Advances in Breast Cancer Research
Vol.3 No.3(2014), Article
ID:47265,11
pages
DOI:10.4236/abcr.2014.33010
GP88 (Progranulin) Confers Fulvestrant (Faslodex, ICI 182,780) Resistance to Human Breast Cancer Cells
Wisit Tangkeangsirisin1,2, Ginette Serrero1,3*
1A&G Pharmaceutical Inc., Columbia, USA
2Present Address: Department of Biopharmacy, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
3Program in Oncology, Greenebaum Cancer Center of the University of Maryland, Baltimore, USA
Email: *gserrero@agpharma.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 6 April 2014; revised 3 May 2014; accepted 1 June 2014
ABSTRACT
The 88 kDa glycoprotein known as GP88, Progranulin or PC cell derived growth factor is an autocrine growth factor with a unique cysteine rich motif that is over expressed in breast cancer whereas it is negative in normal mammary epithelial cells. It has been shown to play a major role in estrogen independence, tamoxifen resistance and tumorigenesis of breast cancer cells. In the present study, we investigated the effect of GP88 overexpression on the response of the human breast cancer MCF-7 cells to the pure estrogen receptor antagonist fulvestrant (ICI 182,780). While fulvestrant effectively inhibited cell proliferation of empty vector transfected cells, it had no inhibitory effect on the proliferation of GP88 overexpressing breast cancer cells. Mouse xenograft experiments in athymic ovariectomized nude mice showed that GP88 over expressing cells were fulvestrant resistant in vivo in contrast to low GP88 expressing cells. We show that the ability of fulvestrant to induce apoptosis determined by measuring cleavage of poly (ADP-ribose) polymerase was inhibited by GP88. Anti-apoptotic activity of GP88 was associated with sustained expression of bcl-2 and bcl-xL after fulvestrant treatment. In contrast, fulvestrant was still able to inhibit the ability of estrogen to stimulate ERE-luciferase reporter gene activity as well as vEGF expression in GP88 over expressing MCF-7 cells similarly to control MCF-7 cells. Collectively, our data suggest that GP88 prevents apoptosis induced by faslodex and contributes to antiestrogen resistance in human breast cancer.
Keywords
Progranulin (GP88), Fulvestrant, Faslodex, breast cancer, anti-estrogen resistance

1. Introduction
Anti-estrogen therapy is the treatment of choice in all stages of estrogen receptor positive (ER+) breast cancer [1] . These agents target either estrogen binding, interaction with its receptor or estrogen synthesis. They include Selective Estrogen Receptor Modulator (SERM) such as tamoxifen and its derivatives, Selective Estrogen Receptor down-regulators (SERD) such as faslodex (fulvestrant or ICI 182, 780) and aromatase inhibitors such as letrozole or anastrozole. Tamoxifen is the first developed antiestrogen that acts by inhibiting the binding of estrogen to its receptor [2] . It has been the most commonly prescribed drug for breast cancer, and is currently used for the treatment of advanced disease as well as adjuvant therapy after surgery for early breast cancer [3] . Fulvestrant (faslodex, ICI 182,780) is a steroidal estrogen receptor (ER) pure antagonist that acts by binding with high affinity to the ER and down-regulating ER expression [4] [5] . It was developed as an alternative anti-estrogen therapy due to the unfavorable agonistic effect of tamoxifen in the endometrium [6] . Unlike tamoxifen, the conformation fulvestrant-ER complex inhibits the activation function of ER, disrupts dimerization and nuclear translocation of the complex and results in ER degradation. Fulvestrant is well-tolerated and produces good response rates when used in patients who develop tamoxifen resistance. In fact, clinical studies have suggested fulvestrant as a second line antiestrogen [7] [8] . Although these agents are effective and show promising clinical results, resistance to these agents can also be observed after a period of treatment [9] [10] . More than half of the patients, who failed previous treatment with tamoxifen, still do not respond to fulvestrant [8] [11] . There have been several mechanisms of action to explain anti-estrogen resistance. Among them, one possible mechanism of anti-estrogen therapy resistance in ER+ tumors [3] [12] has been the constitutive overexpression of autocrine growth/survival factors, and/or the upregulation of growth factor receptor by tumor cells [13] [14] . Such increased autocrine or paracrine growth factor signaling network may bypass the need for ER-mediated growthstimulation in human breast cancer cells, thus rendering anti-estrogen therapy ineffective. For example, clinical studies have reported a decreased efficacy of tamoxifen for tumors overexpressing c-erbB2 (6) and EGFR [15] -[18] . In addition to inhibiting the growth promoting effect of estrogen, anti-estrogen have also been shown to induce programmed-cell death in breast cancer cell lines and in clinical samples [19] -[24] . Failure to undergo apoptosis in response to anti-estrogen would also confer drug resistance [25] Therefore, increase in growth factor signaling that mediates both proliferation signals and anti-apoptotic signals may induce resistance to anti-estrogen therapy.
GP88 is the 88 kDa cysteine-rich glycoprotein autocrine growth factor originally purified from the highly tumorigenic mouse teratoma PC cells by applying a biological screen to mine for drivers of tumorigenesis [26] . GP88, (also known as granulin/epithelin precursor, progranulin, acrogranin or PC-cell derived growth factor) is the largest member of the granulin-epithelin family of growth modulators characterized by 71/2 granulin repeats containing a unique double cysteine rich motif [27] . This protein has been found to have pleiotropic functions in normal and diseased human tissues. In particular, GP88 has been found to be overexpressed in many cancers whereas it is not expressed in the normal tissue counterparts [26] -[28] . GP88 stimulates proliferation, survival and metastasis in several cancer cell types via activation of multiple pathways that include MAP kinase and P-I-3 kinase pathways, FAK kinase [27] . In human breast cancer cells, GP88 expression was stimulated by estradiol in a timeand dose-dependent fashion in estrogen receptor positive cells [29] . In these cells, GP88 was shown to mediate the mitogenic activity of estrogen by stimulating cyclin D1 expression [29] . Inhibition of GP88 expression in estrogen receptor negative MDA-MB-468 cells by antisense transfection led to a complete inhibition of tumorigenesis in nude mice [30] .
Overexpression of the autocrine growth factor GP88 in ER+ breast cancer cells lead to tamoxifen resistance in vitro and in vivo while the cells remained ER+ [31] . In addition, we have shown that GP88 confers resistance to the aromatase inhibitor letrozole in aromatase overexpressing cells MCF7-CA and AC1 cells while estrogen receptor expression remained unchanged [32] . Naturally letrozole resistant cells LTLT and AC1LTR cells overexpressed GP88 whereas inhibition of GP88 by SiRNA restored letrozole responsiveness [32] . Pathological studies with paraffin embedded breast cancer biopsies have shown positive GP88 tissue expression in 60% of ductal carcinoma in situ (DCIS) and 80% of invasive ductal carcinoma (IDC) whereas normal mammary epithelium and benign tumors were GP88 negative [33] In IDC, GP88 expression correlated with parameters of poor prognosis such as tumor grade, proliferation index and p53 expression. Recent studies investigating the correlations of GP88 tumor tissue expression in ER+ IDC with clinical outcomes showed that patients with high GP88 tumor expression (GP88 3+) was associated with a 4-fold increase in recurrence and mortality when compared to patients with low or no GP88 tissue expression [34] . Increased levels of circulating GP88 were found in breast cancer patients when compared to healthy subjects [35] .
Because of the role of GP88 on breast cancer cells and since GP88 confers resistance to a SERM (tamoxifen) and to an aromatase inhibitor (letrozole), it would be interesting to examine the effect of GP88 on anti-estrogen compound with a different mode of action such as the SERD faslodex. The present study focused on investigating the effect of GP88 on the faslodex responsiveness of breast cancer cells in vitro and in vivo.
2. Material and Methods
2.1. Materials
17b-Estradiol (E2) was purchased from Calbiochem (San Diego, CA). G418, Taq polymerase and Superscript II were obtained from Gibco BRL (Gaithersburg, MD). Fulvestrant was purchased from Tocris (Ellisville, MO). Oligonucleotide primers used in the RT-PCR were synthesized by the Biopolymer Core Laboratory of the University of Maryland (Baltimore, MD). Placebo and fulvestrant time-release pellets were manufactured at Innovative Research of America (Sarasota, FL). Enhanced chemiluminescence kit was obtained from Pierce (Rockford, IL). Mouse anti-poly (ADP-ribose) polymerase antibody (anti-PARP) was purchased from Oncogene Research (Boston, MA).
2.2. Cell Lines
MCF-7 cell line was originally obtained from the American Type Culture Collection (Manassas, VA). GP88 overexpressing MCF-7 cells and empty vector MCF-7 control cells were developed in our laboratory as previously described [30] [31] . These cells were cultured in DMEM/F12 supplemented with 5% FBS and 50 mg/ml gentamicin (Sigma) and 400 mg/ml G418 (Gibco).
2.3. Proliferation Assay
Proliferation assay was performed in 6-well tissue culture plates (Costar, Cambridge, MA). 5 × 104 cells were plated in phenol red-free a-MEM (PFMEM) supplemented with 5% charcoal extracted fetal bovine serum (CHX-FBS). Cells were treated with either 1 nM E2 alone or in combination with 10 nM fulvestrant. Control cells were treated with vehicle alone (0.01% DMSO). Medium was changed at day 4. Cell numbers were determined with a hemocytometer. Each time point was performed in triplicate.
2.4. In Vivo Tumorigenesis Assay
All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore. Control and GP88 overexpressing MCF-7 cells (5 × 106 cells per site) were injected subcutaneously in two sites into six-week-old ovariectomized athymic female nude mice (National Cancer Institute, Frederick, MD). E2 pellets (1.7 mg, 60 day release) were implanted subcutaneously in the back, one day before inoculating the cells. The animals received fulvestrant pellets (2.5 mg, 60-day release) or placebo pellets (Innovative Research of America, Sarasota, FL), ten days after the cell inoculation, when the tumors were visible. The width (W) and length (L) of individual tumors were measured using a caliper. Average tumor volume was calculated with the widely used formula: Tumor volume = (W2 × L) × 0.5.
2.5. Determination of mRNA Expression for bcl-2, bcl-xL, bax and VEGF by RT-PCR
Total RNA was isolated with Trizol reagent (Gibco) and was reverse transcribed into single strand cDNA by Superscript II (Gibco). A total of 30 - 35 PCR cycles depending on the gene amplified was performed, followed by electrophoresis on 1% agarose gel. Specific primers for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for bcl-2 and for V-EGF have been previously described [31] [36] .Other specific primers used here were:
for bax: forward primer 5’ GAGCAGATCATGAAGACAGGGG 3’, reverse primer 5’ CTCCAGCAAGGCCCAGCGTC 3’;
for bcl-xL: forward primer 5’ CAGTGAGTGAGCAGGTGTTTTGG 3’, reverse primer 5’ GTTCCACAAAAGTATCCCAGCCG 3’;
2.6. Western Blot Analysis of PARP Cleavage
Cells were seeded at a density of 7 × 105 cells in 60-mm dish in DMEM/F12 supplemented with 5% FBS. After 24 hours, medium was changed to serum-free phenol red-free DMEM/F12 supplemented with vehicle or purified GP88 (400 ng/ml) for another 24 hours. Cells were treated with either vehicle only or factors under investigation for 24 hours. Cell lysates were prepared as described before [31] . 100 mg of protein from eachsample was used for immunoblotting. Intact and cleaved forms of PARP were detected using a mouse monoclonal anti-PARP antibody. The band intensity of PARP cleaved form was determined by densitometric analysis and normalized to the b-actin.
2.7. Statistical Analysis
All experiments were conducted in triplicates and repeated at least twice. Data were analyzed by Student t test for mean comparison and statistical significance. The values are reported as mean ± standard error.
3. Results
3.1. GP88 Overexpression Prevent Growth Inhibitory Effect of Fulvestrant
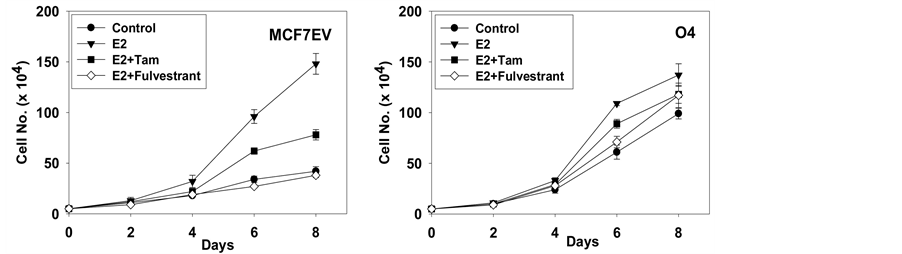
The effect of increasing concentrations of fulvestrant on the proliferation of MCF-7 EV cells and GP88 over expressing cells was first examined by thymidine incorporation assay. Fulvestrant inhibited proliferation of control MCF-7 EV cells in a dose-dependent fashion. Concentrations as low as 0.2 nM, inhibited by 90% thymidine incorporation in MCF-7 EV cells. At similar dose, thymidine incorporation into DNA of GP88 overexpressing cells had been inhibited by only 20%. Even at concentration of 2 nM of fulvestrant, inhibition did not exceed 40% (figure 1(a)). This dose had no effect on the DNA synthesis of GP88 overexpressing cells. Long-term proliferation assay showed that fulvestrant inhibited the proliferative effect of estradiol (E2) in MCF-7 EV cells whereas the proliferation of GP88 overexpressing cells was mostly unchanged (figure 1(b)). As shown in Table 1, the mean doubling time of MCF-7 EV cells treated with E2 and fulvestrant was significantly higher than in cells treated with E2 alone (p < 0.05). On the other hand, the mean doubling time of GP88 overexpressing cells treated with fulvestrant and E2 was similar to the one of cells treated with E2 only, indicating that fulvestrant did not inhibit the growth of cells overexpressing GP88 (p < 0.05).
3.2. GP88 Overexpression Results in Fulvestrant Resistance in Vivo
Since fulvestrant failed to inhibit the proliferation of GP88 overexpressing cells in vitro, its effect on tumorigenesis of ER+ breast cancer cells expressing or not GP88 was then examined in mouse xenografts. Control and GP88 overexpressing MCF-7 cells were injected into female nude mice one day after they had been implanted with E2 pellets, as described in the method section. Ten days later, when the tumors were visible, the mice were randomized and segregated into experimental groups that received either placebo or fulvestrant pellets. The groups were then monitored for an additional 45 days. Tumor size was determined with a caliper and tumor volume determined as described in the method section. As shown in figure 2, fulvestrant significantly inhibited MCF-7 EV tumor growth as evidenced by a 35% inhibition of tumor incidence and a 70% inhibition of mean tumor volume when compared to mice treated with E2 only (p < 0.05), in agreement with the reported inhibitory effect of fulvestrant in MCF-7 tumor growth. In contrast, GP88 overexpressing cells formed larger tumors in mice with or without fulvestrant without any change in tumor incidence (100%) or tumor growth. Monitoring of tumor volume was performed over 45 days after start of treatment (figure 2). In the agreement with our previous experiments [31] , it is clear that GP88 overexpression in breast cancer cells promotes antiestrogen resistance.
3.3. Fulvestrant Inhibits Estrogen Responsive Element Activity in MCF-7EV and O4 Cells
It is well established that fulvestrant acts as a pure estrogen receptor antagonist by inhibiting the formation of E2-ER complexes and its subsequent binding to estrogen responsive element (ERE) within the promoter [7] . Since fulvestrant did not show growth inhibitory effect on GP88 overexpressing MCF-7 cells, we examined the effect of fulvestrant on ERE responsiveness to E2 in these cells when compared to control cells. As shown in figure 3, E2 stimulated ERE-luciferase activity in both MCF-7 EV and O4 cells by 4.4 and 3.9-fold, respectively when com pared to the untreated cells. Fulvestrant treatment resulted in a 95% inhibition of E2 mediated effect on ERE-luciferase in both cells (p < 0.05, compared to untreated groups). These data suggest that fulvestrant still acts as an E2 antagonist of in both cells. Therefore, GP88 overexpression in MCF-7 cells does not change the genomic

Table 1 . Effect of estrogen and fulvestrant on doubling time in breast cancer cell lines.
p < 0.05 compared to control group. The proliferation of Two GP88 overexpressing MCF-7 cell lines and the empty vector transfected control MCF-7 cell line was examined as described in the material and Methods section. Doubling times in each culture conditions were determined from the growth curves of figure 1.
 (a) (b)
(a) (b)
Figure 1. Effect of E2 and fulvestrant on Cell proliferation of MCF-7EV, and GP88 overexpressing cells O4 cells. Long-term growth of MCF-7EV and O4 cells in estrogen–depleted medium. Cells (5 × 104 cells) were cultivated in phenol red-free DMEM/F12 containing 5% charcoal-stripped FBS supplemented with vehicle (˜), with 1 nM17b-estradiol (E2) alone (™) or in combination with 1 mM tamoxifen (q) or 10 nM ICI 182,780 (s). Cell number was counted using hemocytometer. Experiments were performed in triplicates.
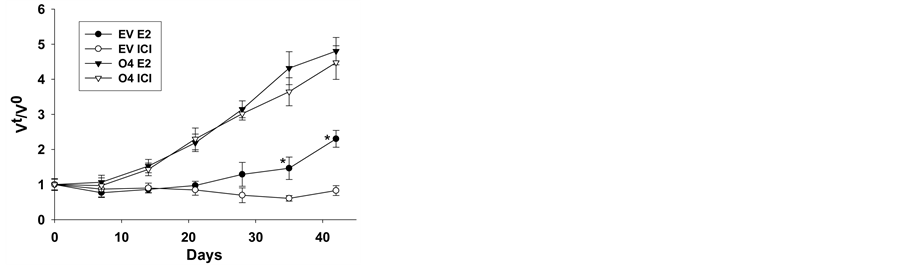
Figure 2. In vivo fulvestrant resistance of MCF-7 EV and O4 cells. Exponentially growing MCF-7 EV or O4 cells (5 × 106 cells/site) were subcutaneously inoculated into the lower abdominal area of ovariectomized athymic nude mice supplemented with E2 pellets. Ten days after cell inoculation, fulvestrant pellets were implanted. Tumor growth was monitored and tumor volume was calculated as described in the method section. Results shown here were plotted as the mean tumor volume at each time point (Vt) over the mean tumor volume at day fulvestrant pellet implanted (V0). Bar; SE.
response to fulvestrant.
3.4. GP88 Prevents Apoptosis Induced by Fulvestrant
It has been documented that fulvestrant, similarly to tamoxifen, induces apoptosis of MCF-7 cells [22] . We determined whether GP88 blocked the apoptotic effect of fulvestrant by using PARP cleavage assay. As shown in Figure 4, incubation with fulvestrant induced PARP cleavage in MCF-7 EV cells. GP88 treatment inhibited PARP cleavage induced by fulvestrant in MCF-7 EV cells (>80% inhibition suggesting that GP88 inhibited fulvestrant-induced apoptosis as one possible mechanism of fulvestrant resistance.
3.5. Failure of Fulvestrant to Downregulate Bcl-2 in GP88 Overexpressing Cells
Bcl-2 is a key regulator for apoptosis in many cell types. Previous reports have suggested that bcl-2 expression was downregulated by fulvestrant treatment leading to activation of apoptosis in MCF-7 cells [22] [37] . Bcl-2 expression was examined in MCF-7 EV and O4 cells treated or not with fulvestrant. Fulvestrant induced the down-regulation of bcl-2 transcript in MCF-7 EV cells in a dose dependent manner (figure 5). In contrast, fulvestrant failed to downregulate bcl-2 in O4 cells even at the highest dose tested (10 nM). Fulvestrant (10 nM) also downregulated the mRNA expression of bcl-xL, an homolog of bcl-2 in MCF-7EV cells by 72%. The reduction of bcl-xL mRNA expression was not observed in O4 cells treated with the same fulvestrant concentration. Bax expression was slightly but not significantly decreased in both cells at all fulvestrant doses tested. These results indicate that fulvestrant-induced apoptosis can be prevented by GP88 overexpression by preventing down regulation of bcl-2, and possibly bcl-xL, mRNA expression.

Figure 3. Effect of E2 and fulvestrant on ERE-luciferase reporter gene activity in MCF-7 EV and O4 cells. Cells were cotransfected with pGL2-ERE-luciferase and b-galactosidase reporter gene constructs. E2 (1 nM) and/or fulvestrant (ICI, 10 nM) were added after transfection. Cell lysates were collected 36 hours later for measuring luciferase activity as described in the method section. The values were normalized to the values of b-galactosidase activity used as internal control to determine transfection efficiency. The data are presented in folds of activation above the control untreated cells.
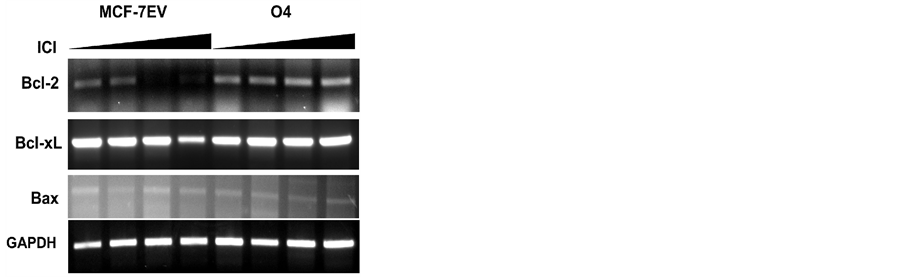
Figure 4. GP88 prevents down regulation of fulvestrant-induced bcl-2 expression. Bcl-2, Bcl-xL and Bax mRNA expression were determined by semi-quantitative RT-PCR of total RNA samples isolated from MCF-7 EV and O4 cells cultivated in estrogen-depleted medium and treated for 24 h with increasing concentrations of fulvestrant (0.1, 1, and 10 nM) as described in the method section. GAPDH mRNA expression was used as internal control for loading. PCR products were resolved by agarose gel electrophoresis and visualized by ethidium bromide staining.
3.6. Fulvestrant Effect on V-EGF Expression in GP88 Overexpressing Cells
It is known that VEGF expression in MCF-7 cells is stimulated by E2 and inhibited by tamoxifen and fulvestrant. We have also shown previously that GP88 stimulated VEGF expression in MCF-7 cells ([36] ). We examined here the effect of fulvestrant on VEGF expression in GP88 overexpressing cells and in MCF-7 control cells. As shown in Figure 6, fulvestrant inhibited E2 effect on VEGF expression in both MCF-7 EV and O4 cells.
4. Discussion
We have shown previously that GP88 overexpression in MCF-7 cells conferred estrogen independence and tamoxifen resistance both in vitro and in vivo [31] [36] . The present study demonstrates that GP88 also confers fulvestrant resistance to MCF-7 cells both in vitro and in vivo. We show here that fulvestrant was ineffective in inhibiting the long-term proliferation of MCF-7 cells that overexpressed GP88 in contrast to what was observed with MCF-7 control cells. More interestingly, fulvestrant failed to inhibit tumor formation of GP88 overexpressing cells injected in nude mice. No change in tumor volume as well as tumor incidence was observed in mice
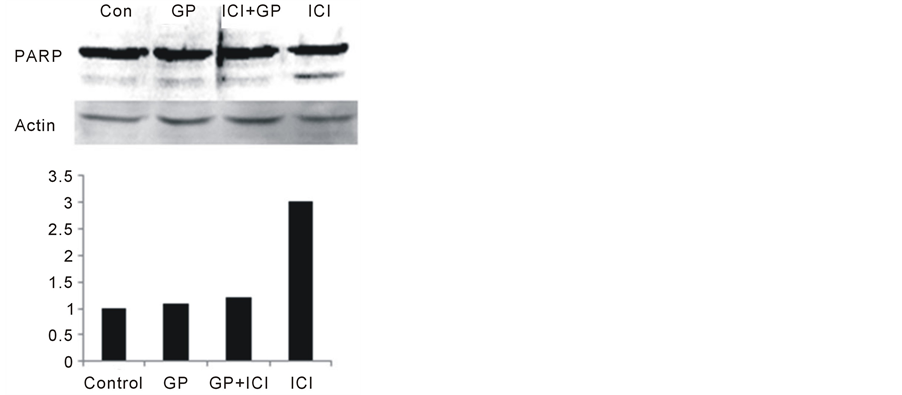
Figure 5. GP88 prevents PARP cleavage and inhibits apoptosis induced by fulvestrant in MCF-7 EV cells. MCF-7 EV cells were cultivated in estrogen-depleted medium and treated with 10 nM fulvestrant (ICI), 1 nM E2 or 400 ng/ml GP88 for 48 hours. Cell lysates were prepared in RIPA buffer containing 6 M urea for the western blot analysis. The level of PARP cleavage was determined by the presence of 85 kDa band (upper panel). Level of a-actin was determined as internal control for equal loading (lower panel).
 (a)
(a)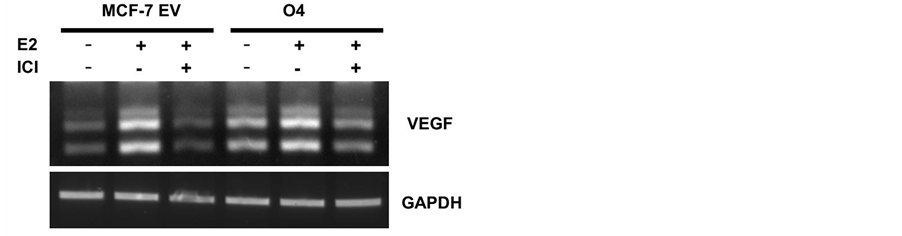 (b)
(b)
Figure 6. Effect of fulvestrant and E2 on VEGF expression in MCF-7EV and O4 cells. Cells were treated as described previously and incubated with 1 nM E2 or 10 nM fulvestrant (ICI) for 5 days. RNA samples were collected. VEGF mRNA expression was compared using RT-PCR. (a) RT-PCR products of VEGF in MCF-7EV and O4 cells. (b) Relative expression of VEGF. VEGF expression was normalized with GAPDH and compared with MCF-7 EV cells treated with vehicle only
injected with O4 cells treated with fulvestrant when compared to mice that received placebo pellets in contrast to what was observed with mice injected with control MCF-7EV cells. It is interesting to note that fulvestrant-resistant MCF-7 variants, isolated by cultivating MCF-7 cells in the continuous presence of fulvestrant displayed a 10-fold increase in GP88 expression as measured by quantitative EIA (data not shown). These data would confirm the close correlation between GP88 expression and resistance to fulvestrant in MCF-7 cells.
In addition to preventing fulvestrant inhibition of cell proliferation, we show here that GP88 prevented apoptosis induced by fulvestrant. This was observed with GP88 added exogenously as well as in GP88 overexpressing cells. GP88 overexpression as well as GP88 added exogenously prevented PARP cleavage induced by fulvestrant and also sustained bcl-2 and bcl-xL expression during fulvestrant treatment. We have reported previously that GP88 inhibited tamoxifen apoptotic effect in MCF-7 cells by preventing tamoxifen induced bcl-2 down regulation [31] . Both tamoxifen and fulvestrant induce apoptosis in human breast cancer cells although fulvestrant is more effective than tamoxifen on inducing apoptosis in MCF-7 cells [22] [24] .
In MCF-7 cells, fulvestrant, similarly to tamoxifen, induced apoptosis via down regulation of bcl-2 [20] -[24] . In addition, fulvestrant can downregulate bcl-xL unlike tamoxifen [20] . This data could explain that fulvestrant has a more potent effect than tamoxifen in stimulating apoptosis. However, the role of bax in fulvestrant treatment remains unclear. While it has been reported that fulvestrant induced bax expression [22] , other reports, in agreement with our results, showed that bax expression remained unchanged [24] .
Even though, GP88 could inhibit the effect of fulvestrant on cell proliferation and apoptosis, we show here that fulvestrant was still able to inhibit the stimulation by estradiol of ERE-luciferase reporter gene activity in GP88 overexpressing cells as well as in MCF-7 control cells. Moreover, fulvestrant maintained its ability to inhibit the expression of the angiogenic factor V-EGF in GP88 over expressing cells as well as in MCF-7 control cells. Both data are in agreement with the fact that ER expression in GP88 over expressing cells remained unchanged compared to MCF-7 control cells [29] . These later results are different from the ones obtained with tamoxifen that not only failed to inhibit VEGF expression in GP88 over expressing cells but also cooperated with GP88 to stimulate VEGF expression [31] [33] . This effect is in agreement with the fact that tamoxifen has been reported as having a weak agonistic effect on VEGF expression [38] . This data is in the agreement with the results from in vitro and in vivo growth study that fulvestrant has no stimulatory effect in GP88 over expressing cells. The results suggested that even though fulvestrant is more effective than tamoxifen, both agents could not counteract the mitogenic effect of GP88. This is unlikely with fulvestrant, since no known agonist effect has been reported yet. Moreover, fulvestrant has been shown to have anti-angiogenic effect in human umbilical vein endothelial cells by inducing apoptosis and preventing the formation of tube-structure in matrigel [39] . Although Progranulin (GP88) has been reported to enhance growth of vascular cells [40] , it is possible that the inhibitory effect of fulvestrant may overcome the proliferative effect of GP88 on vessel growth. Our in vivo experiments have shown that fulvestrant did not promote the growth of GP88 overexpressing tumors in nude mice. In contrast, tamoxifen treatment had resulted in a two-fold increase in tumor growth of GP88 overexpressing cells when compared to mice treated with placebo pellets [31] . Since this effect was observed in vivo and not in vivo, we had hypothesized that it could be due to the increased angiogenesis observed in vivo with tamoxifen and GP88. Based on this, it is possible that no stimulatory effect of fulvestrant on O4 tumor growth was observed because the inhibitory effect of fulvestrant on vessel growth may have neutralized the proliferative effect of GP88. This data would suggest that in ER+ breast tumors that overexpress GP88, fulvestrant would be a better choice of therapy than tamoxifen.
Resistance to anti-estrogen therapies has been observed in cells over expressing other growth factor pathways. GP88 overexpression of several growth factors or growth factor receptors has been found to be associated with tamoxifen resistance, especially in MCF-7 cells [5] . Her-2 receptor overexpression promotes tamoxifen resistance in MCF-7 [28] . However, in this case, the resistance corresponded to a decreased ability of tamoxifen to inhibit tumor growth in vivo rather than a complete loss of tamoxifen response. Enhanced Epidermal Growth Factor Signaling was found in faslodex (fulvestrant resistant) MCF-7 cells [17] . It has been shown that tamoxifen resistance and fulvestrant resistance occurred when fibroblast growth factor (FGF) was overexpressed in MCF-7 cells [13] . Taken together, these results suggest that estrogen independent pathways induced by growth factors may overcome the suppression of estrogen dependent pathway by antiestrogens, allowing cells to grow in estrogen independent condition and abolishing the response to antiestrogen treatment. However, whether GP88 overexpression leads to the resistance to aromatase inhibitor remains to be investigated.
ER and PR status is a good predictor of response to endocrine therapy [41] [42] . Several groups have proposed other additional negative predictors such as kallikrein 10, VEGF, prostate specific antigen, Fas ligand/Fas ratio [42] [43] .
Combined VEGF and TP53 status has been found to predict poor response to tamoxifen therapy in estrogen receptor positive advanced breast cancer [44] . In addition, positive association between expression of transforming growth factor alpha was observed in endocrine unresponsive tumors [45] . Concerning GP88 expression in clinical samples, we have reported that GP88 was expressed in 80% of invasive ductal carcinoma in correlation with poor prognosis such as tumor grade, p53 expression and Ki67 [33] [34] . GP88 expression was found both in ER positive as well as ER negative tumors [33] suggesting that GP88 may play a role in both types of tumors. Since GP88 has been found associated with both tamoxifen resistance and fulvestrant resistance, it would be interesting to hypothesize that its expression may serve as a predictive marker of anti-estrogen response. Clinical studies are necessary to investigate this hypothesis in detail.
References
- Howell, A. (2001) Future Use of Selective Estrogen Receptor Modulators and Aromatase Inhibitors. Clinical Cancer Research, 7, 4402s-4410s; Discussion 11s-12s.
- Cole, M.P., Jones, C.T. and Todd, I.D. (1971) A New Anti-Oestrogenic Agent in Late Breast Cancer. An Early Clinical Appraisal of ICI46474. British Journal of Cancer, 25, 270-275. http://dx.doi.org/10.1038/bjc.1971.33
- Clarke, R., Skaar, T.C., Bouker, K.B., Davis, N., Lee, Y.R., Welch, J.N. and Leonessa, F. (2001) Molecular and Pharmacological Aspects of Antiestrogen Resistance. The Journal of Steroid Biochemistry and Molecular Biology, 76, 71-84.
- Hu, X.F., Veroni, M., De Luise, M., Wakeling, A., Sutherland, R., Watts, C.K. and Zalcberg, J.R. (1993) Circumvention of Tamoxifen Resistance by the Pure Anti-Estrogen ICI 182,780. International Journal of Cancer, 55, 873-876. http://dx.doi.org/10.1002/ijc.2910550529
- Morris, C. and Wakeling, A. (2002) Fulvestrant (“Faslodex”)—A New Treatment Option for Patients Progressing on Prior Endocrine Therapy. Endocrine-Related Cancer, 9, 267-276. http://dx.doi.org/10.1677/erc.0.0090267
- Kellen, J.A. (2001) Raloxifene: Another Selective Estrogen Modulator. In Vivo, 15, 459-460.
- Howell, A., DeFriend, D.J., Robertson, J.F., Blamey, R.W., Anderson, L., Anderson, E., Sutcliffe, F.A. and Walton, P. (1996) Pharmacokinetics, Pharmacological and Anti-Tumour Effects of the Specific Anti-Oestrogen ICI 182780 in Women with Advanced Breast Cancer. British Journal of Cancer, 74, 300-308. http://dx.doi.org/10.1038/bjc.1996.357
- Howell, A., Robertson, J.F., Quaresma, Albano. J., Aschermannova, A., Mauriac, L., Kleeberg, U.R., Vergote, I., Erikstein, B., Webster, A. and Morris, C. (2002) Fulvestrant, formerly ICI 182,780, Is as Effective as Anastrozole in Postmenopausal Women with Advanced Breast Cancer Progressing after Prior Endocrine Treatment. Journal of Clinical Oncology, 20, 3396-3403. http://dx.doi.org/10.1200/JCO.2002.10.057
- Johnston, S.R. (1997) Acquired Tamoxifen Resistance in Human Breast Cancer-Potential Mechanisms and Clinical Implications. Anticancer Drugs, 8, 911-930. http://dx.doi.org/10.1097/00001813-199711000-00002
- Foekens, J.A., Peters, H.A., Grebenchtchikov, N., Look, M.P., Meijer-van Gelder, M.E., Geurts-Moespot, A., van der Kwast, T.H., Sweep, C.G. and Klijn, J.G. (2001) High Tumor Levels of Vascular Endothelial Growth Factor Predict Poor Response to Systemic Therapy in Advanced Breast Cancer. Cancer Research, 61, 5407-5414.
- Osborne, C.K., Pippen, J., Jones, S.E., Parker, L.M., Ellis, M., Come, S., Gertler, S.Z., May, J.T., Burton, G., Dimery, I., Webster, A., Morris, C., Elledge, R. and Buzdar, A. (2002) Double-Blind, Randomized Trial Comparing the Efficacy and Tolerability of Fulvestrant versus Anastrozole in Postmenopausal Women with Advanced Breast Cancer Progressing on Prior Endocrine Therapy: Results of a North American Trial. Journal of Clinical Oncology, 20, 3386-3395. http://dx.doi.org/10.1200/JCO.2002.10.058
- Johnston, S.R., Saccani-Jotti, G., Smith, I.E., Salter, J., Newby, J., Coppen, M., Ebbs, S.R. and Dowsett, M. (1995) Changes in Estrogen Receptor, Progesterone Receptor, and pS2 Expression in Tamoxifen-Resistant Human Breast Cancer. Cancer Research, 55, 3331-3338.
- McLeskey, S.W., Zhang, L., El-Ashry, D., Trock, B.J., Lopez, C.A., Kharbanda, S., Tobias, C.A., Lorant, L.A., Hannum, R.S., Dickson, R.B. and Kern, F.G. (1998) Tamoxifen-Resistant Fibroblast Growth Factor-Transfected MCF-7 Cells Are Cross-Resistant in Vivo to the Antiestrogen ICI 182,780 and Two Aromatase Inhibitors. Clinical Cancer Research, 4, 697-711.
- Nicholson, R.I., Staka, C., Boyns, F., Hutcheson, I.R. and Gee, J.M.W. (2004) Growth-Factor Driven Mechanisms Associated with Resistance to Estrogen Deprivation in Breast Cancer: New Opportunities for Therapy. Endocrine-Related Cancer, 11, 623-641. http://dx.doi.org/10.1677/erc.1.00778
- Carlomagno, C., Perrone, F., Gallo, C., De Laurentiis, M., Lauria, R., Morabito, A., Pettinato, G., Panico, L., D’Antonio, A., Bianco, A.R. and De Placido, S. (1996) c-erb B2 Overexpression Decreases the Benefit of Adjuvant Tamoxifen in Early-Stage Breast Cancer without Axillary Lymph Node Metastases. Journal of Clinical Oncology, 14, 2702-2708.
- Pietras, R.J., Arboleda, J., Reese, D.M., Wongvipat, N., Pegram, M.D., Ramos, L., Gorman, C.M., Parker, M.G., Sliwkowski, M.X. and Slamon, D.J. (1995) HER-2 Tyrosine Kinase Pathway Targets Estrogen Receptor and Promotes Hormone-Independent Growth in Human Breast Cancer Cells. Oncogene, 10, 2435-2446.
- McClelland, R.A., Barrow, D., Madden, T.A., Dutkowski, C.M., Pamment, J., Knowlden, J.M., Gee, J.M. and Nicholson, R.I. (2001) Enhanced Epidermal Growth Factor Receptor Signaling in MCF7 Breast Cancer Cells after Long-Term Culture in the Presence of the Pure Antiestrogen ICI 182,780 (Faslodex). Endocrinology, 142, 2776-2788.
- Okubo, S., Kurebayashi, J., Otsuki, T., Yamamoto, Y., Tanaka, K. and Sonoo, H. (2004) Additive Antitumour Effect of the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Gefitinib (Iressa, ZD1839) and the Antioestrogen Fulvestrant (Faslodex, ICI 182,780) in Breast Cancer Cells. British Journal of Cancer, 90, 236-244. http://dx.doi.org/10.1038/sj.bjc.6601504
- Cameron, D.A., Keen, J.C., Dixon, J.M., Bellamy, C., Hanby, A., Anderson, T.J. and Miller, W.R. (2000) Effective Tamoxifen Therapy of Breast Cancer Involves Both Antiproliferative and Pro-Apoptotic Changes. European Journal of Cancer, 36, 845-851. http://dx.doi.org/10.1016/S0959-8049(00)00013-7
- Zhang, G.J., Kimijima, I., Onda, M., Kanno, M., Sato, H., Watanabe, T., Tsuchiya, A., Abe, R. and Takenoshita, S. (1999) Tamoxifen-Induced Apoptosis in Breast Cancer Cells Relates to Down-Regulation of Bcl-2, but Not Bax and bcl-X(L), without Alteration of p53 Protein Levels. Clinical Cancer Research, 5, 2971-2977.
- Kang, Y., Cortina, R. and Perry, R.R. (1996) Role of c-myc in Tamoxifen-Induced Apoptosis Estrogen-Independent Breast Cancer Cells. Journal of the National Cancer Institute, 88, 279-284. http://dx.doi.org/10.1093/jnci/88.5.279
- Diel, P., Smolnikar, K. and Michna, H. (1999) The Pure Antiestrogen ICI 182780 Is More Effective in the Induction of Apoptosis and Down Regulation of BCL-2 than Tamoxifen in MCF-7 Cells. Breast Cancer Research and Treatment, 58, 87-97. http://dx.doi.org/10.1023/A:1006338123126
- Lim, K.B., Ng, C.Y., Ong, C.K., Ong, C.S., Tran, E., Nguyen, T.T., Chan, G.M. and Huynh, H. (2001) Induction of Apoptosis in Mammary Gland by a Pure Anti-Estrogen ICI 182780. Breast Cancer Research and Treatment, 68, 127-138. http://dx.doi.org/10.1023/A:1011929222555
- Soma?, S., Chaouat, M., Jacob, D., Perrot, J.Y., Rostène, W., Forgez, P. and Gompel, A. (2003) Antiestrogens Are Pro-Apoptotic in Normal Human Breast Epithelial Cells. International Journal of Cancer, 105, 607-612. http://dx.doi.org/10.1002/ijc.11147�
- Clarke, R., Liu, M.C., Bouker, K.B., Gu, Z., Lee, R.Y., Zhu, Y., Skaar, T.C., Gomez, B., O’Brien, K., Wang, Y. and Hilakivi-Clarke, L.A. (2003) Antiestrogen Resistance in Breast Cancer and the Role of Estrogen Receptor Signaling. Oncogene, 22, 7316-7339. http://dx.doi.org/10.1038/sj.onc.1206937
- Bateman, A. and Bennett, H.P. (1998) Granulins: The Structure and Function of an Emerging Family of Growth Factors. Journal of Endocrinology, 158, 145-151. http://dx.doi.org/10.1677/joe.0.1580145
- Serrero, G. (2003) Autocrine Growth Factor Revisited: PC-Cell-Derived Growth Factor (Progranulin), a Critical Player in Breast Cancer Tumorigenesis. Biochemical and Biophysical Research Communications, 308, 409-413. http://dx.doi.org/10.1016/S0006-291X(03)01452-9
- Halper, J. (2010) Growth Factors as Active Participants in Carcinogenesis: A Perspective. Veterinary Pathology, 47, 77-97. http://dx.doi.org/10.1177/0300985809352981
- Lu, R. and Serrero, G. (2001) Mediation of Estrogen Mitogenic Effect in Human Breast Cancer MCF-7 cells by PC-Cell-Derived Growth Factor (PCDGF/Granulin Precursor). Proceedings of the National Academy of Sciences of the United States of America, 98, 142-147. http://dx.doi.org/10.1073/pnas.98.1.142
- Lu, R. and Serrero, G. (2000) Inhibition of PCDGF Expression by Antisense cDNA Transfection Inhibits Tumorigenicity of the Human Breast Carcinoma Cell Line MDA-MB-486. Proceedings of the National Academy of Sciences of the United States of America, 97, 3993-3998.
- Tangkeangsirisin, W., Hayashi, J. and Serrero, G. (2004) PC Cell-Derived Growth Factor Mediates Tamoxifen Resistance and Promotes Tumor Growth of Human Breast Cancer Cells. Cancer Research, 64, 1737-1743. http://dx.doi.org/10.1158/0008-5472.CAN-03-2364
- Abrhale, T., Brodie, A., Sabnis, G., Macedo, L., Tian, C., Yue, B. and Serrero, G. (2011) GP88 (PC-Cell Derived Growth Factor, Progranulin) Stimulates Proliferation and Confers Letrozole Resistance to Aromatase Overexpressing Breast Cancer Cells. BMC Cancer, 11, 231. http://dx.doi.org/10.1186/1471-2407-11-231
- Serrero, G. and Ioffe, O. (2003) Expression of the PC-Cell Derived Growth Factor in benign and Malignant Human Breast Epithelium. Human Pathology, 34, 1148-1154. http://dx.doi.org/10.1016/S0046-8177(03)00425-8
- Serrero, G., Hawkins, D.M., Yue, B., Ioffe, O., Bejarano, P., Phillips, J.T., Head, J.F., Elliott, R.L., Tkaczuk, K.R., Godwin, A.K., Weaver, J.E. and Kim, W. (2012) Progranulin (GP88) Tumor Tissue Expression Is Associated with Increased Risk of Recurrence in Breast Cancer Patients Diagnosed with Estrogen Receptor Positive Invasive Ductal Carcinoma. Breast Cancer Research, 14, R26-R35. http://dx.doi.org/10.1186/bcr3111
- Tkaczuk, K.R., Yue, B., Zhan, M., Tait, N., Yarlagadda, L., Dai, H. and Serrero, G. (2011) Increased Circulating Level of the Survival Factor GP88 (Progranulin) in the Serum of Breast Cancer Patients When Compared to Healthy Subjects. Breast Cancer Basic and Clinical Research, 5, 155-162.
- Tangkeangsirisin, W. and Serrero, G. (2004) PC Cell-Derived Growth Factor (PCDGF/GP88, Progranulin) Stimulates Migration, Invasiveness and VEGF Expression in Breast Cancer Cells. Carcinogenesis, 25, 1587-1592. http://dx.doi.org/10.1093/carcin/bgh171
- Lilling, G., Hacohen, H., Nordenberg, J., Livnat, T., Rotter, V. and Sidi, Y. (2000) Differential Sensitivity of MCF-7 and LCC2 Cells, to Multiple Growth Inhibitory Agents: Possible Relation to High bcl-2/Bax Ratio? Cancer Letters, 161, 27-34. http://dx.doi.org/10.1016/S0304-3835(00)00579-6
- Takei, H., Lee, E.S. and Jordan, V.C. (2002) In Vitro Regulation of Vascular Endothelial Growth Factor by Estrogens and Antiestrogens in Estrogen-Receptor Positive Breast Cancer. Breast Cancer, 9, 39-42. http://dx.doi.org/10.1007/BF02967545
- Soares, R., Guo, S., Russo, J. and Schmitt, F. (2003) Role of the Estrogen Antagonist ICI 182,780 in Vessel Assembly and Apoptosis of Endothelial Cells. Ultrastructural Pathology, 27, 33-39. http://dx.doi.org/10.1080/01913120309946
- He, Z., Ong, C.H., Halper, J. and Bateman, A. (2003) Progranulin Is a Mediator of the Wound Response. Nature Medicine, 9, 225-229. http://dx.doi.org/10.1038/nm816
- Elledge, R.M., Green, S., Pugh, R., Allred, D.C., Clark, G.M., Hill, J., Ravdin, P., Martino, S. and Osborne, C.K. (2000) Estrogen Receptor (ER) and Progesterone Receptor (PgR), by Ligand-Binding Assay Compared with ER, PgR and pS2, by Immuno-Histochemistry in Predicting Response to Tamoxifen in Metastatic Breast Cancer: A Southwest Oncology Group Study. International Journal of Cancer, 89, 111-117.
- Osborne, C.K., Yochmowitz, M.G., Knight 3rd, W.A. and McGuire, W.L. (1980) The Value of Estrogen and Progesterone Receptors in the Treatment of Breast Cancer. Cancer, 46, 2884-2888.
- Luo, L.Y., Diamandis, E.P., Look, M.P., Soosaipillai, A.P. and Foekens, J.A. (2002) Higher Expression of Human Kallikrein 10 in Breast Cancer Tissue Predicts Tamoxifen Resistance. British Journal of Cancer, 86, 1790-1796. http://dx.doi.org/10.1038/sj.bjc.6600323
- Berns, E.M., Klijn, J.G., Look, M.P., Grebenchtchikov, N., Vossen, R., Peters, H., Geurts-Moespot, A., Portengen, H., van Staveren, I.L., Meijer-van Gelder, M.E., Bakker, B., Sweep, F.C. and Foekens, J.A. (2003) Combined Vascular Endothelial Growth Factor and TP53 Status Predicts Poor Response to Tamoxifen Therapy in Estrogen Receptor-Positive Advanced Breast Cancer. Clinical Cancer Research, 9, 1253-1258.
- Nicholson, R.I., McClelland, R.A., Gee, J.M., Manning, D.L., Cannon, P., Robertson, J.F., Ellis, I.O. and Blamey, R.W. (1994) Transforming Growth Factor-Alpha and Endocrine Sensitivity in Breast Cancer. Cancer Research, 54, 1684-1689.
NOTES

*Corresponding author.