Open Journal of Air Pollution
Vol.06 No.02(2017), Article ID:76218,8 pages
10.4236/ojap.2017.62004
An Experimental Study of the Atmospheric Oxidation of a Biogenic Organic Compound (Methyl Jasmonate) in a Thin Water Film as in Fog or Aerosols
Aubrey A. Heath, Kalliat T. Valsaraj*
Cain Department of Chemical Engineering, Louisiana State University, Baton Rouge, LA, USA

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: April 22, 2017; Accepted: May 15, 2017; Published: May 18, 2017
ABSTRACT
Experiments on the reactions of OH radicals in thin films of water were conducted in a photochemical reactor. The OH radical reactivity of a biogenic molecule (methyl jasmonate) was observed to be much larger in thin films of water than in the bulk aqueous phase. The pseudo-first order reaction rate was enhanced by an order of magnitude on a 38-micron film compared to the bulk liquid. However, the first order rate constant increased by 349%. This has implications in atmospheric systems like fog and mist which have large specific surface areas. The enhanced reactivity is attributable to both the partial solvation and faster diffusion at the air-water interface compared to the bulk liquid.
Keywords:
Biogenic Organic, Methyl Jasmonate, Photochemical Surface Reaction

1. Introduction
Aerosol particles contribute to cloud formation in the upper atmosphere and are also implicated in lower atmospheric pollution. Particles that are hygroscopic grow by accumulation of water to make fog in the lower atmosphere. Water is an important component of atmospheric dispersoids such as fog, mist, dew, and rain. Atmospheric water not only retains pollutants via both adsorption and absorption, but also facilitates and participates in their transformation to other species [1] [2] [3] [4] . Specifically, the uptake of organic and inorganic gases leads to transformations in atmospheric water films that lead to more polar and less volatile compounds. When the water finally evaporates, the resulting compounds are left in the atmosphere as secondary organic aerosols (SOA), which act to nucleate water during the next fog or rain event. This is termed the fog- smog cycle [5] . Thus, atmospheric water films play a significant role in the cycling of pollutants via fog or aerosols.
A majority of organic compounds in the atmosphere are biogenic and are derived from plant emissions; these are collectively called biogenic volatile organic compounds (BVOCs) [6] . BVOCs are made up of four groups: viz., isoprene, monoterpenes, other reactive VOCs (ORVOC), and other VOCs (OVOC). ORVOC and OVOC combine to provide about 45% of the BVOC [7] . Among the ORVOC is a class of oxygenated hydrocarbons which are collectively called green leaf volatiles (GLVs). Typically, GLVs are compounds that plants emit under stress or mechanical damage. Although emission rates of GLVs are lower than the combined emission rates of isoprene and monoterpenes, there is information on their reactivity in the atmospheric environment [8] .
GLVs have typically low aqueous solubility and vapor pressure and have been known to partition favorably from the air to fog, mist, dew and rain. It has been shown that both gas phase and aqueous phase oxidation of GLVs can lead to secondary organic aerosols in the atmosphere [8] [9] [10] [11] [12] . The bulk phase oxidation processes are not the only processes that can occur within a droplet; in fact, the oxidation can occur also on the air-water interface of droplets. In some cases, the surface oxidation process can be even more significant than bulk phase oxidation within a droplet, as has been shown in previous cases [1] [2] [13] . The purpose of this new set of experiments was to compare the reaction of methyl jasmonate with hydroxyl radicals at the air-water interface to the same reaction in the bulk aqueous phase by using a thin-film, temperature-controlled reactor. Previous experiments and analysis with other compounds, such as benzene, have shown enhancement of reaction rate as the film thickness decreases due to reactant surface enhancement, large surface diffusion constants, and the incomplete solvent cage effect at the surface [14] . Thus, thinner film environments, like fog droplets, in which conditions are more affected by interfacial properties, can display higher rates of reactivity than bulk phase environments, like rain droplets.
Methyl jasmonate was chosen for this set of experiments because it had been studied in the bulk aqueous phase and for its high Henry’s Law Constant (8000 M atm−1) [12] . The high Henry’s Law Constant indicates that methyl jasmonate partitions strongly into the liquid phase; hence, the experiments should not be significantly affected by evaporation of methyl jasmonate from the aqueous film. Methyl jasmonate does contribute to secondary organic aerosol (SOA) formation in the atmosphere [10] [12] . As such, the overall surface reactivity of methyl jasmonate would help to provide further insight to its significance to SOA formation.
2. Experimental Setup
All experiments in this study were performed in a thin-film, temperature-con- trolled reactor that was fabricated and tested in our laboratory [14] [15] [16] . Using the thin-film reactor has the added benefit in that both bulk-phase and interfacial-phase conditions can be modeled experimentally by a single reactor. Details of the thin-film reactor are explained extensively in previous publications [14] , but some changes were made to the reactor setup for the methyl jasmonate experiments. Figure 1 shows the thin-film reactor schematic and its cross-sec- tional view for the methyl jasmonate experiments.
In this study, it was desired to measure the degradation of methyl jasmonate by the reaction with hydroxyl radicals, generated by the UV-irradiation of hydrogen peroxide. To do this, the thin film reactor was modified to be in a batch configuration (shown in Figure 1(a)) [14] . The thin film reactor is fully insulated and is divided into two sections: the upper portion, which contains the UV-B lights (for UV generation of hydroxyl radicals), and the lower portion, which contains the photoreactor assembly on which the reaction takes place. The four UV-B lights (15 W, 275 - 390 nm, UVP LLC, Upland, CA, USA) in the upper portion generated a substantial amount of heat, so several items were added to counteract this. These items included two Cole-Parmer Polystat Advanced 15 L Heat/Cool Baths (Cole-Parmer Instrument Company, LLC, Vernon Hills, IL, USA) and a vortex tube chiller (Arizona Vortex Tube Manufacturing Company, Wickenburg, AZ, USA). The reactor temperature was kept at a constant 20.0˚C ± 0.5˚C for all experiments in this study.
The photoreactor assembly, located in the lower portion of the reactor, is made up of several items: a 5 cm × 101 cm KG-33 glass tube coated with PFA foil (0.508 mm thickness, McMaster-Carr, Atlanta, GA, USA); a copper tube heat exchanger to ensure temperature consistency along the length of the photoreactor assembly; and a chemically treated borosilicate glass slide, where the reaction physically takes place. The 1000 mm × 40 mm × 2 mm borosilicate glass
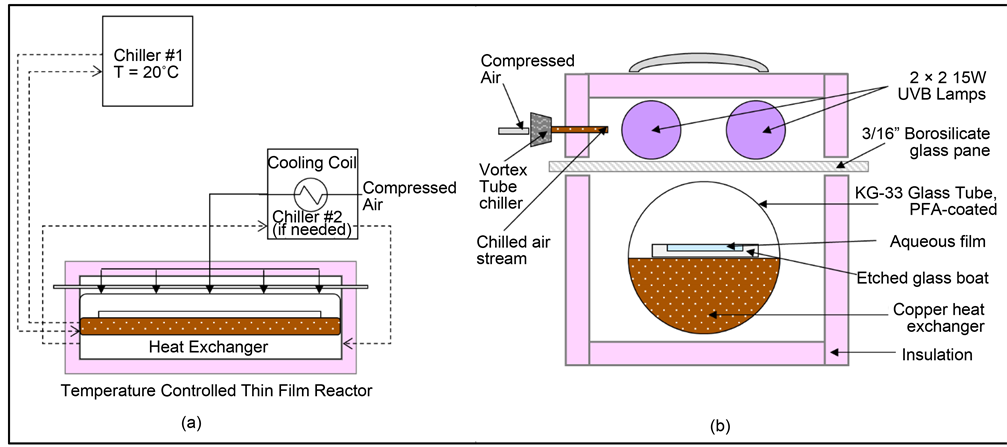
Figure 1. (a) Sketch of the thin film reactor assembly used in photo-oxidation experiments that test the effect of film thickness. Dashed lines represent coolant flow. (b) Cross-section of the thin film reactor [14] .
slide was chemically treated such that it had a hydrophilic inner trough and a hydrophobic outer frame [14] . Thus, the glass slide has a known hydrophilic surface area (SA = 259 cm2). When a known aqueous volume is placed along the glass slide, the surface area to volume ratio (i.e. film thickness) can be obtained (1 mL volume = 38.6 µm). Hence, larger volumes correspond to bulk phase conditions, whereas smaller volumes are used to measure conditions at the air-water interface.
Prior to every experiment, a known volume of a pre-made 2 mM solution of methyl jasmonate (95% purity, Sigma-Aldrich, Milwaukee, WI, USA) in LC-MS grade water (Honeywell, Muskegon, MI, USA) was added directly to the glass slide. A given volume of 1% hydrogen peroxide (EMD Chemicals, Inc., Gibb- stown, NJ, USA) was also added directly to the glass slide via pipette to result in a final hydrogen peroxide concentration of 29.4 mM. Although this concentration is high, the purpose of these experiments was to observe the effect of thinner films on the overall reaction rate. By using the higher oxidant concentration, the methyl jasmonate could degrade much more quickly and the differences in the reaction rate due to the film thickness could be observed. The actual concentration of the 1% hydrogen peroxide solution was obtained by iodometric titration.
Once the slide was loaded with the methyl jasmonate-hydrogen peroxide solution, the slide was inserted into the glass tube, and the reactor was sealed. After the reactor temperature stabilized, the UV lights were turned on for up to three hours of irradiation. Ten different time intervals were tested in total. Each time interval was subsequently tested in duplicate. To test for the effect of evaporation, identical experiments were run where the UVB lights were not turned on. Thus, for every experimental volume and time period, both “dark” and “light” experiments were performed.
After the set experimental time was over, the slide was removed from the reactor and its contents were washed down the slide with HPLC grade acetonitrile (EMD Chemical Inc., Gibbstown, NJ, USA) into a volumetric flask. Using acetonitrile helped decrease the amount of evaporation that could occur during the slide removal process and ensured that all compounds were removed from the slide. Samples from the slide were analyzed immediately by HPLC-UV/DAD (Agilent 1100 model) for methyl jasmonate degradation. The HPLC analysis method is described in detail in a previous study [17] .
3. Results
Previously, we reported extensively on the product formation pathways for the OH radical oxidation of methyl jasmonate in the bulk aqueous phase [12] . Four primary oxidation products (C13H20O4, C13H20O5, C13H22O5, C13H22O6) were identified using HPLC-ESI/MS and the overall reaction rate in the bulk aqueous phase was determined to be (2.83 ± 0.02) × 10−4 min−1. The products resulting from this oxidation were shown to form secondary organic aerosols (SOA) with a yield of (68% ± 8%) [10] . Aqueous phase oxidation processes can occur both in the bulk aqueous phase as well at the air-water interface. Additional work from our laboratory showed that for an aromatic species such as benzene, the surface oxidation rate is enhanced as the surface area to volume ratio of a thin water film increases [14] . This work validated the basic premise that the surface oxidation process is faster than bulk liquid phase oxidation because the surface diffusion process is notably fast and reactants are only partially solvated at the surface.
Figure 2 represents the typical rate of loss of methyl jasmonate from a thin water film of 38 µm, which is the thinnest water film studied in this work. Identical curves were derived for the additional film thicknesses tested in this study. The reaction rate is first order, as shown by the excellent fit to the data, and is represented by the following first order rate equation:
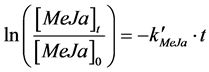 (1)
(1)
where [MeJa]t is the concentration of methyl jasmonate at time t, [MeJa]0 is the concentration of methyl jasmonate at the beginning of the reaction, 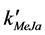 is the apparent first order rate constant, and t is time in minutes.
is the apparent first order rate constant, and t is time in minutes.
Three different film thicknesses were tested in this study, viz., 38.6 μm, 77.2 mm and 193.1 μm. For each of the three thicknesses, dark experiments were also conducted to determine if there was any significant evaporation of MeJa from the thin films during the experiments. Table 1 shows the variation in methyl jasmonate concentration over the span of three hours for each film thickness tested. As shown in Table 1, the concentration of methyl jasmonate did not noticeably change over the three hours, with the standard deviation in concentration ranging from 0.81% for the largest film thickness (δ = 193.1 μm) to 3.19% for the smallest thickness (δ = 38.6 µm). Therefore, for the UV experiments, no corrections were necessary for the rate constants.

Figure 2. Degradation of methyl jasmonate over the span of three hours from the reaction with hydroxyl radicals produced from hydrogen peroxide at 20˚C (δ = 38.6 µm).
Apparent first order rate constants for methyl jasmonate degradation were obtained for all three film thicknesses. Rate constant information for all three film thicknesses is shown in Table 2.
From Table 2, it is apparent that the reaction rate does increase when film thickness decreases. When the film thickness decreased from 193.1 µm to 38.6 µm, the reaction rate increased by 31.9%. It has been shown that film thicknesses of both 77.2 μm and 38.6 µm correspond to the thin film regime [14] . By looking at this data piece-wise, it is observed that the reaction rate increases by 14.4% as the film thickness decreases from 193.1 µm to 77.2 µm and by 15.3% as the film thickness decreases from 77.2 µm to 38.6 µm. Therefore, there is a slightly greater increase in degradation rate within the thin film regime as compared to when the thickness is decreased from a thick film (δ = 193.1 µm) to a thin film (δ = 77.2 µm) scenario. Furthermore, when the experimental data in this study for the smallest film thickness (δ = 38.6 μm) is compared to the bulk aqueous phase rate constant (δ → ¥) obtained in a previous study, the rate constant increases by 348% [12] . This is notable for small droplets such as fog droplets where the average diameter size is only 1 - 10 µm. In these types of environments, surface reactivity is expected to dominate over any bulk aqueous phase reaction.
In a recent work from our group, we showed that an even greater surface enhancement was observed for the reaction of benzene with hydroxyl radicals in a similar experimental setup [14] . The degree of surface enhancement is related not only to the structure of the molecule, but is also related to the relative orientation of the molecule at the surface. Moreover, the functional groups in a molecule determine the reactivity towards the OH radical at the surface. The surface reactivity enhancement is driven by the fact that the reactant molecules are only partially hydrated at the surface and that the surface diffusion is generally fast. Therefore, higher reaction rates are more frequently observed at the surface as compared to the bulk liquid, where the molecules must overcome not only the

Table 1. Concentrations of MeJa in the dark experiments.
Table 2. Apparent first order rate constants for the degradation of methyl jasmonate at 20˚C.
aFrom [12] .
slow bulk diffusion limitations, but also the energy required to break the solvation sheaths for the reactants [13] .
4. Conclusion
This set of experiments displayed the surface enhancement of the reaction of methyl jasmonate with hydroxyl radicals. Thus, reaction rates are increased when the film thickness is reduced. For a more critical look into this reaction, it would be recommended to perform additional experiments at even smaller film thicknesses. Obtaining these rate constants would allow for the derivation of a function that describes to what extent the change in film thickness affects the reaction rate [13] [14] [18] . Likewise, it would be beneficial to run these experiments at temperatures typical of various fog events and in solutions of multiple ionic strengths to simulate conditions of the natural environment. In order to determine to what extent the film thickness affects the SOA yield, the oxidative products and their quantities produced in this reaction would also have to be identified, which was beyond the scope of this study. However, the products identified in a previous study for the bulk aqueous phase reaction had higher molecular weights, lower vapor pressures, and higher HLCs than methyl jasmonate [12] . These factors are critical for SOA formation. By identifying the yields of these products in this reaction at various film thicknesses, more adequate conclusions concerning SOA formation could be obtained.
Acknowledgements
This work was supported, in part, by the National Science Foundation (Grant No: AGS-1106559).
Cite this paper
Heath, A.A. and Valsaraj, K.T. (2017) An Experimental Study of the Atmospheric Oxidation of a Biogenic Organic Compound (Methyl Jasmonate) in a Thin Water Film as in Fog or Aerosols. Open Journal of Air Pollution, 6, 44-51. https://doi.org/10.4236/ojap.2017.62004
References
- 1. Donaldson, D.J. and Valsaraj, K.T. (2010) Adsorption and Reaction of Trace Gas-Phase Organic Compounds on Atmospheric Water Film Surfaces: A Critical Review. Environmental Science & Technology, 44, 865-873.
https://doi.org/10.1021/es902720s - 2. Valsaraj, K.T. (2012) A Review of Aqueous Aerosol Surface Chemistry in the Atmsopheric Context. Open Journal of Physical Chemistry, 2, 58-66.
https://doi.org/10.4236/ojpc.2012.21008 - 3. Rossignol, S., Tinel, L., Bianco, A., Passananti, M., Brigante, M., Donaldson, D.J. and George, C. (2016) Atmospheric Photochemistry at a Fatty Acid-Coated Air-Water Interface. Science, 353, 699-702.
https://doi.org/10.1126/science.aaf3617 - 4. Bjorneholm, O., Hansen, M.H., Hodgson, A., Liu, L.M., Limmer, D.T., Michaelides, A., Pedevilla, P., Rossmeisl, J., Shen, H., Tocci, G., Tyrode, E., Walz, M.M., Werner, J. and Bluhm, H. (2016) Water at Interfaces. Chemical Reviews, 116, 7698-7726.
https://doi.org/10.1021/acs.chemrev.6b00045 - 5. Munger, J.W., Jacob, D.J., Waldman, J.M. and Hoffman, M.R. (1983) Fogwater Chemistry in an Urban Atmosphere. Journal of Geophysical Research, 88, 5109-5121.
https://doi.org/10.1029/JC088iC09p05109 - 6. Goldstein, A.H. and Galbally, I.E. (2007) Known and Unexplored Organic Constituents in the Earth’s Atmosphere. Environmental Science & Technology, 41, 1514-1521.
https://doi.org/10.1021/es072476p - 7. Guenther, A., Hewitt, C.N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., McKay, W.A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J. and Zimmerman, P. (1995) A Global Model of Natural Volatile Organic Compound Emissions. Journal of Geophysical Research, 100, 8873-8892.
https://doi.org/10.1029/94JD02950 - 8. Hamilton, J.F., Lewis, A.C., Carey, T.J., Wenger, J.C., Garcia, E.B.I. and Munoz, A. (2009) Reactive Oxidation Products Promote Secondary Organic Aerosol Formation from Green Leaf Volatiles. Atmospheric Chemistry & Physics, 9, 3815-3823.
https://doi.org/10.5194/acp-9-3815-2009 - 9. Mentel, T.F., Kleist, E., Andres, S., Dal Maso, M., Hohaus, T., Kiendler-Scharr, A., Rudich, Y., Springer, M., Tillmann, R., Uerlings, R., Wahner, A. and Wildt, J. (2013) Secondary Aerosol Formation from Stress-Induced Biogenic Emissions and Possible Climate Feedbacks. Atmospheric Chemistry & Physics., 13, 8755-8770.
https://doi.org/10.5194/acp-13-8755-2013 - 10. Richards-Henderson, N.K., Hansel, A.K., Valsaraj, K.T. and Anastasio, C. (2014) Aqueous Oxidation of Green Leaf Volatiles by Hydroxyl Radical as a Source of SOA: Kinetics and SOA Yields. Atmospheric Environment, 95, 105-112.
- 11. Richards-Henderson, N.K., Pham, A.T., Kirk, B.B. and Anastasio, C. (2014) Secondary Organic Aerosol from Aqueous Reactions of Green Leaf Volatiles with Organic Triplet Excited States and Singlet Molecular Oxygen. Environmental Science & Technology, 49, 268-276.
https://doi.org/10.1021/es503656m - 12. Hansel, A.K., Ehrenhauser, F.S., Richards-Henderson, N.K., Anastasio, C. and Valsaraj, K.T. (2015) Aqueous-Phase Oxidation of Green Leaf Volatiles by Hydroxyl Radical as a Source of SOA: Product Identification from Methyl Jasmonate and Methyl Salicylate Oxidation. Atmospheric Environment, 102, 43-51.
- 13. Valsaraj, K.T. (2009) Trace Gas Adsorption Thermodynamics at the Air-Water Interface: Implications in Atmospheric Chemistry. Pure Applied Chemistry, 81, 1889-1901.
https://doi.org/10.1351/PAC-CON-08-07-06 - 14. Heath, A.A. and Valsaraj, K.T. (2015) Effects of Temperature, Oxygen Level, Ionic Strength, and pH on the Reaction of Benzene with Hydroxyl Rradicals at the Air-Water Interface in Comparison to the Bulk Aqueous Phase. Journal of Physical Chemistry A, 119, 8527-8536.
https://doi.org/10.1021/acs.jpca.5b05152 - 15. Ehrenhauser, F.S. (2012) Photochemical Reaction Products of Polycyclic Aromatic Hydrocarbons Adsorbed at an Air-Water Interface. PhD Dissertation, Louisiana State University, Baton Rouge.
- 16. Chen, J. and Valsaraj, K.T. (2007) Uptake and UV-Photooxidation of Gas-Phase Polyaromatic Hydrocarbons on the Surface of Atmospheric Water Films. 2. Effects of Dissolved Surfactants on Naphthalene Photooxidation. Journal of Physical Chemsitry A, 111, 4289-4296.
https://doi.org/10.1021/jp068394p - 17. Vempati, H. (2014) Physico-Chemical Properties of Green Leaf Volatiles. Master’s Thesis, Louisiana State University, Baton Rouge.
- 18. Raja, S., Yaccone, F. S., Ravikrishna, R. and Valsaraj, K. T. (2002) Thermodynamic Parameters for the Adsorption of Aromatic Hydrocarbon Vapors at the Gas-Water Interface. Journal of Chemical & Engineering Data, 47, 1213.
https://doi.org/10.1021/je025520j