International Journal of Otolaryngology and Head & Neck Surgery
Vol.3 No.2(2014), Article ID:43581,5 pages DOI:10.4236/ijohns.2014.32014
Neurofibroma of the Pinna
Satinder Singh, Swati Tandon*, Asish Lahiri, Shalabh Sharma
Sir Ganga Ram Hospital, Delhi, India
Email: satindersingh123@rediffmail.com, *drswatitandon86@gmail.com, lasish@yahoo.co.uk, drshalabh68@yahoo.co.in
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 21 December 2013; revised 20 January 2014; accepted 19 February 2014
ABSTRACT
Neurofibromas are relatively common lesions of the nervous system, but only a few cases involving the pinna have been reported. Isolated neurofibroma of pinna without neurofibromatosis has not been reported in the literature so far. Neurofibromas may develop anywhere in the body, including cranial and peripheral nerves. The type of impairment associated with neurofibromas depends largely on the site of the lesion. Cutaneous lesions, especially in the head and neck, generally cause deformity, whereas lesions that affect deeper systems (e.g., the auditory and ocular systems) are more likely to cause functional impairment. Usually found in individuals with neurofibromatosis, we report a case of neurofibroma of pinna in a patient without neurofibromatosis.
Keywords:Neurofibroma; Pinna; Neurofibromatosis

1. Introduction
Neurofibromas are peripheral nerve sheath tumours that arise from peripheral nerve elements and are frequently seen in neurofibromatosis type 1. Neurofibromatosis type 1 is caused by a disruptive mutation in NF1 gene that codes for protein neurofibromin. In contrast to schwanommas, another type of tumor arising from schwann cells, neurofibromas incorporate many additional type of cells. Neurofibromas have invasive and angiogenic properties suggesting that these are genetically altered cells with tumourogenic properties [1] .
2. Clinical History
A 35-year-old lady presented to the otolaryngology outpatient department of our hospital with complaint of a painful swelling over the left pinna since past 6 months (Figure 1). It was insidious in onset and slowly increasing in size. There was no history of bleeding from the swelling and no history of such lesions elsewhere in the
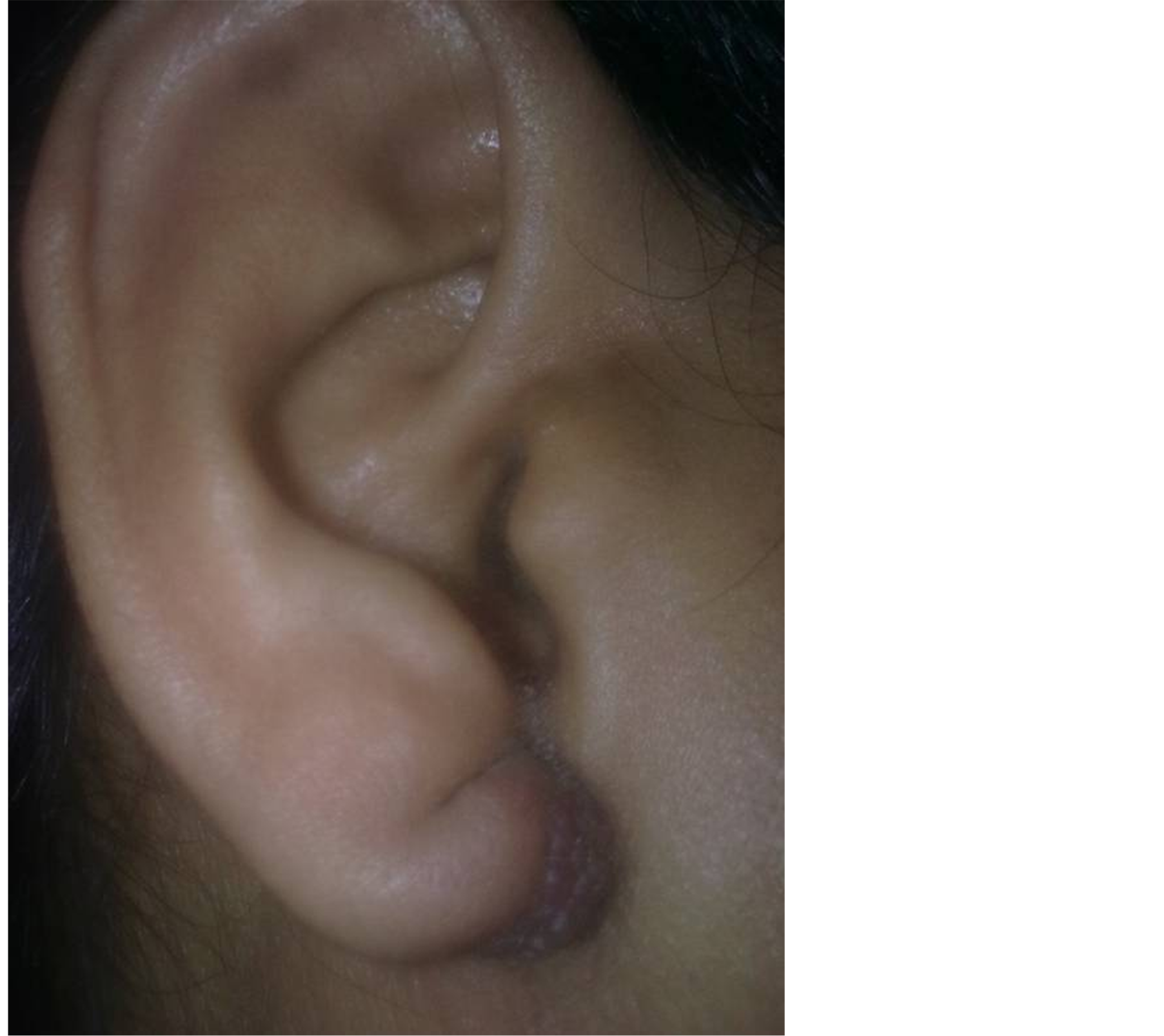
Figure 1. Preoperative photograph showing the lesion on pinna.
body. There was no significant family history. Examination of pinna showed single 2 × 2 cm reddish brown swelling with an irregular surface seen on medial and lateral surface of the lobule. It was firm in consistency, tender, did not bleed on touch, and overlying skin was involved. Nose and throat examination was normal. There was no significant finding on systemic examination.
Fine needle aspiration cytology was suggestive of neurogenic tumour. The lesion was excised under local anesthesia with 0.5 cm margin of the healthy tissue and wound was closed primarily. The wound healed within a week (Figure 2). Histopathological examination showed a lesion composed of spindle shaped cells arranged in short fascicles, whorling and storiform patterns. Nuclei were ovoid to elongated, vesicular and show mild pleomorphism. No atypia or necrosis was seen. The lesion was infiltrating the dermal collagen and entrapping the skin appendages. Subcutaneous fat was free (Figure 3). On immunohistochemistry tumor cells were strongly positive for S-100, suggestive of neurofibroma (Figure 4).
3. Discussion
Neurofibromatosis is one of the most common inherited disorders and can affect anyone, regardless of family history, race, gender or ethnic background [1] . There are two types of Neurofibromatosis, type 1 (NF1) and type 2 (NF2). Neurofibromatosis type 1 is a common autosomal dominant condition with high frequency of peripheral nerve sheath tumours called neurofibromas [1] . NF1 is caused by changes in gene carried on chromosome 17. There are two types of neurofibromas: Dermal and Plexiform. Dermal neurofibromas usually develop during
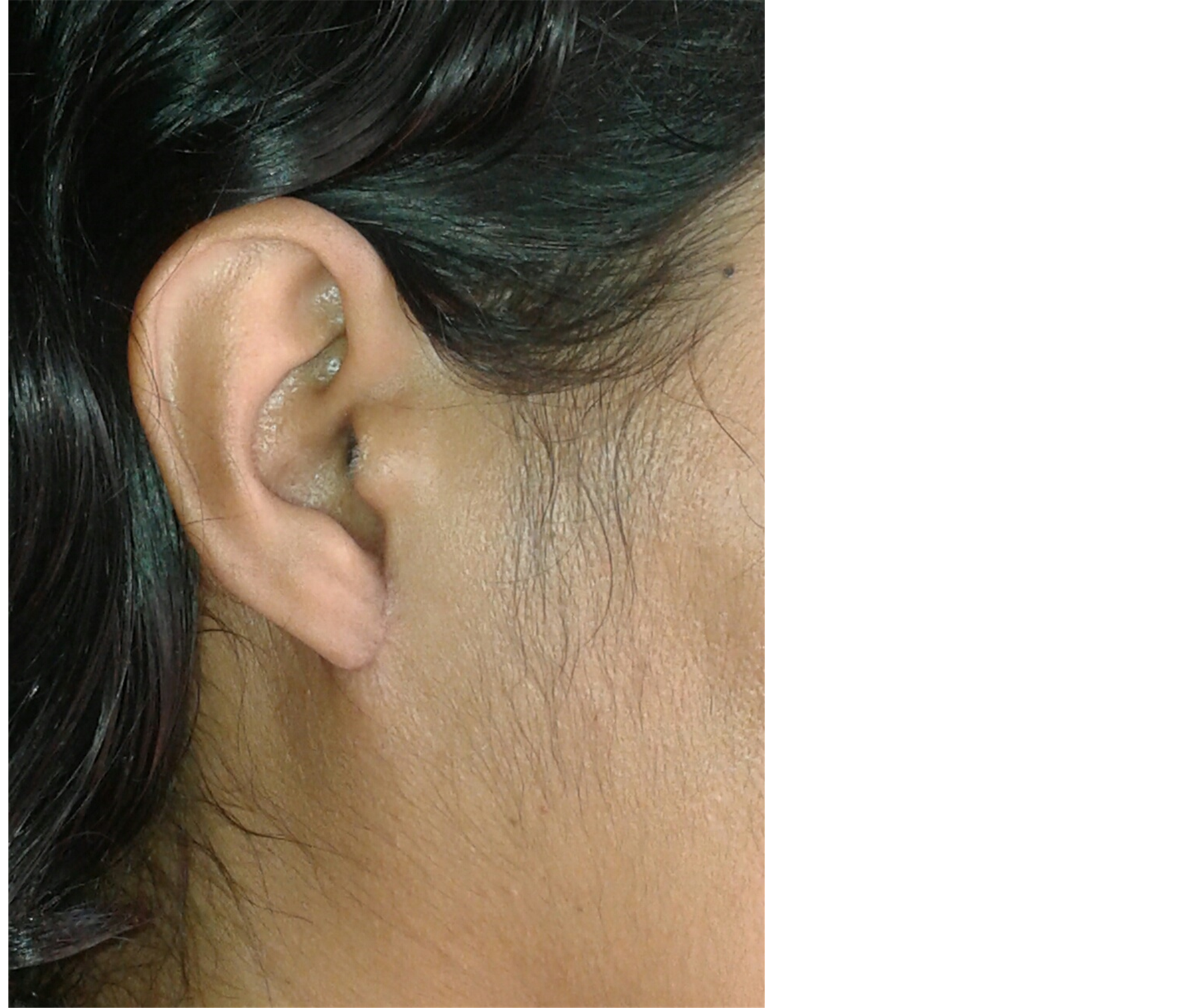
Figure 2. Postoperative photograph.
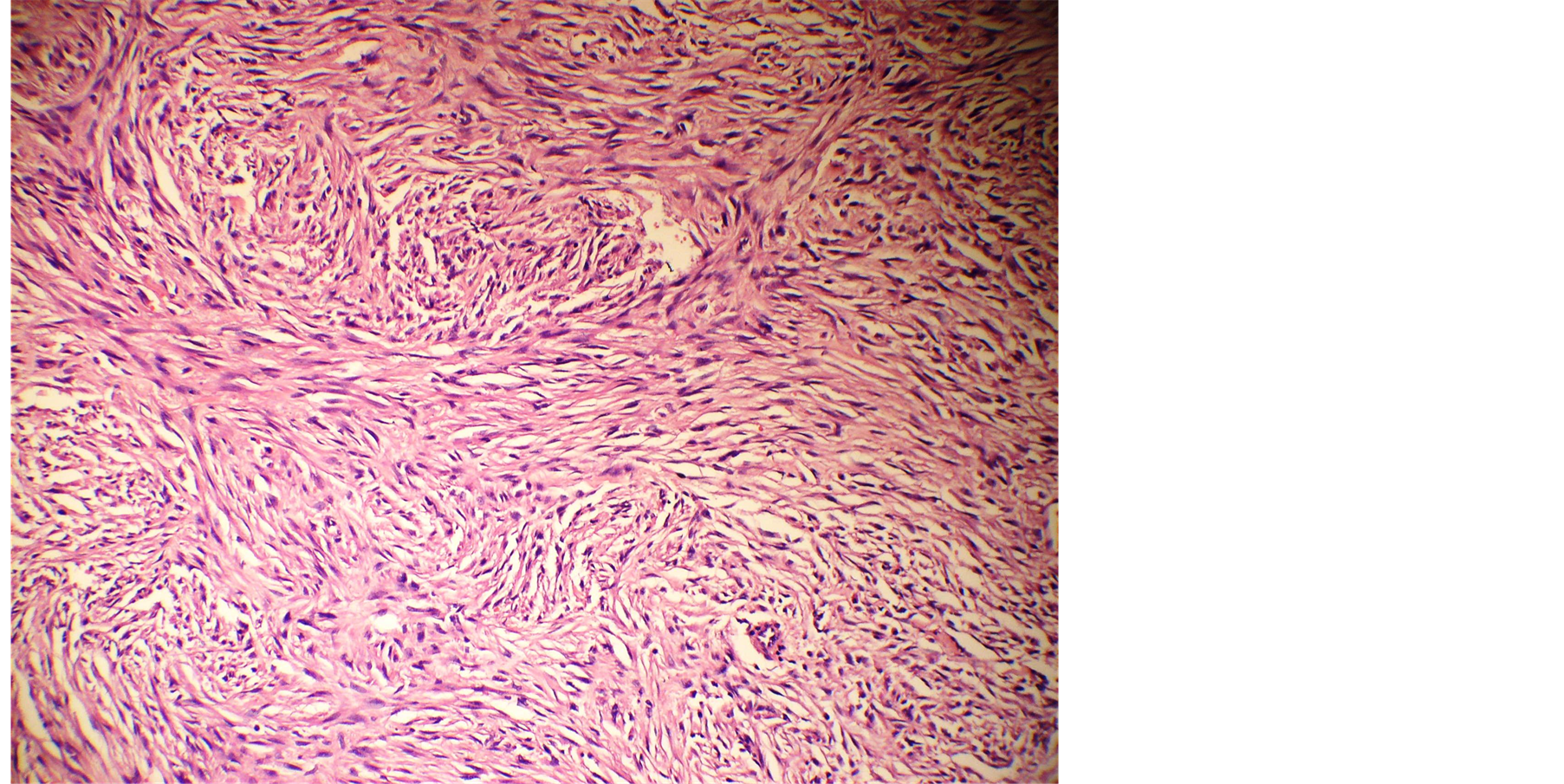
Figure 3. Microphotograph showing spindle shaped cells arranged in short fascicles, whirling and storioform pattern.
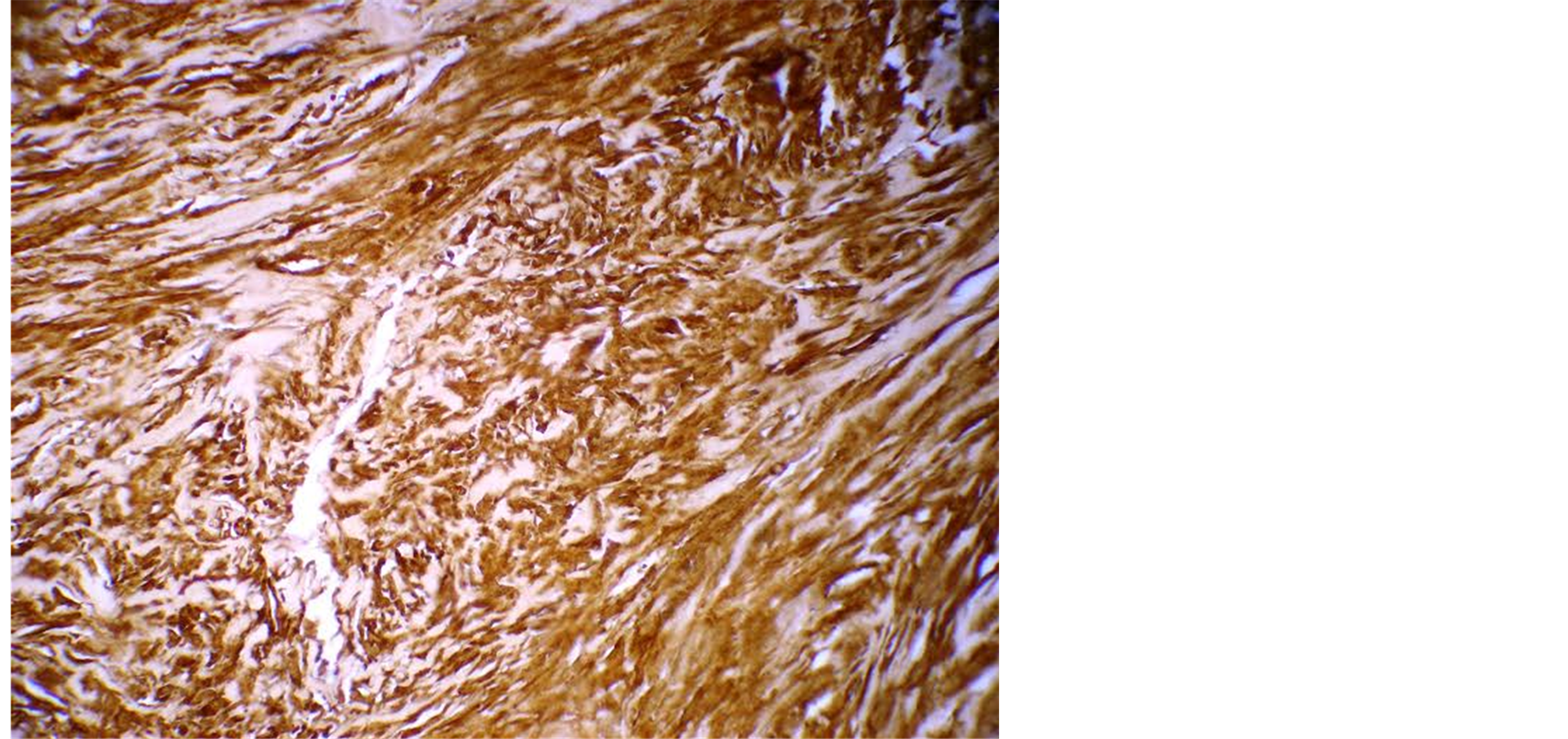
Figure 4. Tumour cells positive for S-100.
adolescence and adulthood. These small tumours involve terminal nerves and may be numerous, yet have no apparent risk of malignant transformation. In contrast, plexiform neurofibromas are usually congenital, typically involve deep or named nerves, can become very large and may cause serious functional impairment. Risk of malignant transformation is 6% in these tumours [2] .
Neurofibromas may occur in isolation or as a part of Von Recklinghausen’s syndrome in combination with café au lait skin patches, lisch’s nodules, axillary freckling and fibroma molluscum [2] . The disease may be inherited as an autosomal dominant trait with variable penetrance in 50% of patients or it may occur as a result of spontaneous mutation [3] .
Unlike schwanommas, which consist of schwann cells, neurofibromas show marked cellular heterogeneity. Nevertheless, schwann cells are major cell type amplified in neurofibromas and typically comprise 40% - 80% of tumour cells. Additionally, there is substantial population of interspersed fibroblastic or perineural cells, along with various vascular and imflammatory elements embedded in an extensive extracellular matrix. Because of this cellular heterogeneity histogenesis of neurofibromas has been controversial. In our case, immunohistochemistry showed tumour cells which were strongly positive for S-100 suggestive of neurofibroma.
The change in genetic material that causes NF1 and NF2 can be inherited or it may occur as a result of spontaneous mutations. In our patient, there was no family history.
Most tumours caused by NF need no treatment, but tumours that are painful, disfiguring, growing rapidly or impairing function need treatment. In main trunk neurofibroma, nerve autografting with interposition is therapy of choice with good chance of functional recuperation [3] . Large lesion may require reconstruction and elevation of pinna with dermal strips or marlex mesh [4] . Most often, the redistribution of local and adjacent tissue allows satisfactory reconstruction [5] . In our case we were able to achieve complete excise of the tumour and primary closure was achieved after rotation of surrounding tissue.
Shaida A.M. et al. [6] reported a case of pinna neurofibroma with otitis externa, hearing loss and cosmetic deformity. In our case cosmetic deformity and pain were the chief complaints and we were able to obtain a satisfactory result.
4. Conclusion
Neurofibromas are commonly seen in patients with neurofibromatosis. In a few cases of solitary neurofibroma, depending upon the size and site of lesion, complete excision of the tumour may be possible without causing significant functional and cosmetic deformity, as was achieved in our case.
References
- David Muir et al. (2003) Tumorigenic Properties of Neurofibromin-Deficient Neurofibroma Schwann Cells. American Journal of Pathology, 158, 501-513.
- Griffith, B.H., et al. (1972) Von Rcklinnghausen’s Disease in Children. Plastic Reconstruction Surgery, 49, 647-653. http://dx.doi.org/10.1097/00006534-197206000-00012
- Kemph et al. (1995) Diagnostic and Clinical Outcome of Neurogenic Tumours in the Head and Neck Area. ORL: Journal for Oto-Rhino-Laryngology, 57, 273-278. http://dx.doi.org/10.1159/000276757
- Trevisani, T.P., Pohl, A.L. and Matloub, H.S. (1982) Neurofibroma of the Ear. Function and Aesthetics. Plastic Reconstructive Surgery, 70, 217-219. http://dx.doi.org/10.1097/00006534-198208000-00019
- Menick, F.J. (1990) Reconstruction of the Ear after Tumour Excision. Clinics in Plastic Surgery, 17, 405-415.
NOTES

*Corresponding author.