10.1021/cr00066a003
- Zassinovich, G., Mestroni, G. and Gladiali, S. (1992) Asymmetric Hydrogen Transfer Reactions Promoted by Homogeneous Transition Metal Catalysts. Chemical Reviews, 92, 1051-1069. http://dx.doi.org/10.1021/cr00013a015
- Trocha-Grimshaw, J. and Henbest, H.B. (1967) Catalysis of the Transfer of Hydrogen from Propan-2-ol to αβ-Unsaturated Ketones by Organoiridium Compounds. A Carbon-Iridium Compound Containing a Chelate Keto-Group. Chemical Communications, 1967, 544. http://dx.doi.org/10.1039/c19670000544
- Jin, W., Wang, L. and Yu, Z.A. (2012) Highly Active Ruthenium(II) Pyrazolyl-Pyridyl-Pyrazole Complex Catalyst for Transfer Hydrogenation of Ketones. Organometallics, 31, 5664-5667. http://dx.doi.org/10.1021/om300602w
- Ghoochany, L.T., Farsadpour, S., Sun, Y. and Thiel, W.R. (2011) New N,N,N-Donors Resulting in Highly Active Ruthenium Catalysts for Transfer Hydrogenation at Room Temperature. European Journal of Inorganic Chemistry, 2011, 3431-3437. http://dx.doi.org/10.1002/ejic.201100459
- Ye, W., Zhao, M., Du, W., Jiang, Q., Wu, K., Wu, P. and Wu, Z. (2011) Highly Active Ruthenium(II) Complex Catalysts Bearing an Unsymmetrical NNN Ligand in the (Asymmetric) Transfer Hydrogenation of Ketones. Chemistry—A European Journal, 17, 4737-4741. http://dx.doi.org/10.1002/chem.201002039
- Hashiguchi, S., Fujii, A., Takehara, J., Ikariya, T. and Noyori, R. (1995) Asymmetric Transfer Hydrogenation of Aromatic Ketones Catalyzed by Chiral Ruthenium(II) Complexes. Journal of the American Chemical Society, 117, 7562-7563. http://dx.doi.org/10.1021/ja00133a037
- Noyori, R. and Hashiguchi, S. (1997) Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Accounts of Chemical Research, 30, 97-102. http://dx.doi.org/10.1021/ar9502341
- Fujii, A., Hashiguci, S., Uematsu, N., Ikariya, T. and Noyori, R. (1996) Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Ketones Using a Formic Acid-Triethylamine Mixture. Journal of the American Chemical Society, 118, 2521-2522. http://dx.doi.org/10.1021/ja954126l
- Matsumura, K., Hashiguchi, S., Ikariya, T. and Noyori, R. (1997) Asymmetric Transfer Hydrogenation of α,β-Acetylenic Ketones. Journal of the American Chemical Society, 119, 8738-8739. http://dx.doi.org/10.1021/ja971570a
- Yamada, I. and Noyori, R. (2000) Asymmetric Transfer Hydrogenation of Benzaldehydes. Organic Letters, 2, 3425-3427. http://dx.doi.org/10.1021/ol0002119
- Miyagi, M., Takehara, J., Collet, S. and Okano, K. (2000) Practical Synthesis of (S)-1-(3-Trifluoromethylphenyl)ethanol via Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation. Organic Process Research & Development, 4, 346-348. http://dx.doi.org/10.1021/op000019m
- Zhang, J., Blazecka, P.G., Brunedl, M.M. and Huang, Y.J. (2009) Ru-TsDPEN with Formic Acid/Hünig’s Base for Asymmetric Transfer Hydrogenation, a Practical Synthesis of Optically Enriched N-Propyl Pantolactam. The Journal of Organic Chemistry, 74, 1411-1414. http://dx.doi.org/10.1021/jo802380j
- Lennon, I.C. and Ramsden, J.A. (2005) An Efficient Catalytic Asymmetric Route to 1-Aryl-2-imidazol-1-yl-ethanols. Organic Process Research & Development, 9, 110-112. http://dx.doi.org/10.1021/op049838n
- Hansen, K.B., Chilenski, J.R., Desmond, R., Devine, P.N., Grabowski, E.J.J., Heid, R., Kubryk, M., Mathre, D.J. and Varsolona, R. (2003) Scalable, Efficient Process for the Synthesis of (R)-3,5-bistrifluoromethylphenyl Ethanol via Catalytic Asymmetric Transfer Hydrogenation and Isolation as a DABCO Inclusion Complex. Tetrahedron: Asymmetry, 14, 3581-3587. http://dx.doi.org/10.1016/j.tetasy.2003.08.043
- Ikariya, T. and Blacker, A.J. (2007) Asymmetric Transfer Hydrogenation of Ketones with Bifunctional Transition Metal-Based Molecular Catalysts. Accounts of Chemical Research, 40, 1300-1308. http://dx.doi.org/10.1021/ar700134q
- Eisenstein, O. and Crabtree, R.H. (2013) Outer Sphere Hydrogenation Catalysis. New Journal of Chemistry, 37, 21-27. http://dx.doi.org/10.1039/c2nj40659d
- Noyori, R., Yamakawa, M. and Hashiguchi, S. (2001) Metal-Ligand Bifunctional Catalysis: A Nonclassical Mechanism for Asymmetric Hydrogen Transfer between Alcohols and Carbonyl Compounds. The Journal of Organic Chemistry, 66, 7931-7944. http://dx.doi.org/10.1021/jo010721w
- Yamakawa, M., Ito, H. and Noyori, R. (2000) The Metal-Ligand Bifunctional Catalysis: A Theoretical Study on the Ruthenium(II)-Catalyzed Hydrogen Transfer between Alcohols and Carbonyl Compounds. Journal of the American Chemical Society, 122, 1466-1478. http://dx.doi.org/10.1021/ja991638h
- Zhao, B., Han, Z. and Ding, K. (2013) The N-H Functional Group in Organometallic Catalysis. Angewandte Chemie International Edition, 52, 4744-4788. http://dx.doi.org/10.1002/anie.201204921
- Noyori, R. and Ohkuma, T. (2001) Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemoand Stereoselective Hydrogenation of Ketones. Angewandte Chemie International Edition, 40, 40-73.
- Soni, R., Cheung, F.K., Clarkson, G.C., Martins, J.E.D., Graham, M.A. and Wills, M. (2011) The Importance of the N-H Bond in Ru/TsDPEN Complexes for Asymmetric Transfer Hydrogenation of Ketones and Imines. Organic & Biomolecular Chemistry, 9, 3290-3294. http://dx.doi.org/10.1039/c1ob05208j
- Casey, C.P. and Johnson, J.B. (2003) Kinetic Isotope Effect Evidence for a Concerted Hydrogen Transfer Mechanism in Transfer Hydrogenations Catalyzed by [p-(Me2CH)C6H4Me]Ru-(NHCHPhCHPhNSO2C6H4-p-CH3). The Journal of Organic Chemistry, 68, 1998-2001. http://dx.doi.org/10.1021/jo0205457
- Handgraaf, J.W. and Meijer, E.J. (2007) Realistic Modeling of Ruthenium-Catalyzed Transfer Hydrogenation. Journal of the American Chemical Society, 129, 3099-3103. http://dx.doi.org/10.1021/ja062359e
- Baratta, W., Ballico, M., Baldino, S., Chelucci, G., Herdtweck, E., Siega, K., Magnolia, S. and Rigo, P. (2008) New Benzo[h]quinoline-Based Ligands and Their Pincer Ru and Os Complexes for Efficient Catalytic Transfer Hydrogenation of Carbonyl Compounds. Chemistry—A European Journal, 14, 9148-9160. http://dx.doi.org/10.1002/chem.200800888
- Haack, K.J., Hashiguchi, S., Fujii, A., Ikariya, T. and Noyori, R. (1997) The Catalyst Precursor, Catalyst, and Intermediate in the RuII-Promoted Asymmetric Hydrogen Transfer between Alcohols and Ketones. Angewandte Chemie International Edition in English, 36, 285-288.http://dx.doi.org/10.1002/anie.199702851
- Everaere, K., Mortreux, A., Bulliard, M., Brussee, J., van der Gen, A., Nowogrocki, G. and Carpentier, J.F. (2001) (β-Amino alcohol)(arene)ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Functionalized Ketones-Scope, Isolation of the Catalytic Intermediates, and Deactivation Processes. European Journal of Organic Chemistry, 2001, 275-291.
- Pelagatti, P., Carcelli, M., Calbiani, F., Cassi, C., Elviri, L., Pelizzi, C., Rizzotti, U. and Rogolino, D. (2005) Transfer Hydrogenation of Acetophenone Catalyzed by Half-Sandwich Ruthenium(II) Complexes Containing Amino Amide Li-gands. Detection of the Catalytic Intermediates by Electrospray Ionization Mass Spectrometry. Organometallics, 24, 5836-5844. http://dx.doi.org/10.1021/om050519+
- Kenny, J.A., Versluis, K., Heck, A.J.R., Walsgrove, T. and Wills, M. (2000) The Detection of Intermediates in the Ruthenium(II) Catalysed Asymmetric Hydrogenation of Ketones Using Electrospray Ionisation Mass Spectrometry. Chemical Communications, 2000, 99-100. http://dx.doi.org/10.1039/a908098h
- Perry, R.H., Brownell, K.B., Chingin, K., Cahill III, T.J., Waymouth, R.M. and Zare, R.N. (2012) Transient Ru-Methyl Formate Intermediates Generated with Bifunctional Transfer Hydrogenation Catalysts. Proceedings of the National Academy of Sciences of the United States of America, 109, 2246-2250.http://dx.doi.org/10.1073/pnas.1118934109
- Perry, R.H., Splendore, M., Chien, A., Davis, N.K. and Zare, R.N. (2011) Detecting Reaction Intermediates in Liquids on the Millisecond Time Scale Using Desorption Electrospray Ionization. Angewandte Chemie International Edition, 50, 250-254. http://dx.doi.org/10.1002/anie.201004861
- Zhang, X., Guo, X., Chen, Y., Tang, Y., Lei, M. and Fang, W. (2012) Mechanism Investigation of Ketone Hydrogenation Catalyzed by Ruthenium Bifunctional Catalysts: Insights from a DFT Study. Physical Chemistry Chemical Physics, 14, 6003-6012. http://dx.doi.org/10.1039/c2cp23936a
- Wiseman, R.V., de Vries, J.G., Deelman, B.J. and Heeres, H.J. (2006) Kinetic Studies on the Asymmetric Transfer Hydrogenation of Acetophenone Using a Homogeneous Ruthenium Catalyst with a Chiral Amino-Alcohol Ligand. Organic Process Research & Development, 10, 423-429. http://dx.doi.org/10.1021/op050231w
- Guo, X.J., Tang, Y.H., Zhang, X. and Lei, M. (2011) Concerted or Stepwise Hydrogen Transfer in the Transfer Hydrogenation of Acetophenone Catalyzed by Ruthenium-Acetamido Complex: A Theoretical Mechanistic Investigation. The Journal of Physical Chemistry A, 115, 12321-12330. http://dx.doi.org/10.1021/jp2046728
- Geldbach, T.J. and Dyson, P.J. (2004) A Versatile Ruthenium Precursor for Biphasic Catalysis and Its Application in Ionic Liquid Biphasic Transfer Hydrogenation: Conventional vs Task-Specific Catalysts. Journal of the American Chemical Society, 126, 8114-8115. http://dx.doi.org/10.1021/ja048886k
- Sandee, A.J., Petra, D.G.I., Reek, J.N.H., Kramer, P.C.J. and van Leeuwen, P.W.N.M. (2001) Solid-Phase Synthesis of Homogeneous Ruthenium Catalysts on Silica for the Continuous Asymmetric Transfer Hydrogenation Reaction. Chemistry—A European Journal, 7, 1202-1208.
- Ikariya, T., Hashiguchi, S., Murata, K. and Noyori, R. (2005) Preparation Of Optically Active (R,R)-Hydrobenzoin from Benzoin or Benzyl [1,2-Ethanediol, 1,2-diphenyl-, (1R,2R)-]. Organic Syntheses, 82, 10-17. http://dx.doi.org/10.15227/orgsyn.082.0010
- High Pressure Liquid Chromatography (HPLC) Equipped with a Plus Autosampler, SurveyorTM LC Pump Plus and Surveyor PDA Plus Detector. The HPLC Instrument Was Controlled and the Data Analyzed Using the ChromQuestTM 5.0 Data System, with UV-Vis Detector, Chiral Pak AD-H Column and Hexane: 2-Propanol Mixtures Were Used for Elution.
- Kirkland, A.I., Edwards, P.P., Jefferson, D.A. and Duff, D.G. (1990) Chapter 8. The Structure, Characterization, and Evolution of Colloidal Metals. Annual Reports Section “C” (Physical Chemistry), 87, 247-304.
- Toshima, N., Harada, M., Yonezawa, T., Kushihashi, K. and Asakura, K. (1991) Structural Analysis of Polymer-Protected Palladium/Platinum Bimetallic Clusters as Dispersed Catalysts by Using Extended X-Ray Absorption Fine Structure Spectroscopy. The Journal of Physical Chemistry, 95, 7448-7453. http://dx.doi.org/10.1021/j100172a061
- Finney, E.E. and Finke, R.G. (2008) Nanocluster Nucleation and Growth Kinetic and Mechanistic Studies: A Review Emphasizing Transition-Metal Nanoclusters. Journal of Colloid and Interface Science, 317, 351-374. http://dx.doi.org/10.1016/j.jcis.2007.05.092
- Henglein, A. and Giersig, M. (2000) Reduction of Pt(II) by H2: Effects of Citrate and NaOH and Reaction Mechanism. The Journal of Physical Chemistry B, 104, 6767-6772. http://dx.doi.org/10.1021/jp000801o
- Creighton, J.A. and Eadon, D.G. (1991) Ultraviolet-Visible Absorption Spectra of the Colloidal Metallic Elements. Journal of the Chemical Society, Faraday Transactions, 87, 3881-3891. http://dx.doi.org/10.1039/ft9918703881
- The UV-Vis Spectrum Was Distracted in the Presence of Acetophenone and Some of the Information Is Not Detected So This Test Was Run with Cyclohexanone.
- Zhang, Y., Yu, J., Niu, H. and Liu, H. (2007) Synthesis of PVP-Stabilized Ruthenium Colloids with Low Boiling Point Alcohols. Journal of Colloid and Interface Science, 313, 503-510. http://dx.doi.org/10.1016/j.jcis.2007.05.005
- Gaikwad, A.V. and Rothenberg, G. (2006) In-Situ UV-Visible Study of Pd Nanocluster Formation in Solution. Physical Chemistry Chemical Physics, 8, 3669-3675. http://dx.doi.org/10.1039/b604665g
- Aiken III, J.D. and Finke, R.G. (1999) A Review of Modern Transition-Metal Nanoclusters: Their Synthesis, Characterization, and Applications in Catalysis. Journal of Molecular Catalysis A: Chemical, 145, 1-44. http://dx.doi.org/10.1016/S1381-1169(99)00098-9
- Gibbs, S.J. and Johnson Jr., C.S. (1991) A PFG-NMR Experiment for Accurate Diffusion and Flow Studies in the Presence of Eddy Current. Journal of Magnetic Resonance, 93, 395-402. http://dx.doi.org/10.1016/0022-2364(91)90014-K
- Morris, K.F. and Johnson Jr., C.S. (1992) Diffusion-Ordered Two-Dimensional Nuclear Magnetic Resonance Spectroscopy. Journal of the American Chemical Society, 114, 3139-3141. http://dx.doi.org/10.1021/ja00034a071
- Johnson Jr., C.S. (1999) Diffusion Ordered Nuclear Magnetic Resonance Spectroscopy: Principles and Applications. Progress in Nuclear Magnetic Resonance Spectroscopy, 34, 203-256. http://dx.doi.org/10.1016/S0079-6565(99)00003-5
- Stejskal, E.O. and Tanner, J.E. (1965) Spin Diffusion Measurements: Spin Echoes in the Presence of a Time Dependent Field Gradient. The Journal of Chemical Physics, 42, 288-292. http://dx.doi.org/10.1063/1.1695690
- Price, W.S. (1997) Pulsed-Field Gradient Nuclear Magnetic Resonance as a Tool for Studying Translational Diffusion: Part 1. Basic Theory. Concepts in Magnetic Resonance, 9, 299-336.
- Nicolay, K., Braun, K.P.J., de Graaf, R.A., Dijkhuizen, R.M. and Kruiskamp, M.J. (2001) Diffusion NMR Spectroscopy. NMR in Biomedicine, 14, 94-111. http://dx.doi.org/10.1002/nbm.686
- Cohen, Y., Avram, L. and Frish, L. (2005) Diffusion NMR Spectroscopy in Supramolecular and Combinatorial Chemistry: An Old Parameter—New Insights. Angewandte Chemie International Edition, 44, 520-554. http://dx.doi.org/10.1002/anie.200300637
- Favier, I., Teuma, E. and Montserrat, G. (2009) Palladium and Ruthenium Nanoparticles: Reactivity and Coordination at the Metallic Surface. Comptes Rendus Chimie, 12, 333-545. http://dx.doi.org/10.1016/j.crci.2008.10.017
- Uccello-Barretta, G., Evangelisti, C., Raffa, P., Balzano, F., Nazzi, S., Martra, G., Vitulli, G. and Salvadori, P. (2009) The Control of the Growth of Pt Clusters in Solution: A Way to Prepare Pt Particles of Tailored Size. Journal of Organometallic Chemistry, 694, 1813-1817. http://dx.doi.org/10.1016/j.jorganchem.2009.01.010
- Everaere, K., Scheffler, J.L., Mortreux, A. and Carpentier, J.F. (2001) Stereoselective Synthesis of 3-Substituted Phtalides via Asymmetric Transfer Hydrogenation Using Well-Defined Ruthenium Catalysts under Neutral Conditions. Tetrahedron Letters, 42, 1899-1901. http://dx.doi.org/10.1016/S0040-4039(01)00076-4
- Pàmies, O. and Bäckvall, J.E. (2001) Studies on the Mechanism of Metal-Catalyzed Hydrogen Transfer from Alcohols to Ketones. Chemistry—A European Journal, 7, 5052-5058.
- Toubiana, J. and Sasson, Y. (2012) The True Catalyst in Hydrogen Transfer Reactions with Alcohol Donors in the Presence of RuCl2(PPh3)3 is Ruthenium(0) Nanoparticles. Catalysis Science & Technology, 2, 1644-1653. http://dx.doi.org/10.1039/c2cy00514j
- Pery, T., Pelzer, K., Mathes, J., Buntkowski, G., Philippot, K., Limbach, H.H. and Chaudret, B. (2005) Direct NMR Evidence for the Presence of Mobile Surface Hydrides on Ruthenium Nanoparticles. ChemPhysChem, 6, 605-607. http://dx.doi.org/10.1002/cphc.200400621
- Gual, A., Axet, M.R., Philippot, K., Chaudret, B., Denicourt-Nowicki, A., Roucoux, A., Castillon, S. and Claver, C. (2008) Diphosphite Ligands Derived from Carbohydrates as Stabilizers for Ruthenium Nanoparticles: Promising Catalytic Systems in Arene Hydrogenation. Chemical Communications, 2008, 2759-2761. http://dx.doi.org/10.1039/b802316f
- Hostetler, M.J., Wingate, J.E., Zhong, C.J., Harris, J.E., Vachet, R.W., Clark, M.R., Londono, J.D., Green, S.J., Stokes, J.J., Wignall, G.D., Glish, G.L., Porter, M.D., Evans, N.D. and Murray, R.W. (1998) Alkanethiolate Gold Cluster Molecules with Core Diameters from 1.5 to 5.2 nm: Core and Monolayer Properties as a Function of Core Size. Langmuir, 14, 17-30. http://dx.doi.org/10.1021/la970588w
- Pan, C., Pelzer, K., Philippot, K., Chaudret, B., Dassenoy, F., Lecante, P. and Casanove, M.J. (2001) Ligand-Stabilized Ruthenium Nanoparticles: Synthesis, Organization, and Dynamics. Journal of the American Chemical Society, 123, 7584-7593. http://dx.doi.org/10.1021/ja003961m
- Jansat, S., Picurelli, D., Pelzer, K., Philippot, K., Gómez, M., Muller, G., Lecante, P. and Chaudret, B. (2006) Synthesis, Characterization and Catalytic Reactivity of Ruthenium Nanoparticles Stabilized by Chiral N-Donor Ligands. New Journal of Chemistry, 30, 115-122. http://dx.doi.org/10.1039/b509378c
- Han, D., Li, X., Zhang, H., Liu, Z., Hu, G. and Li, C. (2008) Asymmetric Hydroformylation of Olefins Catalyzed by Rhodium Nanoparticles Chirally Stabilized with (R)-BINAP Ligand. Journal of Molecular Catalysis A: Chemical, 283, 15-22. http://dx.doi.org/10.1016/j.molcata.2007.12.008
- Axet, M.R., Castillón, S., Claver, C., Philippot, K., Lecante, P. and Chaudret, B. (2008) Chiral Diphosphite-Modified Rhodium(0) Nanoparticles: Catalyst Reservoir for Styrene Hydroformylation. European Journal of Inorganic Chemistry, 2008, 3460-3466. http://dx.doi.org/10.1002/ejic.200800421
- Jansat, S., Gómez, M., Philippot, K., Muller, G., Guiu, E., Claver, C., Castillón, S. and Chaudret, B. (2004) A Case for Enantioselective Allylic Alkylation Catalyzed by Palladium Nanoparticles. Journal of the American Chemical Society, 126, 1592-1593.
- Jiang, H., Yang, C., Li, C., Fu, H., Chen, H., Li, R. and Li, X. (2008) Heterogeneous Enantioselective Hydrogenation of Aromatic Ketones Catalyzed by Cinchonaand Phosphine-Modified Iridium Catalysts. Angewandte Chemie International Edition, 47, 9240-9244.
- Kitaev, V. (2008) Chiral Nanoscale Building Blocks—From Understanding to Applications. Journal of Materials Chemistry, 18, 4745-4749. http://dx.doi.org/10.1039/b808054b
- Barbaro, P., Dal Santo, V. and Liguori, F. (2010) Emerging Strategies in Sustainable Fine-Chemical Synthesis: Asymmetric Catalysis by Metal Nanoparticles. Dalton Transactions, 39, 8391-8402. http://dx.doi.org/10.1039/c002051f
- Mori, K. and Yamashita, H. (2010) Progress in Design and Architecture of Metal Nanoparticles for Catalytic Applications. Physical Chemistry Chemical Physics, 12, 14420-14432. http://dx.doi.org/10.1039/c0cp00988a
- Yao, H. (2008) Optically Active Gold Nanoclusters. Current Nanoscience, 4, 92-97.
- Gautier, C. and Burgi, T. (2009) Chiral Gold Nanoparticles. ChemPhysChem, 10, 483-492. http://dx.doi.org/10.1002/cphc.200800709
- Guerro-Martínez, A., Alonso-Gómez, J.L., Auguié, B., Cid, M.M. and Liz-Márzan, L.M. (2011) From Individual to Collective Chirality in Metal Nanoparticles. NanoToday, 6, 381-400. http://dx.doi.org/10.1016/j.nantod.2011.06.003
- Roy, S. and Pericás, M.A. (2009) Functionalized Nanoparticles as Catalysts for Enantioselective Processes. Organic & Biomolecular Chemistry, 7, 2669-2677. http://dx.doi.org/10.1039/b903921j
- Sánchez-Castillo, A., Noguez, C. and Garzón, I.L. (2010) On the Origin of the Optical Activity Displayed by Chiral-Ligand-Protected Metallic Nanoclusters. Journal of the American Chemical Society, 132, 1504-1505. http://dx.doi.org/10.1021/ja907365f
- Noguez, C. and Garzón, I.L. (2009) Optically Active Metal Nanoparticles. Chemical Society Reviews, 38, 757-771. http://dx.doi.org/10.1039/b800404h
- Bochicchio, D. and Ferrando, R. (2012) Structure and Thermal Stability of AgCu Chiral Nanoparticles. The European Physical Journal D, 66, 115-122. http://dx.doi.org/10.1140/epjd/e2012-30054-0
- Mori, K., Kondo, Y. and Yamashita, H. (2009) Synthesis and Characterization of FePd Magnetic Nanoparticles Modified with Chiral BINAP Ligand as a Recoverable Catalyst Vehicle for the Asymmetric Coupling Reaction. Physical Chemistry Chemical Physics, 11, 8949-8954. http://dx.doi.org/10.1039/b910069e
- Govorov, A.O., Gun’ko, Y.K., Slocik, J.M., Gérard, V.A., Fan, Z. and Naik, R.R. (2011) Chiral Nanoparticle Assemblies: Circular Dichroism, Plasmonic Interactions, and Exciton Effects. Journal of Materials Chemistry, 21, 16806-16818. http://dx.doi.org/10.1039/c1jm12345a
- Zhu, M., Qian, H., Meng, X., Jin, S., Wu, Z. and Jin, R. (2011) Chiral Au25 Nanospheres and Nanorods: Synthesis and Insight into the Origin of Chirality. Nano Letters, 11, 3963-3969. http://dx.doi.org/10.1021/nl202288j
Appendix
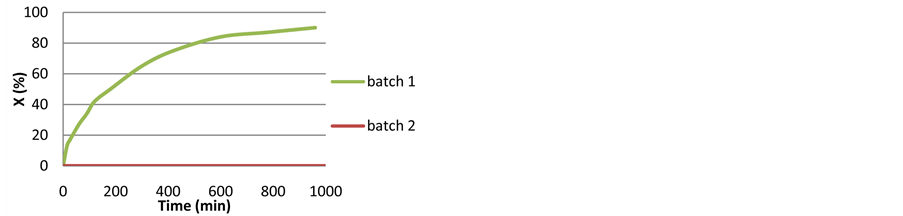
Figure S1. Kinetic profiles of Scheme 1 (2 consecutive runs). Reagents and conditions: acetophenone (4 mmol), 4 (0.01 mmol), (RR) TsDPEN (0.04 mmol), KOH (0.1 mmol), 2-propanol (40 ml), 30˚C, under N2.
 (a) (b)
(a) (b)
Figure S2. . CTH reaction at reflux temperature (82˚C). (a) Scheme 1 with catalyst 4 + KOH (three consecutive batches); (b) Scheme 1 with catalyst 4 + TsDPEN + KOH (four consecutive batches). Reagents and conditions: ketone (4 mmol), 4 (0.01 mmol), TsDPEN (0.04 mmol), KOH (0.1 mmol), isopropanol (40 ml), 82˚C under N2.
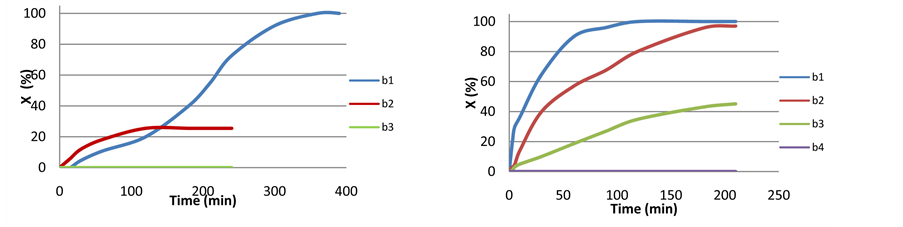
Figure S2(a) displays the kinetic profiles of Scheme 1 at 82˚C in the presence of 4 + KOH in the absence of any auxiliary ligand (3 consecutive runs). Scrutinizing Figure S2(a) (first batch), an induction period of 10 minutes is clearly observed. This is followed by a typical autocatalytic kinetics where the rate attains maximum TOF (121 min−1) only after 200 minutes. 100% conversion is accomplished after 400 minutes. It is apparent that the formation of the active catalyst is not instant in this case and it takes 3.5 hours to arrive at its maximum performance.
In the second batch, carried out immediately after the first run, there is no induction period nor a sigmoidal profile, but the maximum rate observed is lower (TOF = 80 min−1) and after two hours with conversion of only 25% the reaction totally stops. The third batch did not work at all. Overall the catalyst’s lifetime under these conditions is approximately 10 hours.
With catalyst 4 + TsDPEN was applied at 82˚C, no induction period and no autocatalysis were observed in Scheme 1 (Figure S2(b)). Evidently the active catalytic species is formed instantly in the presence of TsDPEN under these conditions. Three productive batches were performed (4 hours break at 30˚C between runs), with maximum TOF of 730 min−1 and complete conversion after 2 hours in the first, 285 min−1 and 100% conversion after 3.5 hours in the second and 100 min−1 and deactivation at 44% conversion after 4.5 hours in the third cycle. The lifetime of the catalyst is approximately 12 hours.
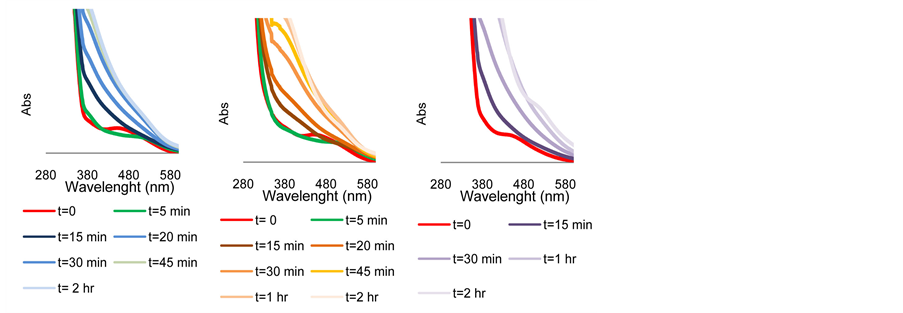
Figure S3. UV-Visible spectral analyses of Scheme 1 at 30˚C in 2-propanol as function of time (a) Complex 4 + TsDPEN + KOH + Acetophenone; (b) Complex 4+ TsDPEN + KOH+ Cyclohexanone; (c) Complex 4 + TsDPEN + KOH + Acetophenone under N2.
 (a) (b) (c)
(a) (b) (c)
 (d) (e) (f)
(d) (e) (f)
Figure S4. TEM coupled by Electron Diffraction and High Resolution Image and STEM analyses of samples taken after 30 min pretreatment in 2-propanol at 82˚C. (a) TEM image of Ru nanoclusters in mixture of catalyst 4 + Tsdpen at 82˚C; (b) Same confirmed by High Resolution Image; (c) Same by Electron Diffraction that reveal the fundamental state of Ruthenium (0); (d) and (e) Presence of Ru nanoparticles in mixture of catalyst 4 + KOH detected by (f) Same confirmed by High Resolution Image.

Figure S5. CD spectra of complex 4 + TsDPEN + KOH in 2-propanol after heating to 82˚C.
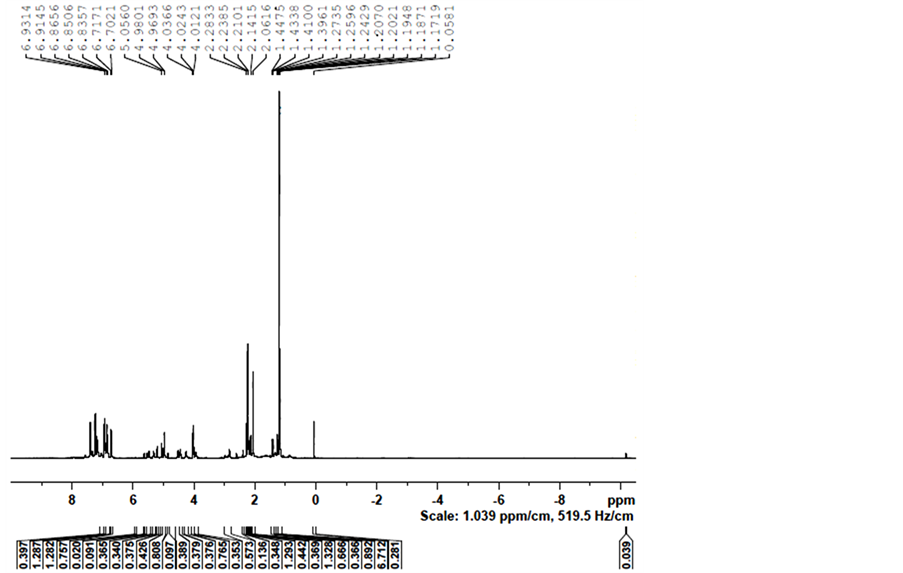
Figure S6. H-NMR spectra of the complex formed by mixing 4 and TsDPEN in 2-propanol at 82˚C.
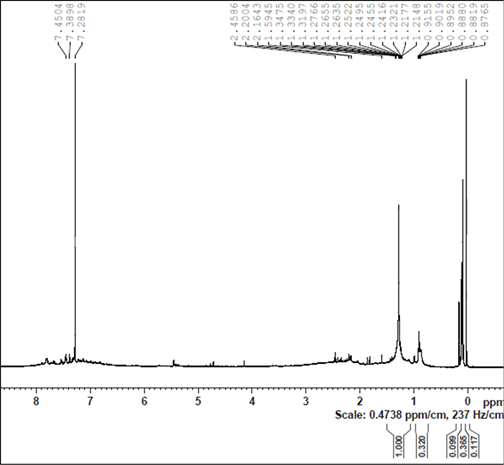
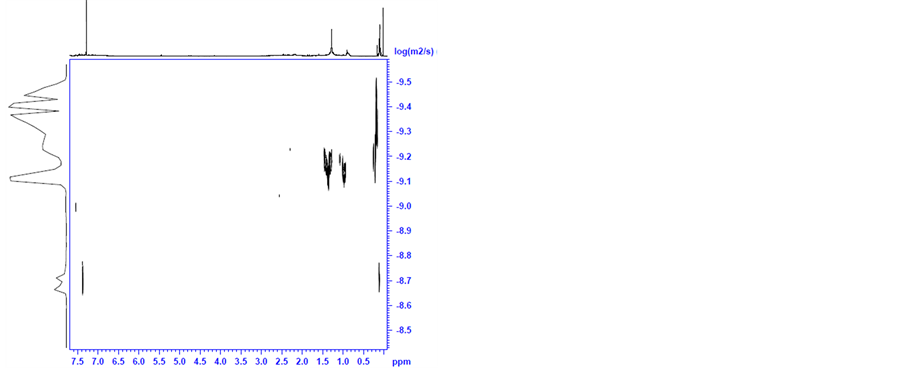 (a) (b)
(a) (b)
Figure S7. DOSY (a) and H-NMR (b) spectra of the ruthenium species, prepared by heating 4 and TsDPEN in 2-propanol to 82˚C under nitrogen for 30 minutes followed by addition of KOH at 82˚C (after drying, in chloroform-d).
After heating the above solution to 82˚C for 1 hour the H-NMR spectrum of the solution shows that the TsDPEN and the cymene ligands are totally converted (Figure S7(b)). This is evidently accompanied with loss of chirality. Based on the DOSY spectra we measured D= 7.1 × 10−10 m2 s−1 which corresponds to diameter of 1.13 nm (Figure S7(a)).
 (a) (b)
(a) (b)
Figure S8. (a) UV-visible spectra of complex 4 + TsDPEN + KOH in Scheme 1 mixture after 1 hour as function of pH. Reaction is observed only where the absorption line is flat. (b) UV-visible spectra of the same at low pH mixture at pH = 6.4 as function of time.

NOTES

*Corresponding author.