Open Journal of Synthesis Theory and Applications
Vol. 2 No. 1 (2013) , Article ID: 27193 , 25 pages DOI:10.4236/ojsta.2013.21002
Synthesis, Structural Study and Spectroscopic Characterization of a Quinolin-8-Yloxy Derivative with Potential Biological Properties
1Faculty of Biochemistry, Chemistry and Pharmacy of the National University of Tucumán, Tucumán, R. Argentina
2Chemistry Rosario Institute (CONICET-UNR), Faculty of Biochemistry and Pharmacists Sciences, Rosario, R. Argentina
Email: *sbrandan@fbqf.unt.edu.ar
Received November 14, 2012; revised December 16, 2012; accepted December 25, 2012
Keywords: (5-Chloro-Quinolin-8-Yloxy) Acetic Acid; Vibrational Spectra; Molecular Structure; Force Field; DFT Calculations
ABSTRACT
We have prepared the (5-chloro-quinolin-8-yloxy) acetic acid and characterized it by using infrared, Raman and multidimensional nuclear magnetic resonance spectroscopies. The density functional theory (DFT) together with the 6-31G* and 6-311++G** basis sets were used to study its structure and vibrational properties. Three stable conformations of the compound were theoretically determined in gas phase and probably these conformations are present in the solid phase. The harmonic vibrational wavenumbers for the optimized geometries were calculated at the same theory levels. For a complete assignment of the observed bands in the vibrational spectra, the DFT calculations were combined with Pulay’s scaled quantum mechanical force field (SQMFF) methodology in order to fit the theoretical wavenumber values to the experimental ones. Besides, the force constants of the three conformers of (5-chloro-quinolin-8-yloxy) acetic acid were calculated and compared with those obtained by us for the 2-(quinolin-8-yloxy) acetic acid. In addition, the characteristics of the electronic delocalization of those structures were performed by using natural bond orbital (NBO), while the corresponding topological properties of electronic charge density are analysed by employing Bader’s atoms in molecules theory (AIM).
1. Introduction
Heterocyclic compounds that contain the (quinolin-8- yloxy) moiety exhibit a wide range of biological properties [1-5], such as the 2-(quinolin-8-yloxy) acetohydrazones that have antiamoebic activities [5] for which, the structural and vibrational study of these types of compounds are of great chemical and pharmaceutical importance. Recently, a complete vibrational analysis of all observed bands in the vibrational spectra of 2-(quinolin- 8-yloxy)-acetic acid [6] was performed by means of the DFT calculations combined with Pulay’s scaled quantum mechanical force field (SQMFF) methodology [7-9]. In that study, the normal mode calculations were accomplished by using a generalized valence force field (GVFF) [8,9] considering three different structures for the anhydrous and monohydrated compounds. In the present work, we report an experimental and theoretical vibrational study of (5-chloro-quinolin-8-yloxy) acetic acid (CQA) by means of the B3LYP calculations using 6-31G* and 6-311++G** basis sets. For a complete assignment of the compound, the DFT calculations were combined with the SQMFF methodology [7-9] in order to fit the theoretical wavenumber values to the experimental ones. So far, the crystal and molecular structure of (5-chloro-quinolin-8- yloxy) acetic acid has not been determined. First, we have synthesized and characterized the compound and then, the optimized geometries and frequencies for the normal modes of vibration of CQA considering three different stable structures were calculated in order to carry out a complete assignment of all observed bands in the infrared and Raman spectra. Here, the normal mode calculations and the force fields for those structures were obtained by using the transferable scaling factors of Rauhut and Pulay [9] while the harmonic force constants were subsequently scaled to reproduce as well as possible the experimental frequencies. Thus, a complete assignment of the compound in terms of the potential energy distribution was performed. Furthermore, when the forces constants values of this compound were compared with those corresponding to the 2-(quinolin-8-yloxy)- acetic acid [6] numerous changes in the structural, topological and vibrational properties attributed to the chloro atom were observed. Additionaly, the nature of the different rings and bonds of the three studied structures of (5-chloro-quinolin-8-yloxy) acetic acid were analyzed by means of the NBO studies [10-12] while the topological properties of electronic charge density were determined employing the Bader’s atoms in molecules theory (AIM) [13].
2. Experimental Methods
2.1. Synthesis
(5-chloroquinolin-8-yloxy) acetic acid was obtained according to Cho et al. [1] using the following procedure.
2.1.1. (5-Chloroquinolin-8-Yloxy) Acetic Acid Methyl Ester
A mixture of 5-chloro 8-hydroxyquinoline (3.6 g, 20 mmol), methyl bromoacetate (3.7 g, 24 mmol) and K2CO3 (5.52 g, 40 mmol) in acetone (50 mL) was heated for 3 h under reflux, filtered and concentrated in vacuo. The residue was partitioned between ethyl acetate and brine, and the organic layer was dried with MgSO4, filtered, and concentrated in vacuo. The residue was purified by crystallization from isopropyl ether to give (5-chloroquinolin-8-yloxy) acetic acid methyl ester 3584 g (71%) m.p. 102.1˚C - 102.3˚C.
2.1.2. (5-Chloroquinolin-8-Yloxy) Acetic Acid (CQA)
A mixture of ester ((1.26 g, 5 mmol) and LiOH·H2O (352 mg, 8.40 mmol) in 75 mL of THF/CH3OH/H2O (1:1:1, 6) was stirred and heated at reflux for 1 h and concentrated. The aqueous layer was washed with ether and adjusted to pH 3 with 1 N HCl. The precipitate was filtered and dried to give (5-chloroquinolin-8-yloxy) acetic acid (1085 g, 91%) m.p. 219.5˚C - 221˚C.
2.2. NMR Spectra
1H NMR (300 MHz, CDCl3) δ: 4.92 (2H, s, O-CH2); 7.08 (1H, d, J = 8.48, H7), 7.64 (1H, d, J = 8.48, H6); 8,47 (1H, d, J = 8.58, H4); 7.70 (1H, dd, J = 4.18, J = 8.58, H3); 8.94 (1H, d, J = 4.18, H2).
13C NMR (75 MHz, CDCl3) δ: 65.94 (CH2); 110.54 (C7); 121.69 (C5); 123.53 (C3); 126.66 (C4a); 127.07 (C6); 132.64 (C4); 140.55 (C8a); 150.28(C2); 153.54 (C8); 170.34 (CO2H) (Atom numbering according to naphthalene).
2.3. Infrared and Raman Spectra
The infrared spectrum of the solid substance was recorded in KBr pellets in the wavenumbers range from 4000 to 400 cm−1 with a FT-IR Perkin Elmer spectrometer, provided with a Globar source and a DGTS detector. FT-Raman spectrum of the crystalline solid was recorded on a Bruker RFA 106/S FT-Raman instrument using the 1064 nm excitation line from an Nd: YAG laser in the region of 4000 - 0 cm−1. Two hundred scans were accu mulated at 4 cm−1 resolution using a laser power of 150 mW.
3. Computational Details
The potential energy curves associated with the internal rotation described by the C21-C18-O17-C10 dihedral angle for CQA were studied at the B3LYP/6-31G* and 6-311++G** theory levels. With both calculations, two conformations stable, named, CI and CII, according to the position of the OH group in relation to the N atom of the ring were obtained. Another plane structure was also considered (CIII) in agreement with the experimental structure of the 8-(carboxymethoxy) quinolinium nitrate monohydrate compound [14]. The structures and labelling of the atoms for all conformers of CQA can be seen in Figure 1. The electronic charge density topological analysis for those structures were performed by using the AIM200 program package [15] while the NBO calculations were obtained by means of the NBO 3.1 [16] program, as implemented in the GAUSSIAN 03 package [17]. The natural internal coordinates for the compound have been defined as those reported in the literature [7,8,18] and are listed in the Supporting Material as Table S1. The resulting force fields were transformed to “natural” internal coordinates by using the MOLVIB program [19,20]. Then, following the SQMFF procedure [7-9,21], the harmonic force fields for those structures were evaluated at the B3LYP/6-31G* level. The potential energy distribution components (PED) higher than or equal to 10% were subsequently calculated with the resulting SQMFF. The nature of all vibration modes was carried out by means of the Gauss View program [22].
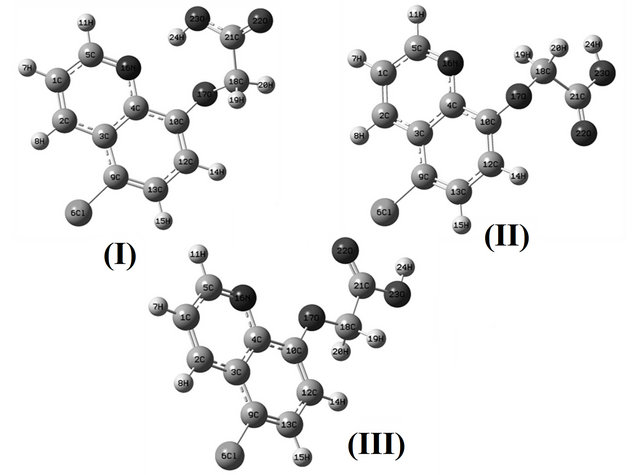
Figure 1. Theoretical structures and labelling of the atoms for the stable CI, CII and CIII conformers of (5-chloro-quinolin-8-yloxy) acetic acid.
4. Results and Discussion
4.1. Geometry
Table S2 (Supporting Material) shows the comparison of the total energies for the compound, and the corresponding dipole moment values for the CI, CII and CIII conformers of C1 symmetries. With both methods, the energy of the CI is lower than the other ones, as was observed in the compound without chlorine [6], and the potential energy difference between the CI and CII forms, by using the B3LYP/6-31G* and B3LYP/6-311++G** methods, are −43.80 and −46.43 kJ/mol, respectively. When the CI conformer is compared with the planar ones the potential energy difference values slightly by using both basis sets decrease up to −27.80 and −32.36 kJ/mol, respectively.
Also, in this case, the high values of the dipolar moments for the CI structures could probably explain its stabilities, as was observed in similar molecules [6,23-25].
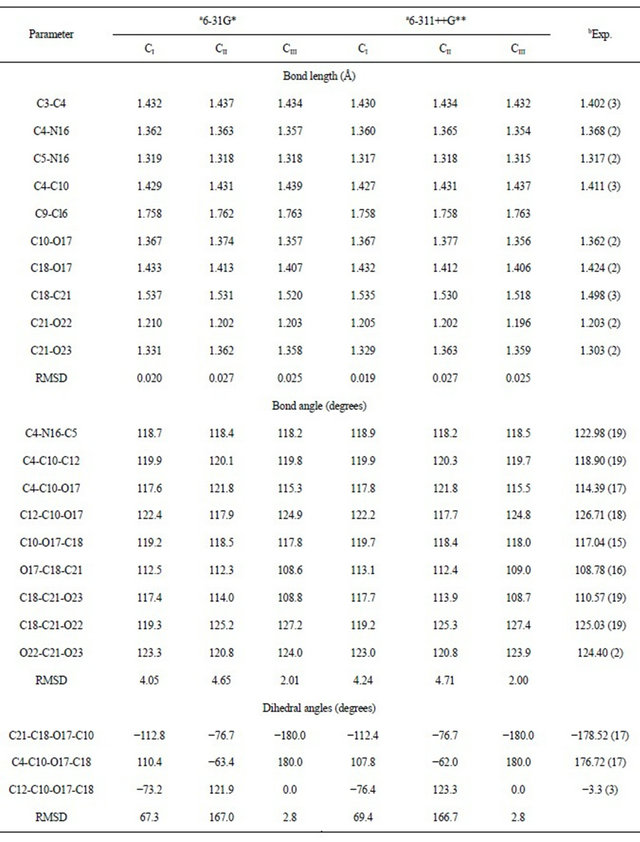
Table 1 shows a comparison of the calculated geometrical parameters for all structures of CQA, with the ones corresponding to that observed from X-ray diffraction for the 8-(carboxymethoxy) quinolinium nitrate monohydrate compound [14] by means of the root mean of square deviations (rmsd) values. Note that the 6-311++G** basis set reproduce reasonably well the theoretical bond lengths for the CI structure (0.019 Å) and the bond angles for the CIII structure (2.00˚), as is expected because this last conformer is planar as that compared compound [14]. Besides, the calculation predicts that the CI and CIII conformers are the most stable and probably both are present in the crystalline state.
The stabilities of the CII and CIII structures of CQA, in relation to the CI structure, were investigated by using the natural atomic charges [26-29] and the results are given in Table S3. Note that the natural charges values (NPA) corresponding to the C21 atoms have the most positive values in all structures while the most negative values correspond to the O23 atoms. In this compound, as in 2-(quinolin-8-yloxy)-acetic acid [6], the higher NPA charges observed on the O17 and O22 atoms in the CII structure justified the repulsion between those atoms. This way, the CII structure is more unstable than the corresponding to the CI and CIII conformers. The bond orders, expressed by Wiberg’s index for all conformers of CQA are given in Table S4. The bond order values for the O23 atoms in all conformers have the lowest values (belong to the COO groups) than the other ones (the O17 and O22 atoms belongs to the heterocyclic rings), while the bond orders for the N16 atoms have higher values.
4.2. NBO Study
The stability of the three structures of CQA was also investigated by means of NBO calculations [10-12]. The second order perturbation energies E(2) (donor ® acceptor) that involve the most important delocalization all conformers of CQA are given in Tables S5, S6. The contributions of the stabilization energies for the DETs®s* charge transfers are similar to those obtained for the 2-(quinolin-8-yloxy)-acetic acid compound [6].
Here, the delocalizations DETLP®s* for the CII structure has lower values than the DETs®s* delocalizations, and the calculated total energy values favours to the CI and CIII conformers, which structures are the most stable in the gas phase. Thus, the total energy values clearly show the higher stability of the CI conformer however, by using the 6-311++G** basis set, the CII and CIII conformers have the same ones approximately total energy values.
4.3. AIM Analysis
Furthermore, the three structures of CQA were analysed by means of Bader’s charge electron density topological analysis [13]. The calculated electron density, (r) and the Laplacian values, Ñ2r(r) in the bond critical points (BCPs) and ring critical points (RCPs) for those structures are shown in Table S7. The BCP has the typical properties of the closed–shell interaction and for this, the value of ρ(r) is relatively low, the relationship |l1|/l3 is <1 and Ñ2r(r) is positive indicating that the interaction is dominated by the charge contraction away from the interatomic surface toward each nucleus. Note that the results are clearly dependent of the size basis set. Thus, the analysis shows two BCPs and RCPs in the CI structure when the 6-31G* basis set is employed but, only one BCP and RCP when the 6-311++G** basis set is used. On the contrary, for the CII structure with both basis sets only one BCP and RCP are observed while for the CIII structure there is one BCP with the 6-31G* basis set. These results, togheter to the NBO analysis, justify the stabilities of the CI and CII structures due to the presence of short intramolecular O17-H24 and N16-H24 bonds, in the first case and only a N16-H19 bonds in the second one, as observed in Table S7.
4.4. Vibrational Analysis
The Figures 2, 3 show the registered infrared and Raman spectra for the compound in solid phase. In this study, in accordance with the NBO and AIM results, the three structures for the compound in gas phase were considered. The CQA’s structures have C1 symmetries and 66 normal vibration modes, all active in the infrared and Raman spectra. Probably, the three species are present in the solid phase because the comparison of each vibrational spectrum with the corresponding experimental one is very different among them, as observed in Figure 4, however, a comparison between the average calculated
Table 1. Calculated geometrical parameters for the conformers of (5-chloro-quinolin-8-yloxy) acetic acid.
infrared spectra (from B3LYP/6-31G* level for the CI, CII and CIII conformers) by using average wavenumbers and intensities with the corresponding experimental one demonstrate a good correlation, as observed in Figure S1.
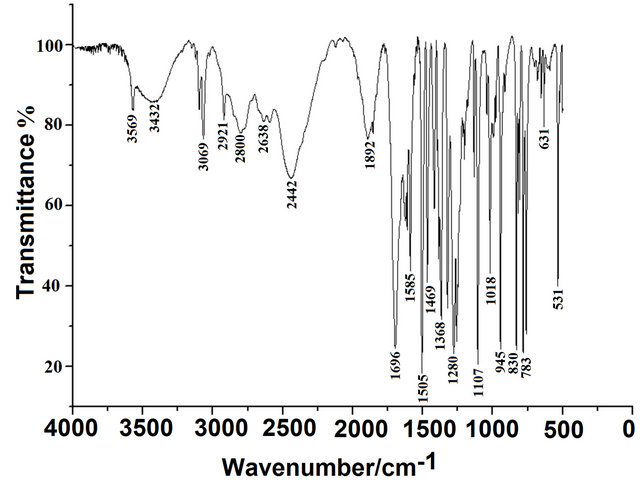
Figure 2. Experimental infrared spectrum of the solid (5- chloro-quinolin-8-yloxy) in KBr pellets.
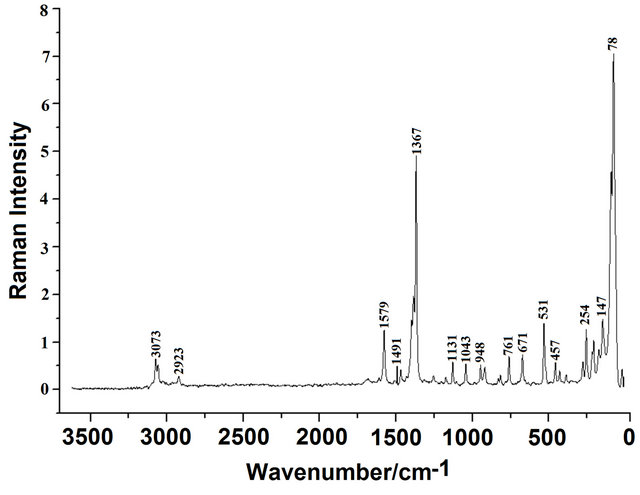
Figure 3. Experimental raman spectrum of the solid (5- chloro-quinolin-8-yloxy)of in a glass capillary.
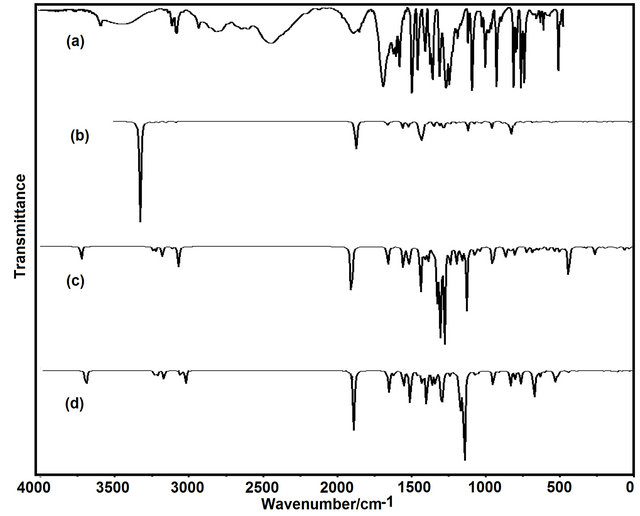
Figure 4. Comparison between: a) the experimental infrared spectrum of the solid (5-chloro-quinolin-8-yloxy) with, the theoretical spectra corresponding to the b) conformer CI; c) conformer CII, and; d) conformer CIII by using B3LYP/6-31G* level.
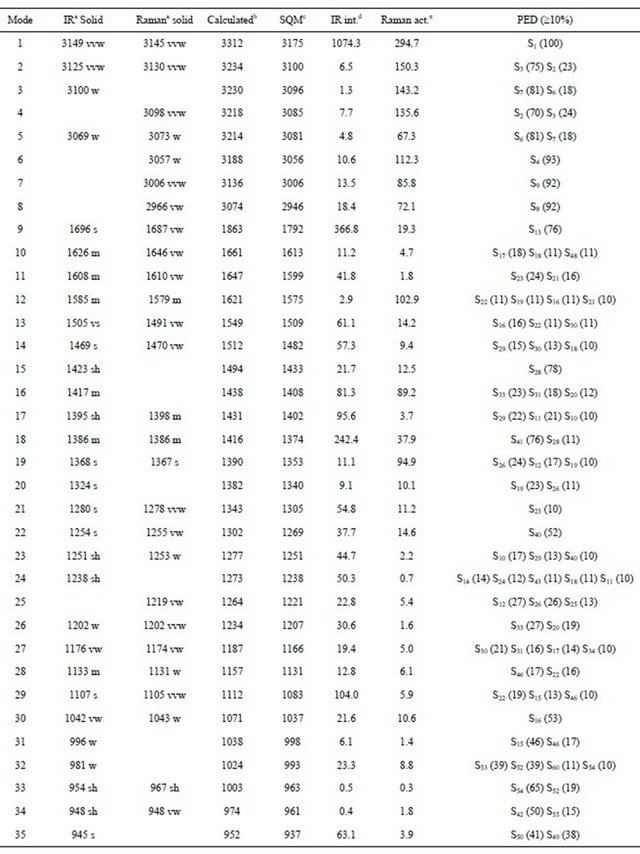
Thus, the resulting IR spectrum reproduces rather well some bands of the experimental spectrum, especially in the 2000 - 400 cm−1 region. The assignment of the experimental bands to the expected normal vibration modes were made on the basis of the PED in terms of symmetry coordinates and taking into account the corresponding assignment of related molecules [6,30-34]. Tables 2, S8-S10 show the experimental and calculated frequentcies, potential energy distribution based on the 6-31G* basis set, and assignment for the CI, CII and CIII structures of CQA. In a similar way as observed in the molecular packing of the 2-(2’-furyl)-1H-imidazole [23], 2-(2’- furyl)-4,5-1H-dihydroimidazole [27] and tolazoline hydrochloride molecules [35,36], the broad band with several shoulders between 2800 and 1900 cm−1 can be attributed to the N-H and O-H hydrogen bondings formed by the spatial arrangement of molecules in the lattice crystal.
Here, the B3LYP/6-31G* calculations were considered because the used scale factors are defined for this basis set. The SQM force fields for this compound can be obtained upon request. Below we discuss the assignment of the most important groups.
4.4.1. Assignments
4.4.1.1. OH Modes The weak bands in the IR spectra at 3569, 3432 and 3149 cm−1, in accordance to the values reported for similar molecules [30,33,34], can be assigned to the O-H stretching vibrations of the three conformers, as observed in Table 2. The width and shape of these bands shows clearly the typical spectroscopic signature of an H-bond interaction (inter or intramolecular). In accordance to that reported value for the benzoic acid [33,34] and to the theoretical calculations, the corresponding deformation modes of this group are assigned, for those conformers, to the strong IR bands at 1368, 1280 and 1254 cm−1, for the CI, CIII and CII conformers, respectively. The out-ofplane deformation mode (tOH or gOH) in the 4-hydroxybenzoic acid dimer [28] is calculated at 877 cm−1 while in CQA that mode is assigned for the CI conformers to the strong band in the IR spectrum at 760 cm−1 and to the very weak band in the same spectrum at 504 cm−1 for the CIII conformer while for the CII conformers that mode is assigned to the shoulder in the Raman spectrum at 409 cm−1. This last band in the Cp conformer of 2-(quinolin-8-yloxy)-acetic acid [6] is assigned to the weak IR band at 497 cm−1.
4.4.1.2. CH Modes The C-H stretching modes can be clearly assigned, for their positions, to the group of bands in the 3125 - 3025 cm−1 region of the IR and Raman spectra. The in-plane deformation modes of the phenyl ring, in similar compounds are observed between 1267 and 1042 cm−1 [31] while in this compound those modes are assigned to the IR bands between 1505 and 1176 cm−1, as observed in Table 2. On the other hand, the six expected out-of-plane
Table 2. Observed and calculated wavenumbers (cm−1) and assignment for the (5-chloro-quinolin-8-yloxy) acetic acid.
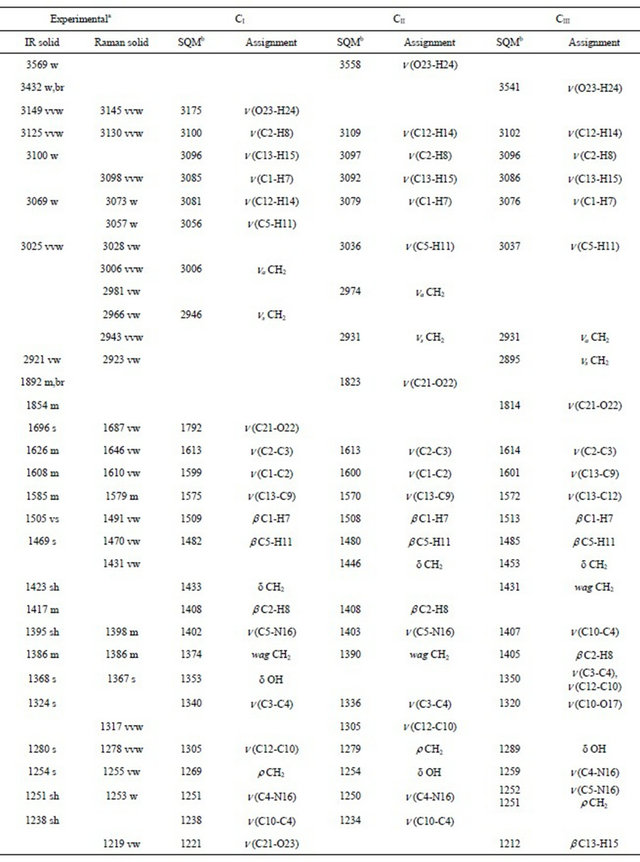
Continued
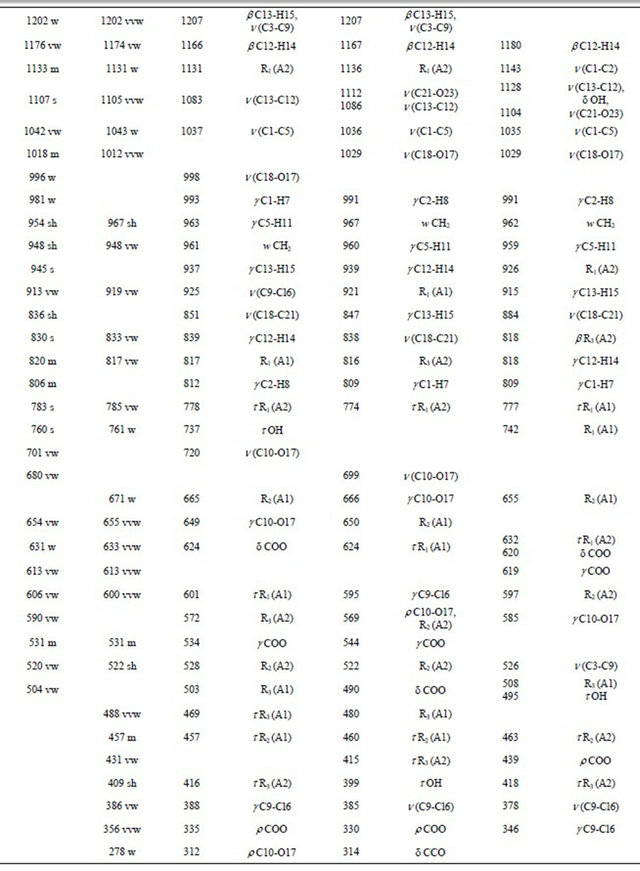
Continued
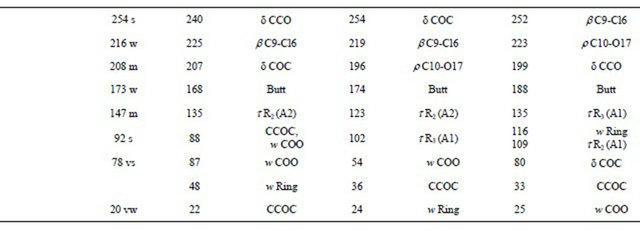
deformations of C-H group for the three conformers of CQA are assigned to the IR and Raman bands located between 981 and 806 cm−1.
4.4.1.3. CH2 Modes The group of bands in the 3006 - 2923 cm−1 region of the Raman spectra are assigned to the antisymmetric and symmetric stretching modes of this group in agreement to similar compounds [26,32]. The scissoring modes for the three conformers of CQA can be assigned to the shoulder in the IR spectrum and to the weak Raman band respectively at 1423 and 1431 cm−1. The shoulder in the IR spectrum and the weak Raman band at 1423 cm−1 and the band of medium intensity at 1386 cm−1 are assigned to the wagging modes of this group, as predicted by calculation, while the strong bands and the shoulder at 1280, 1254 and 1251 cm−1 are assigned to the rocking modes. The twisting mode for all CQA conformers are associated with the shoulders in the IR spectrum at 954 and 948 cm−1 as was observed in the 2-(quinolin-8-yloxy)-acetic acid [6].
4.4.1.4. COO Modes The C=O stretching mode, for the CI conformer of CQA is calculated at 1792 cm−1, it is at lower wavenumber than the other conformers (1823 and 1814 cm−1, CII and CIII conformers, respectively). On the other hand, for the benzoic acid the C=O stretching mode is predicted between 1786 and 1608 cm−1 while the C-O stretching mode are assigned between 1359 and 1334 cm−1 [33,34]. Hence, the IR bands of the media intensities and the strong IR band at 1892, 1854 and 1696 cm−1 are clearly assigned to the C=O stretching modes, as predicted by the calculations while the the Raman and IR bands respectively at 1219 and 1107 cm−1 are assigned to the C-O stretching mode corresponding to the three conformers of CQA. In the 4-hidroxybenzoic acid dimer [30] the two g(COO) modes are predict at 772 cm−1, whereas the two d(COO) modes at 771 and 620 cm−1. Here, those modes are predict at different wavenumbers, hence the IR bands at 613 and 531 cm−1 are associated with the g(COO) modes while the IR bands at 631 and 504 cm−1 are assigned to the d(COO) modes, as observed in Table 2. The rocking modes of COO groups are predicted between 439 and 335 cm−1, as in similar compounds [30,33,34], hence, they are assigned to the IR bands at 431 and 356 cm−1. The SQM clearly predict the twisting modes between 87 and 25 cm−1, accordingly, these modes are associated to the very strong and very weak Raman bands at 78 and 20 cm−1, respectively.
4.4.1.5. Skeletal Modes Here, the skeletal stretching modes in the three CQA’s conformers are predicted strongly mixed among them (Tables S8-S10). In agreement to the values reported for similar molecules [30-34] and our theoretical results, the IR bands of the media intensities at 1626, 1608 and 1585 cm−1 are mainly associated to a C=C stretchings. Also, the IR bands at 1368, 1324, 1202, 1133, 1107, 1042, 913 and 830 cm−1, and the shoulders in the same spectrum at 1395, 1238 and 836 cm−1 are associated to the C-C stretching modes, as observed in Table 2. The strong band and the shoulders in the IR spectrum respectively at 1254, 1395 and 1251 cm−1 are associated to the C-N stretching modes. A very important observation in this compound is related to the C-Cl stretchings mode because for the CI conformer is predicting at higher wavenumber than the other ones. Thus, the IR band at 813 cm−1 is assigned to that mode for the CI conformer while the very weak Raman band at 386 cm−1 are assigned to those modes for the remains conformers. The six phenyl ring deformations and torsions modes for the three CQA’s conformers, as the calculation predicts and taking into account the assignments for similar molecules [30-34], are assigned in the expected regions, as observed in Table 2. Finally, the butterfly modes are predicted by the calculations between 188 and 168 cm−1, for this, they are assigned to the weak Raman band at 173 cm−1, as observed in Table 2.
4.5. Force Field
The calculated forces constants for the three CQA’s conformers are given in Table 3 and they were calculated by means of the scaling procedure of Pulay et al. [7,8] with the MOLVIB program [19,20]. The C=O stretching force constants values for the CII and CIII conformers (14.16 and 14.10 mdyn·Å−1, respectively) are higher than the corresponding to the CI structure (13.62 mdyn·Å−1) because the calculated C=O distances in those conformers are lower (1.202 and 1.203 Å, respectively) than the corresponding to CI conformer (1.210 Å). Also, these same reasons justify the higher f(C-O) force constant value in the CI structure (6.33 mdyn·Å−1) in relation to the others (see Table 1). The formation of a H bond (OH-O) in the CI structure through the OH group and the lower C-O-H angle value in this structure (112.4˚) justifies the lower f(nOH) force constant value in this structure (6.11 mdyn·Å−1) in reference to the others. The differences between the f(O=C=O), f(H-C-H) and f(C-O-H) force constant values for the CI structure in relation to the others are also attributed to the geometrical parameters. On the other hand, the analysis of the force constants for all conformers of this compound with the values for 2-(quinolin-8-yloxy) acetic acid [6] show that the values
Table 3. Comparison of scaled internal force constants for the three conformers of (5-chloro-quinolin-8-yloxy) acetic acid.
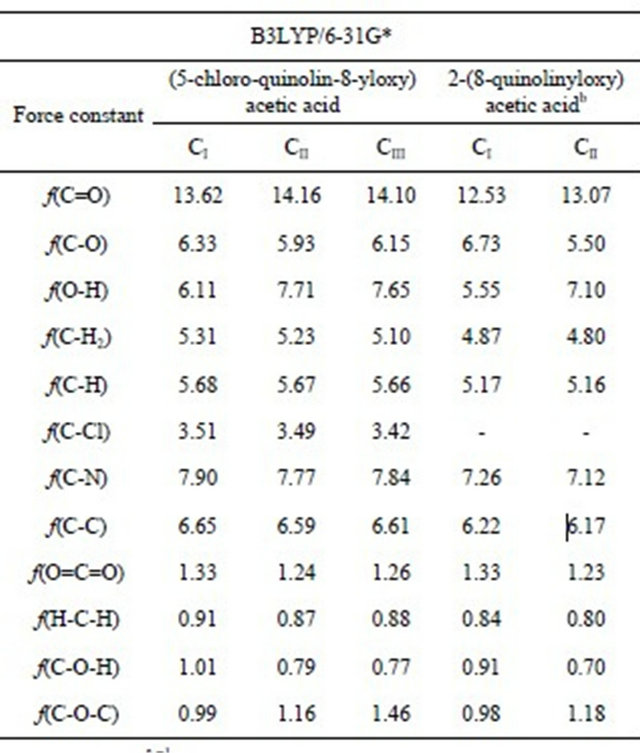
Units are mdyn·Å−1 for stretching and stretching/stretching interaction and mdyn·Å·rad−2 for angle deformations; aThis work; bAnhydrous from Ref [6].
are higher in CQA, with exeption of the f(C-O) force constant. An explanation can be probably due to that the chloro atom increase the topological properties of the RCPs in the CI conformer and as conesquence decrease the O-H distance increasing the C-O distance in this conformer in relation to the 2-(quinolin-8-yloxy) acetic acid [6].
4.6. HOMO-LUMO Energy Gap
The frontier molecular HOMO and LUMO orbitals were calculated for 2-(quinolin-8-yloxy) acetic acid and compared with the corresponding values for the (5-chloroquinolin-8-yloxy) acetic acid [37], as observed in Table S11. The results show clearly that both orbitals are mainly localized on the rings, indicating that the HOMOLUMO are mostly the π-antibonding type orbitals and that the values of the energy separation between those orbitals are higher in the 2-(quinolin-8-yloxy) acetic acid than in CQA. This large HOMO-LUMO gap for the 2- (quinolin-8-yloxy) acetic acid automatically means high excitation energies for many excited states, a good stability and a high chemical hardness. For these reasons, the presence of a Cl atom in CQA increases the reactivity compared to the 2-(quinolin-8-yloxy) acetic acid.
4.7. NMR Analysis
Tables 4, 5 show a comparison between the experimental and calculated chemical shifts for the 1H and 13C nuclei, respectively. The calculated chemical shifts for the H nuclei show a reasonable agreement in relation to experimental values with observed RMSD values between 0.578 and 0.174 ppm, while the chemical shifts for the carbon nuclei show higher RMSD values (7.325 and 0.254 ppm). The calculated 1H chemical and 13C shifts show a good concordance for the three conformers when the 6-311++G** basis set is used. Thus, Table 4 shows that the calculated 13C chemical shifts with the GIAO method using the 6-311++G** basis set are in accordance with the experimental values. In general, the calculated shifts for the 13C nuclei are higher than the corresponding experimental values.
5. Conclusion
The (5-chloro-quinolin-8-yloxy) acetic acid was synthesized and characterized by infrared, Raman and NMR spectroscopic techniques. The presence of CI, CII and CIII conformers was detected in both spectra, and a complete assignment of the vibrational modes was accomplished. The B3LYP/6-31G* and B3LYP/6-311++G** calculations suggest the existence of three conformers for CQA in the gas phase and, probably, the three are present in the solid state. An SQM/B3LYP/6-31G* force field was obtained for the three structures of CQA after adjusting the force constants obtained theoretically to minimize the difference between the observed and calculated wavenumbers. Also, the principal force constants for the stretching and deformation modes of CQA were determined. The NBO and AIM analysis confirm the O-H and N-H bonds in the three conformers of CQA while the HOMO-LUMO study shows that the Cl atom increases the reactivity of CQA, as compared with 2-(quinolin-8-yloxy) acetic acid.
6. Acknowledgements
This work was supported with grants from CIUNT (Consejo de Investigaciones, Universidad Nacional de Tucumán) and CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas, R. Argentina). The authors thank Prof. Tom Sundius for his permission to use MOLVIB and the Dr. Jorge Güida for the Raman spectrum.
REFERENCES
- S. Y. Cho, J. H. Ahn, J. D. Ha, S. K. Kang, J. Y. Baek, S. S. Han, E. Y. Shin, S. S. Kim, K. R. Kim, H. G. Cheon and J. K. Choi, “Protein Tyrosine Phosphatase 1B Inhibitors: Heterocyclic Carboxylic Acids,” Bulletin of the Korean Chemical Society, Vol. 24, No. 10, 2003, pp. 1455-1464. doi:10.5012/bkcs.2003.24.10.1455
- B. P. Kennedy and C. Ramachandran, “Protein Tyrosine Phosphatase-1B in Diabetes,” Biochemical Pharmacology, Vol. 60, No. 7, 2000, pp. 877-883. doi:10.1016/S0006-2952(00)00305-1
- N. Moller, L. Iversen, H. Andersen, J. McCormack, “Protein Tyrosine Phosphatases (PTPs) as Drug Targets: Inhibitors of PTP-1 for the Treatment of Diabetes,” Current Opinion in Drug Discovery & Development, Vol. 3, No. 5, 2000, pp. 527-540.
- A. Hubele, “Use of Quinoline Derivatives for the Protection of Cultivated Plants from Herbicides,”US Patent No. 4902340, 1990.
- F. Hayat, A. Salahuddin, J. Zargan and A. Azam, “Synthesis, Characterization, Antiamoebic Activity and Cytotoxicity of Novel 2-(Quinolin-8-Yloxy) Acetohydrazones and Their Cyclized Products (1,2,3-Thiadiazole and 1,2,3-Selenadiazole Derivatives),” European Journal of Medicinal Chemistry, Vol. 45, No. 12, 2010, pp. 6127-6134. doi:10.1016/j.ejmech.2010.09.066
- G. R. Argañaraz, E. Romano, J. Zinczuk and S. A. Brandán, “Structural and Vibrational Study of 2-(Quinolin- 8-yloxy)-Acetic Acid Based on FT-IR-Raman Spectroscopy and DFT Calculations,” Journal of Chemistry and Chemical Engineering, Vol. 5, No. 8, 2011, pp. 747-758.
- P. Pulay, G. Fogarasi, F. Pang and E. J. Boggs, “Systematic ab Initio Gradient Calculation of Molecular Geometries, Force Constants, and Dipole Moment Derivatives,” Journal of the American Chemical Society, Vol. 101, No. 10, 1979, pp. 2550-2560. doi:10.1021/ja00504a009
- P. Pulay, G. Fogarasi, G. Pongor, J. E. Boggs and A. Vargha, “Combination of Theoretical ab Initio and Experimental Information to Obtain Reliable Harmonic Force Constants. Scaled Quantum Mechanical (QM) Force Fields for Glyoxal, Acrolein, Butadiene, Formaldehyde, and Ethylene,” Journal of the American Chemical Society, Vol. 105, No. 24, 1983, pp. 7037-7047. doi:10.1021/ja00362a005
- G. Rauhut and P. Pulay, “Transferable Scaling Factors for Density Functional Derived Vibrational Force Fields,” The Journal of Physical Chemistry, Vol. 99, No. 39, 1995, pp. 3093-3100. doi:10.1021/j100039a056
- A. E. Reed, L. A. Curtis and F. Weinhold, “Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint,” Chemical Review, Vol. 88, No. 6, 1988, pp. 899-926. doi:10.1021/cr00088a005
- J. P. Foster and F. J. Weinhold, “Natural Hybrid Orbitals,” Journal of the American Chemical Society, Vol. 102, No. 24, 1980, pp. 7211-7218. doi:10.1021/ja00544a007
- A. E. Reed and F. Weinhold, “Natural Localized Molecular Orbitals,” Chemical Physics, Vol. 83, No. 4, 1985, pp. 1736-1740.
- R. F. W. Bader, “Atoms in Molecules: A Quantum Theory,” Oxford University Press, Oxford, 1990.
- F. Sun, L. Chen, H. C. Fang, X. M. Lin and Y. P. Cai, “8-(Carboxymethoxy)Quinolinium Nitrate Monohydrate,” Acta Crystallographica, Vol. 64, 2008, p. 1641.
- F. Biegler-Köning, J. Schönbohm and D. Bayles, “AIM 2000—A Program to Analyze and Visualize Atoms in Molecules,” Journal of Computational Chemistry, Vol. 22, No. 5, 2001, pp. 545-559.
- E. D. Glendening, J. K. Badenhoop, A. D. Reed, J. E. Carpenter and F. Weinhold, “Natural Bond Orbital: NBO 3.1,” University of Wisconsin, Madison, 1996.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, “Gaussian 03, Revision B.01,” Gaussian, Inc., Pittsburgh, 2003.
- G. Fogarasi, X. Zhou, P. Taylor and P. Pulay, “The Calculation of Ab Initio Molecular Geometries: Efficient Optimization by Natural Internal Coordinates and Empirical Correction by Offset Forces,” Journal of the American Chemical Society, Vol. 114, No. 21, 1992, pp. 8191-8201. doi:10.1021/ja00047a032
- T. Sundius, “Molvib—A Flexible Program for Force Field Calculations,” Journal of Molecular Structure, Vol. 218, No.1-2, 1990, pp. 321-326. doi:10.1016/0022-2860(90)80287-T
- T. Sundius, “Scaling of Ab Initio Force Fields by MOLVIB,” Vibrational Spectroscopy, Vol. 29, No. 1-2, 2002, pp. 89-95. doi:10.1016/S0924-2031(01)00189-8
- F. Kalincsák and G. Pongor, “Extension of the Density Functional Derived Scaled Quantum Mechanical Force Field Procedure,” Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 58, No. 5, 2002, pp. 999-1011. doi:10.1016/S1386-1425(01)00572-8
- A. B. Nielsen and A. J. Holder, “Gauss View, User’s Reference,” GAUSSIAN, Inc., Pittsburgh, 1997-1998.
- A. E. Ledesma, S. A. Brandán, J. Zinczuk, O. Piro, J. J. L. González, A. B. Altabef, “Structural and Vibrational Study of 2-(2’-Furyl)-1H-Imidazole,” Journal of Physical Organic Chemistry, Vol. 21, No. 12, 2008, pp. 1086-1097. doi:10.1002/poc.1449
- E. Romano, A. B. Raschi, A. Benavente and S. A. Brandán, “Structural Analysis, Vibrational Spectra and Coordinated Normal of 2R-(−)-6-Hydroxytremetone,” Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 84, No. 1, 2011, pp. 111-116. doi:10.1016/j.saa.2011.09.011
- S. A. Brandán, G. Benzal, et al., “Theoretical and Experimental Atudy of the Vibrational Apectra of 1,5-Simethylcytosine,” Vibrational Spectroscopy, Vol. 46, No. 2, 2008, pp. 89-99. doi:10.1016/j.vibspec.2007.11.001
- A. E. Ledesma, J. Zinczuk, A. B. Altabef, J. J. López-González and S. A. Brandán, “Synthesis and Vibrational Analysis of N-(2’-Furyl)-Imidazole,” Journal of Raman Spectroscopy, Vol. 40, No. 8, 2009, pp. 1004- 1010.
- J. Zinczuk, A. E. Ledesma, S. A. Brandán, O. E. Piro, J. J. López-González, A. B. Altabef, “Structural and Vibrational Study of 2-(2’-Furyl)-4,5-1H-Dihydroimidazole,” Journal of Physical Organic Chemistry, Vol. 22, No. 12, pp. 1166-1167. doi:10.1002/poc.1572
- S. A. Brandán, E. Erog˘lu, A. E. Ledesma, O. Oltulu and O. B. Yalçınkaya, “A New Vibrational Study of Acetazolamide Compound Based on Normal Coordinate Analysis and DFT Calculations,” Journal of Molecular Structure, Vol. 993, No. 1-3, 2011, pp. 225-231. doi:10.1016/j.molstruc.2010.11.012
- L. C. Bichara, H. E. Lanús, C. G. Nieto and S. A. Brandán, “Density Functional Theory Calculations of the Molecular Force Field of l-Ascorbic Acid, Vitamin C,” The Journal of Physical Chemistry A, Vol. 114, No. 14, 2010, pp. 4997-5004. doi:10.1021/jp912251g
- S. A. Brandán, F. M. López and M. Montejo, J. J. L. González, A. B. Altabef, “Theoretical and Experimental Vibrational Spectrum Study of 4-Hydroxybenzoic Acid as Monomer and Dimer,” Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 75, No. 5, 2010, pp. 1422-1434. doi:10.1016/j.saa.2010.01.012
- A. E. Ledesma, C. Contreras, J. Svoboda, A. Vektariane and S. A. Brandán, “Theoretical Structures and Experimental Vibrational Spectra of Isomeric Benzofused thieno [3,2-b] Furan Compounds,” Journal of Molecular Structure, Vol. 967, No. 1-3, 2010, pp. 159-165. doi:10.1016/j.molstruc.2009.12.050
- C. D. Contreras and M. Montejo, J. J. L. González, J. Zinczuk and S. A. Brandán, “Structural and Vibrational Analyses of 2-(2-Benzofuranyl)-2-Imidazoline,” Journal of Raman Spectroscopy, Vol. 42, No. 1, 2011, pp. 108-116. doi:10.1002/jrs.2659
- J. Antony, G. V. Helden, G. Meijer and B. Schmidt, “Anharmonic Midinfrared Vibrational Spectra of Benzoic Acid Monomer and Dimer,” Journal of Chemical Physics, Vol. 123, No. 1, 2005, pp. 14305-14311. doi:10.1063/1.1947191
- C. D. Contreras, A. E. Ledesma, H. E. Lanús, J. Zinczuck and S. A. Brandán, “Hydration of L-Tyrosine in Aqueous Medium. An Experimental and Theoretical Study by Mixed Quantum Mechanical/Molecular Mechanics Methods,” Vibrational Spectroscopyc, Vol. 57, No. 1, 2011, pp. 108- 115.
- S. Ghose and J. K. Dattagupta, “Crystal Structure of Tolazoline Hydrochloride (Priscoline), an α-Adrenergic Antagonist,” Journal of the Chemical Society, Perkin Transactions 2, Vol. 200, No. 1-2, 1989, pp. 599-601.
- C. D. Contreras, A. E. Ledesma, J. Zinczuk and S. A. Brandán, “Vibrational Study of Tolazoline Hydrochloride by Using FTIR-Raman and DFT Calculations,” Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 79, No. 5, 2011, pp. 1710-1714. doi:10.1016/j.saa.2011.05.041
- E. Romano, M. V. Castillo, J. L. Pergomet, J. Zinczuk and S. A. Brandán, “Synthesis and Structural and Vibrational Analysis of (5,7-Dichloro-Quinolin-8-Yloxy) Acetic Acid,” Journal of Molecular Structure, Vol. 1018, No. 27, 2012, pp. 149-155. doi:10.1016/j.molstruc.2012.03.013
Supplement
Table S1. Definition of natural internal coordinates for (5-chloro-quinolin-8-yloxy) acetic acid compound.
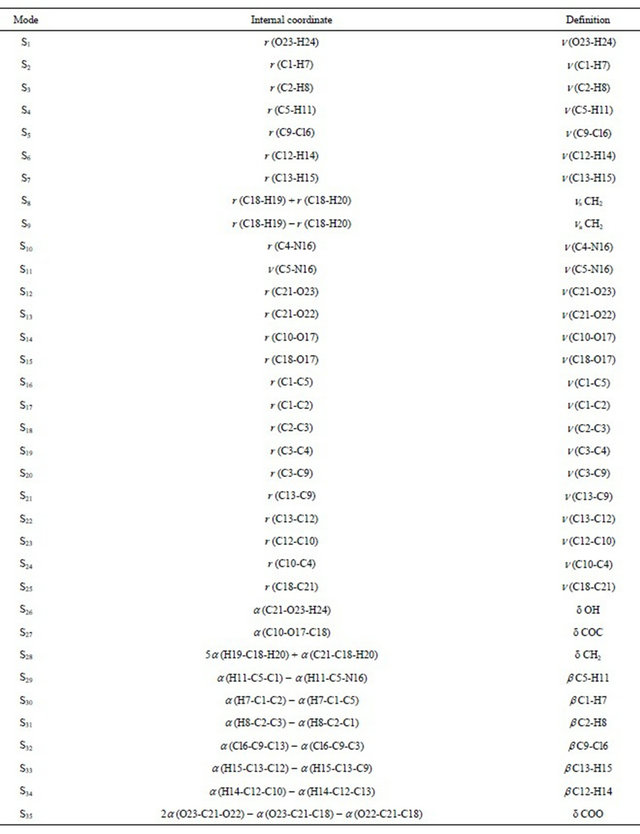 Continued
Continued

Abbreviations:n, stretching; b, deformation in the plane; g, deformation out of plane; wag, wagging; t , torsion; bR, deformation ring; tR, torsion ring; r, rocking; twis, twisting; a, angular deformation; d, deformation; a, antisymmetric; s, symmetric; A1, Ring 1; A2, Ring 2.
Table S2. Calculated total energy (E) and dipolar moments for the structures of (5-chloro-quinolin-8-yloxy) acetic acid at different theory levels.

Table S3. Atomic charges (NPA) for the conformers of (5-chloro-quinolin-8-yloxy) acetic acid at different theory levels.
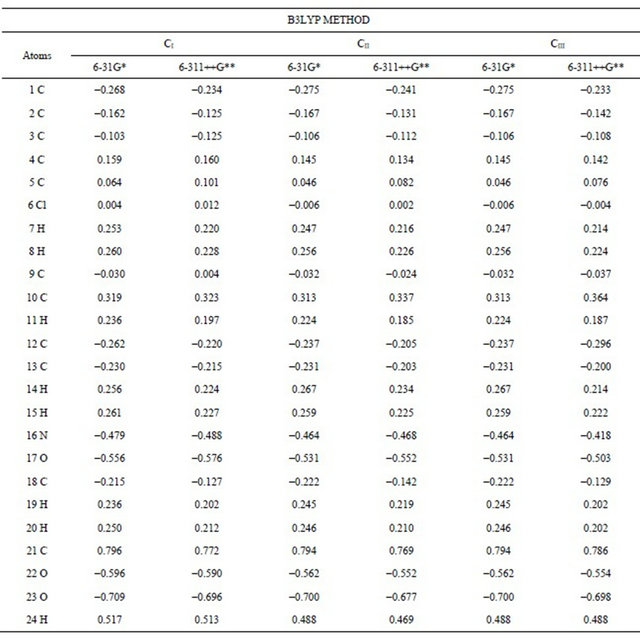 Table S4. Wiberg index bond for the conformers of (5-chloro-quinolin-8-yloxy) acetic acid at different theory levels.
Table S4. Wiberg index bond for the conformers of (5-chloro-quinolin-8-yloxy) acetic acid at different theory levels.
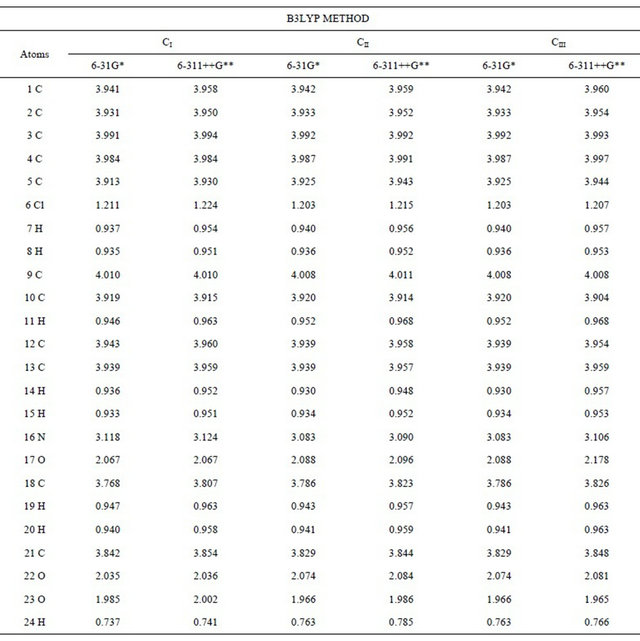
Table S5. Main delocalization energy (in kJ/mol) for the CI conformer of (5-chloro-quinolin-8-yloxy) acetic acid at different theory levels.
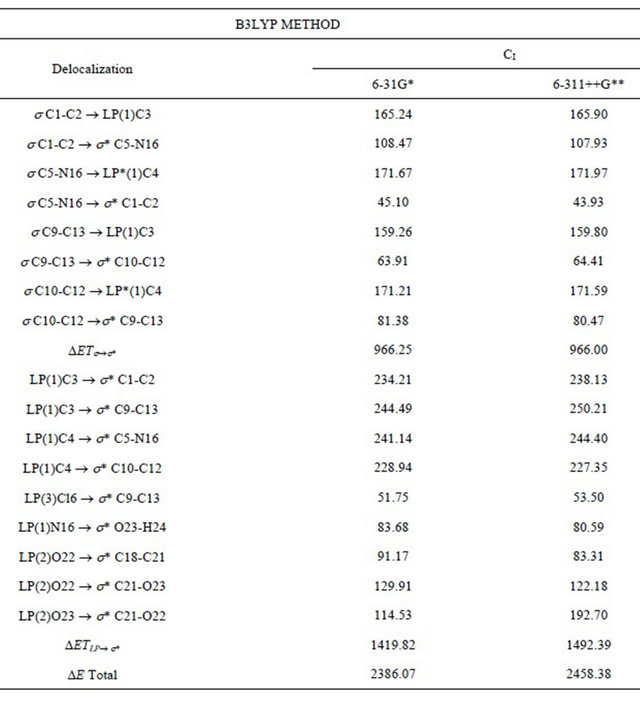
Table S6. Main delocalization energy (in kJ/mol) for the CII and CIII conformers of (5-chloro-quinolin-8-yloxy) acetic acid at different theory levels.
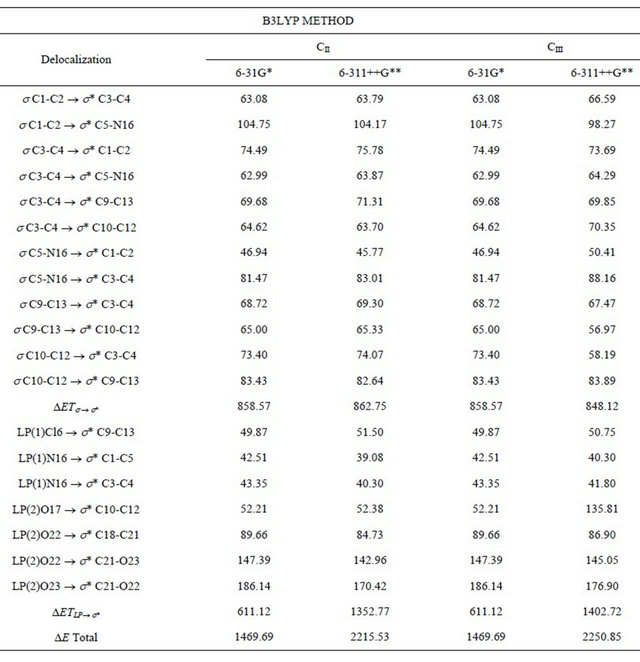
Table S7. An analysis of the bond and ring critical points (BCP, RCP) for the conformers of (5-chloro-quinolin-8-yloxy) acetic acid at different theory levels.
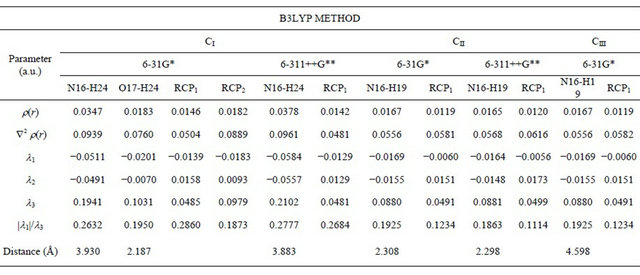
Table S8. Observed and calculated wavenumbers (cm−1), potential energy distribution and assignment for the CI conformer of (5-chloro-quinolin-8-yloxy) acetic acid.
Continued
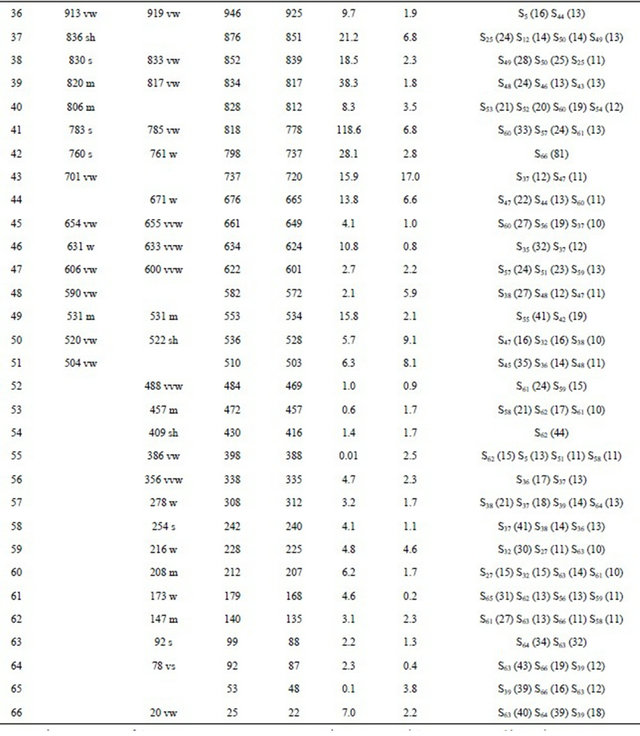
aThis work; bDFT B3LYP/6-31G*; cFrom scaled quantum mechanics force field; dUnits are km∙mol−1; eRaman activities in Å4 (amu)−1.
Table S9. Observed and calculated wavenumbers (cm−1), potential energy distribution and assignment for the CII conformer of (5-chloro-quinolin-8-yloxy) acetic acid.

Continued
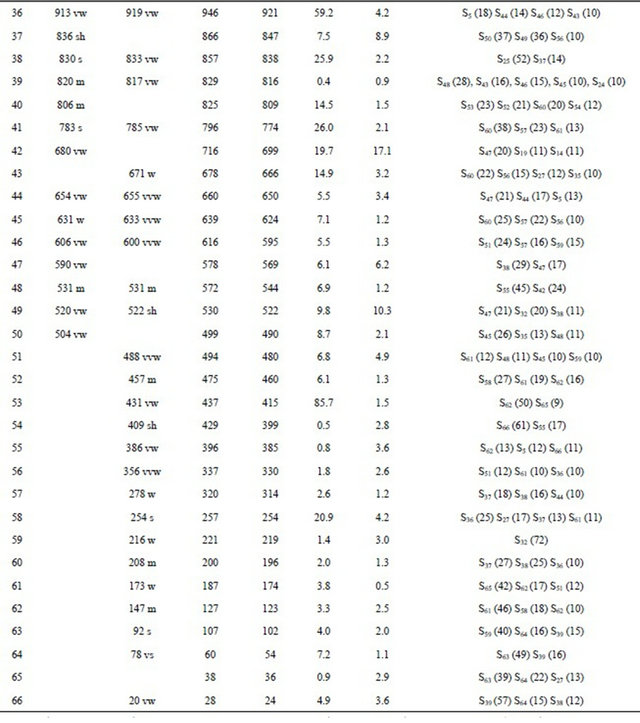
aThis work; bDFT B3LYP/6-31G*; cFrom scaled quantum mechanics force field; dUnits are km∙mol−1; eRaman activities in Å4 (amu) −1.
Table S10. Observed and calculated wavenumbers (cm−1), potential energy distribution and assignment for the CIIÏ con-former of (5-chloro-quinolin-8-yloxy) acetic acid.

Continued
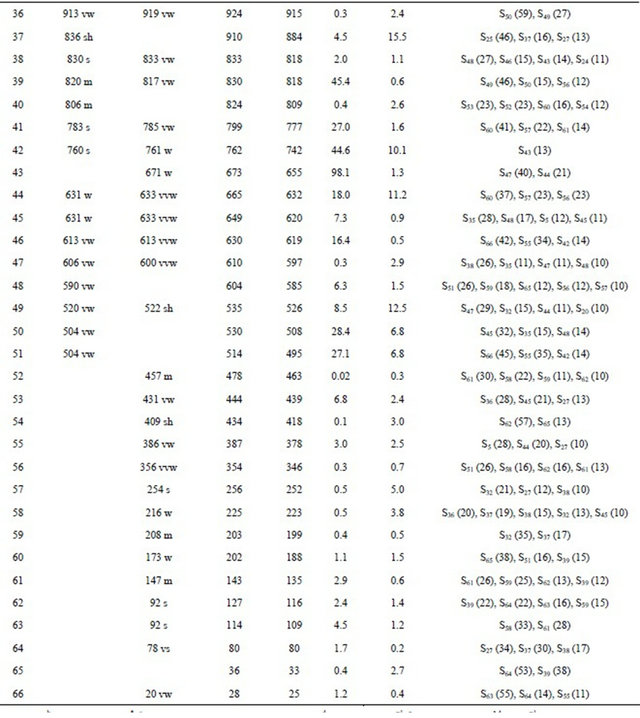
aThis work; bDFT B3LYP/6-31G*; cFrom scaled quantum mechanics force field; dUnits are km·mol−1; eRaman activities in Å4 (amu) −1.
Table S11. The frontier molecular HOMO and LUMO orbitals for both conformers of (5-chloro-quinolin-8-yloxy) acetic acid.
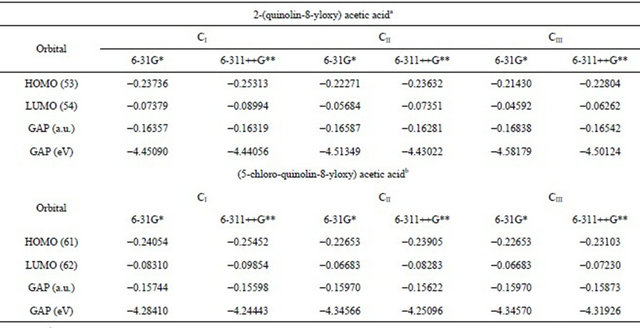
aThis work, bFrom Ref [37].
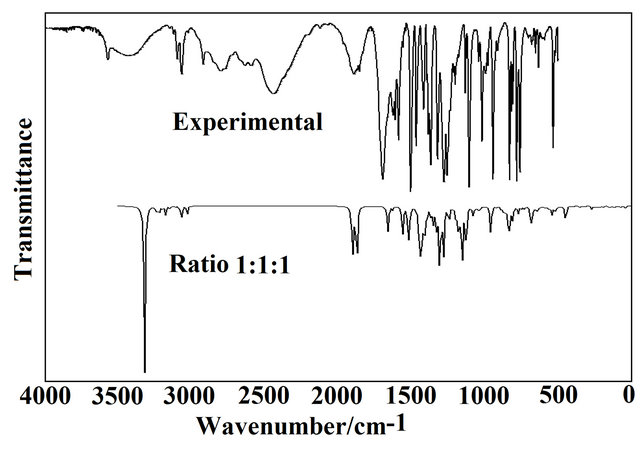
Figure S1.. Comparison between the infrared experimental spectrum of (5-chloro-quinolin-8-yloxy) acetic acid (upper) with the calculated infrared spectra (bottom) for CI, CII and CIII conformers from B3LYP/6-31G* wavenumbers and intensities using Lorentzian band shapes (for a population relation CI, CII and CIII of 1:1:1 for each conformer).
NOTES
*Corresponding author.