Advances in Infectious Diseases
Vol.2 No.4(2012), Article ID:25227,5 pages DOI:10.4236/aid.2012.24015
Screening of Potent Inhibitor of H1N1 Influenza NS1 CPSF30 Binding Pocket by Molecular Docking
![]()
1School of Life Science, Liaoning University Shenyang, China; 2Research Center for Computer Simulating and Information Processing of Bio-Macromolecules of Liaoning, Shenyang, China.
Email: *liuhongsheng@lnu.edu.cn
Received July 15th, 2012; revised August 17th, 2012; accepted September 2nd, 2012
Keywords: CPSF30 Binding Pocket; Molecular Docking; Influenza A H1N1
ABSTRACT
The swine flu, H1N1 virus was outbroken in Mexico and the United States in April 2009 and then rapidly spread worldwide. The World Health Organization declared that the outbreak of influenza is caused by a new subtype of influenza H1N1 influenza virus. And researchers have isolated some oseltamivir resistance strains in 2009 swine flu which makes the imminency of research and development of new anti influenza drug. The CPSF30 binding pocket of effector domain in NS1 protein is very important in the replication of influanza A virus and is a new attractive anti flu drug target. But up to now there is no antiviral drug target this pocket. Here we employ molecular docking to screening of about 200,000 compounds. We find four novel compounds with high binding energy. Binding comformation analysis revealed that these small molecules can interact with the binding pocket by some strong hydrophobic interaction. This study find some novel small molecules can be used as lead compounds in the development of new antiinfluenza drug based on CPSF30 pocket.
1. Introduction
Influenza A virus can cause higher mortality than seasonal flu, is a very serious epidemic respiratory infections disease. And this virus have caused many serious influenza pandemics in history, such as the 1918 Spanish flu and 1957 Asian flu, resulting in several million deaths [1,2]. Recently, a new strain of influenza A virus was identified and commonly referred as “swine flu” in the outbreak of H1N1 influenza A virus in Mexico and the United states in April 2009. And then the WHO (World Health Organization) enhanced the Ifluenza A virus warning to phase 6, meaning that the 2009 H1N1 flu become a serious global pandemic. More unfortunately, several Oseltamivir (Tamiflu) resistance strain have been isolated from them [3]. Therefore, there is an urgent need to find new anti-viral compounds.
Nonstructural 1 (NS1) protein of influenza A virus (NS1A) is a nonstructural protein encoded by segment 8 of influenza virus A [4]. The protein is a 230 - 237 amino acid residue small multifunctional protein comprising two domains [5]: The N-terminal (1 - 70) RNA-Binding Domain (RBD) which binds double-stranded RNA (dsRNA) in a non-sequence specific manner [4,6-8] and inhibit the interferon (IFN)-α/β-induced 2’-5’-oligo A synthetase/RNase L pathway [6] and C-terminal (86 - 230/237) Effector Domain (ED) which can binds several host cellular proteins and is the 30-kDa subunit of Cleavage and Polyadenylation Specificity Factor (CPSF30), thereby inhibit 3’-end processing of cellular pre-mRNAs, including IFN-β pre-mRNA [9,10].
The CPSF30 binding pocket in the ED of NS1A is an attractive target for antiviral drug development as it is very important for virus replication. Several experiments have proved that the second and the third zinc finger (F2F3) of CPSF30 can bind efficiently to the NS1A ED and inhibit the activity of CPSF30 [11]. The NS1-CPSF30 complex is essential for suppression of the key anti-viral response of host and the efficient replication of virus. The X-ray crystal structure of NS1A ED in complex with the F2F3 fragment revealed the detail of the CPSF30 binding pocket and the interaction between NS1 ED and CPSF30. The CPSF30 binding pocket is a largely hydrophobic pocket and contains several highly conserved residues and these conserved residues can interect with the aromatic residues of F3 zinc finger (Tyr97, Phe98 and Phe102) [12]. Based on the fact of the importence of CPSF30 binding pocket and the structural of the NS1A-CPSF30, small molecules mimicking the three aromatic residues of F3 are predicted to inhibit viral replication by inhibiting CPSF30 binding to NS1A.
A recent study showed that several chemicals (Figure 1) that can bind to NS1A and inhibit the virus replication, but the binding region of these chemicals were not determined [13]. In 2011, they found a new inhibitor JJ3297 which has a similar structural scaffold with NSC125044 can block multi-cycle replication in an RnaseL-dependent manner and also can not determine the bind region [14]. So it is urgent to find new chemicals which can bind to CPSF30 binding pocket and inhibit the virus replication.
The main focus in this research is to discover potential compounds that can directly bind to the NS1 CPSF30 binding pocket. We docked about 200,000 compounds to CPSF30 binding pocket by the molecular docking technique, and find some potential inhibitors which have high binding affinity. In this study we obtained 4 best-binding compounds which provide an important reference for discovering new influenza virus drugs.
2. Materials and Methods
2.1. 3-Dimensional Structure of NS1A-ED
We can find a lot of structure of NS1 effector domain in PDB (Protein Data Bank), but there is only one structure complexed with F2F3 and only one structure come from 2009 A(H1N1), they are 2RHK and 3M5R respectively. The RMS of the tow structure is 0.4 Å, thus we can say they are approximately the same. So we selected 2RHK to study the interaction mechanism between NS1 and F2F3, selected 3M5R as the structure used in molecular docking.
2.2. Preprocessing the Small Molecules
The original virtual screening library comprises about 200,000 compounds, including the Maybridge Screening
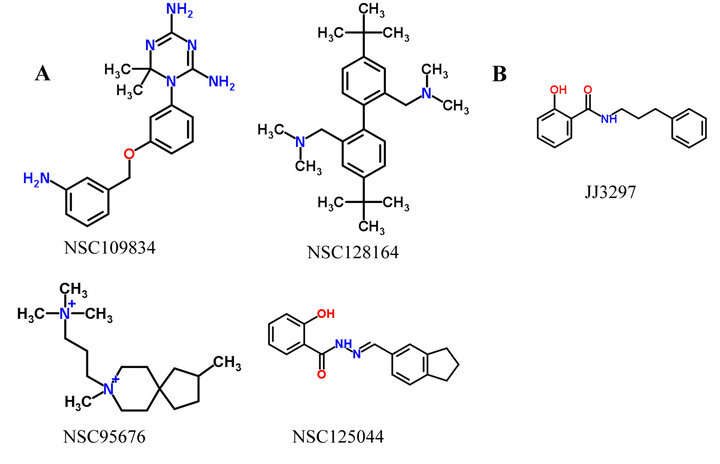
Figure 1. Five potent NS1 inhibitor. (A) Four potent inhibitor Basu et al. found in 2008, and can not determine the binding region; (B) One new potent inhibitor like NSC 125044 has the activity of inhibit virus replacation.
and NCI Plated Compounds (approximately 140,000 compounds). We used OpenBabel [15] to generate the threedimensional atomic coordinates of Maybridge Screening collection, and use XlogP3 [16] to screen drug-like compounds based on Lipinski’s Rule of five [17]. And the final compounds library comprises remain 164,745 compounds and has been add Gasteiger atomic charges, merged non-polar hydrogen. The detailed procedure of preprocessing these small molecules has been described in our previous paper [18].
2.3. Molecular Docking
AutoDock [19,20] docking with a grid-point method which must calculate the interaction energy between the probe atoms and receptor-binding sites before docking. The docking region and the center of docking region was setted based on the key residues of CPSF30 binding pocket. Then the AutoGrid program calculated the gridpoint energy of every atom type in the compounds library. Then each compound was docked into the CPSF30 binding pocket of NS1 one by one using unix shell script. During the docking procedure, ligands were flexible and the receptor was fixed. Lamarckian genetic algorithm was used to optimise, for each compounds in the library, 10 docking runs were performed with the initial population of 150 individuals. Maximum number of generations and energy evaluation were set to 27,000 and 250,000 respectively. Evaluation function is semi-empirical free energy evaluation function.
To get a more accurate screening results, we chose 4000 compounds in a high binding energy in the first docking to make a secondary docking, the parameters are the same as the first run except the value of maximum number of evaluation was set to 2,500,000 and the docking runs was set to 30 for more accurate dock.
After all the docking were finished we selected the molecules promising candidates according to the following criteria: 1) cluster conformation analysis of the molecules, select the high degree concentration molecules; 2) select the molecules which have a ensemble binding mode similar to F3 fragment; 3) observe the hydrogen bonds between compound and protein, and select the molecules whose hydrogen bond formation with key residues of CPSF30 binding pocket.
3. Results and Discussion
3.1. Conserved Residues of CPSF30 Binding Pocket
It is important to define receptor-docking area accurately in computer-aided drug designing to screen out activated compounds and the binding site shoud better be conserved. Therefore, we collected all 125 reviewed NS1 protein sequences of influenza A virus in UniProtKB. Then all the sequences were aligned by ClustalW2.0 [21]. The overview of the multiple sequence alignment shown in Figure 2, reveals that the sequence conservation of residues around 100 and 186 is higher than others and there was a low conservation in C-terminal. Combined with detail sequence alignment data and the residue distribution of F2F3 in NS1A CPSF30 binding pocket (Figure 3) we found the residues Q121, G183, G184, W187 located in the CPSF30 binding site are the same in all 125 strains, some residues like N188, K110, I117, I119, V180 are highly conserved and usually replaced by their analogical amino acid. In the view of above, the five highly conserved residues Q121, G183, G184, W187, K110, and N118 are chosen as the active site of the docking region.
3.2. The Interaction Model between F2F3 Fragment and NS1 ED
NS1A effect domain CPSF30 binding pocket is a very attractive antiinfluenza drug targets. Small molecules mimicking the three aromatic residues (Tyr97, Phe98 and Phe102) of F3 are predicted to inhibit viral replication by inhibiting CPSF30 binding to NS1A. Therefore, the small molecules we screen out should be more similar to the three aromatic residues in structure or the interaction between NS1. So, we need to figure out the interaction model between Tyr97, Phe98, Phe102 and NS1 ED. We analysis the interaction between Tyr97, Phe98, Phe102 and NS1 in NS1A-F2F3 complex (2RHK) by LigPlot [22] software. The result shown in Figure 4.
The main interaction between NS1 and the three residues is Hydrophobic contact as we can see in Figure 4, This is identical with the largely hydrophobic feature
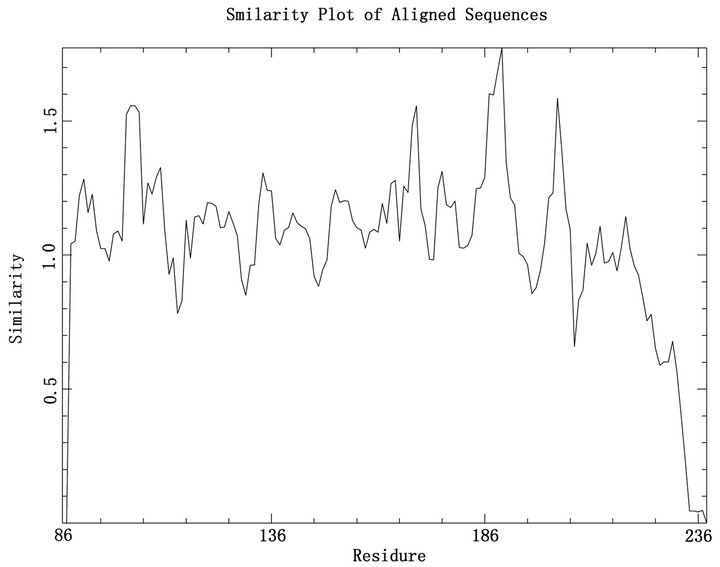
Figure 2. Overview of the multiple sequence alignment for the 125 NS1 sequences. This figure was made by plotcon of the EMBOSS [23] package, the degree of conservation within a window of 4 residues.
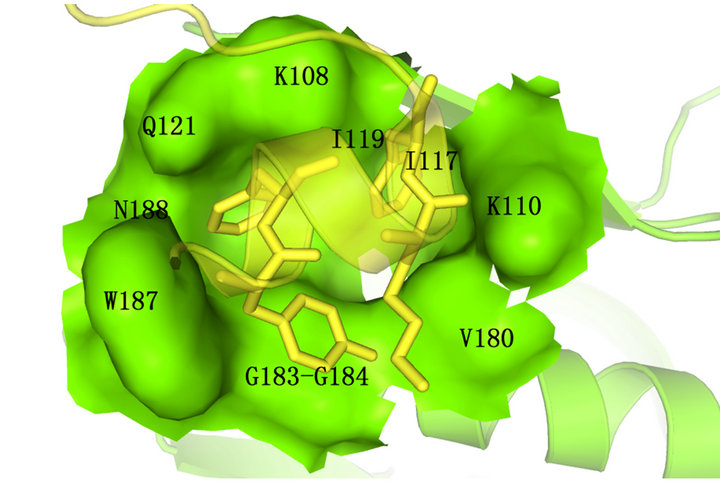
Figure 3. The bresidue distribution of F2F3 in NS1A CPSF 30 binding pocket. F2F3 fragment was shown in cartoon and stick format and colored yellow,NS1 effector domain was colored green ( drawn by PyMOL [24]).
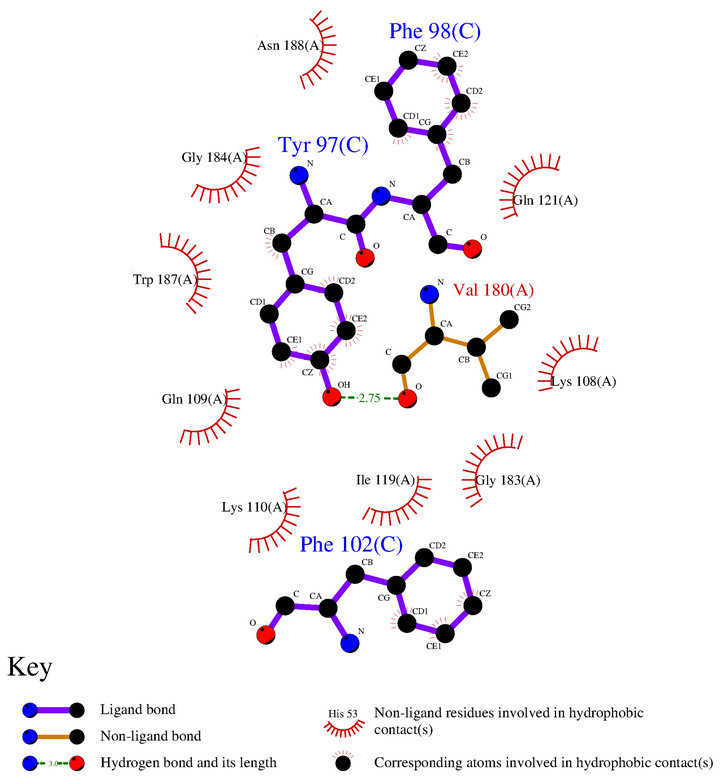
Figure 4. The interaction model of 3 aromatic residues Tyr97, Phe98, Ph102 of F3 fragment with NS1.
of NS1. Only one hydrogen bond was found between phenolichydroxyl of Tyr97 and hydroxy of Val180 which is located in the edge of the pocket. Therefore, we can predict that the small molecule inhibitor should contain a large hydrophobic group which can plug in the inside of hydrophobic binding pocket and also should have hydrophilic groups to form hydrogen bonds with conserved hydrophilic residues in the edge of the pocket.
3.3. Molecular Docking
After determined the conserved residues of CPSF30 binding pocket, we docked all the compounds in our compounds library and five potential inhibitors into this binding pocket and calculated the binding energy. All the molecules are sorted by binding energy. Then 51 molecules were identified as they have higher binding affinity than others in the two docking runs. And then we surveyed these 51 molecules by three criteria described in materials and methods, 6 molecules were finally identified as candidate inhibitors. The binding energy of the 6 molecules was ranged from –9.2 kcal/mol to –10.0 kcal/ mol, which is better than the binding energy of 5 potential inhibitors (ranged from –4.9 kcal/mol to –6.4 kcal/ mol). The chemical structures of these 6 molecules are presented along with their names and binding energy in Figure 5. These 6 molecules have the same feature that they all contain at least one hydrophobic group and also contain some nitrogen or oxygen; such molecules can not only bind to the hydrophobic binding pocket of NS1 closely but also form hydrogen bonds with the hydrophilic residues in the edge of the pocket. Also we can find a common feature that the molecules were comprised of tree hexatomic ring and some atoms joint the rings and also some modifiers in the rings. Thus, they have similar structures.
3.4. Binding Conformation of the Candidate Inhibitors
Furthermore, we analysed the interaction model of these 6 molecules. As shown in Figure 6. The hydrophobic groups of the 6 molecules are bind to the deepest pocket and form a strong hydrophobic interaction with NS1, which is similar to the three aromatic residues (Tyr97, Phe98 and Phe102) of F3. The difference is that some of the nitrogen and oxygen of these 6 molecules are formed more than one hydrogen bonds with Q121, R108 or K110, but there is only one hydrogen bond between F3 and NS1 ED, and this could strengthen the binding affinity by hydrogen bond network.
These facts shown that the 6 candidate inhibitors are
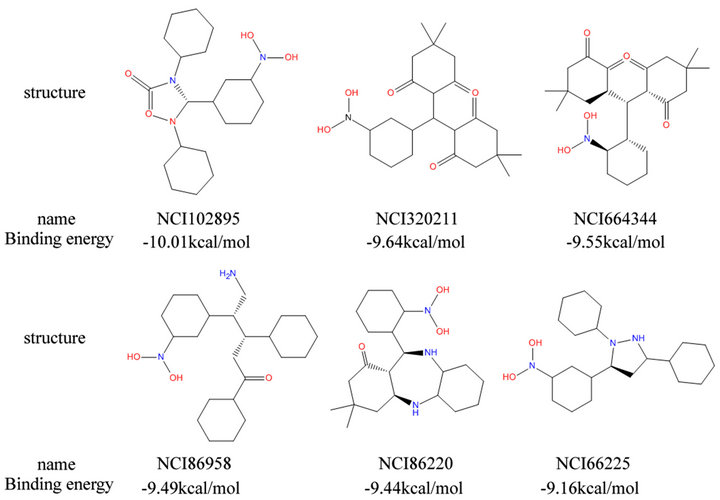
Figure 5. Six candidate inhibitor we identified.
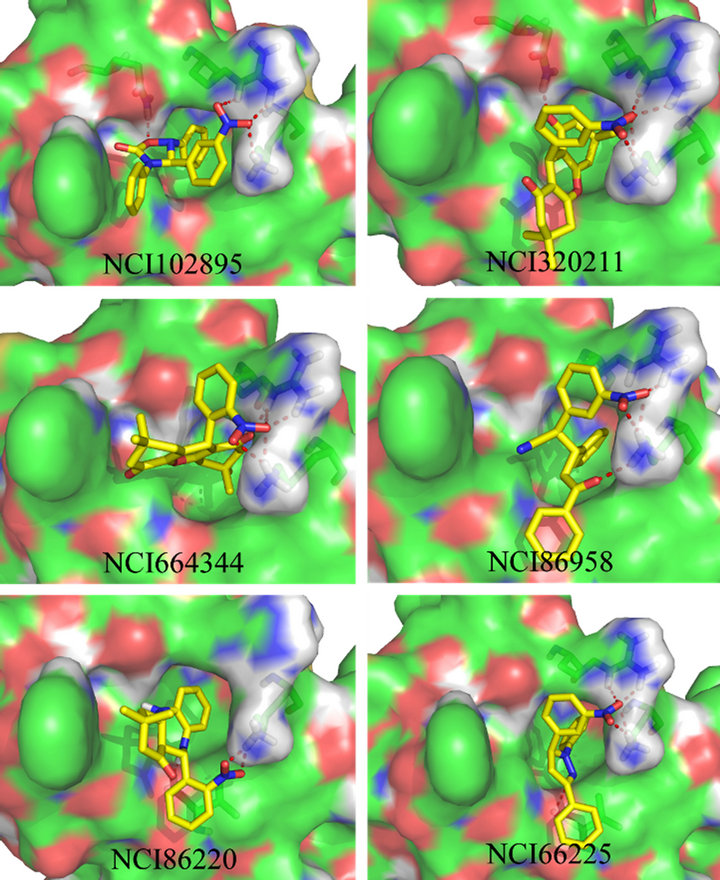
Figure 6. The protein-ligand conformation of the six candidate inhibitor. The small molecules were shown in stick format and carbon atoms were colored yellow others were colored by atom type, red for oxygen, blue for nitrogen, the hydrogen bonds were shown in dotted red lines.
similar to the three aromatic residues of F3 and may have the ability to bind to CPSF30 pocket and form hydrogen bond with the residues in the edge of the pocket. Thereby, they have the ability to inhibit NS1 bind to host cell CPSF30, the free CPSF30 can increase the production of IFN-β mRNA and then IFN-β inhibit the replication of influenza A virus.
4. Conclusion
In this study, we screened more than 200,000 small molecules by structure-based molecular docking and identified six molecules which have abilities to bind to CPSF30 binding pocket strongly and analyzed their chemical structures and binding mode. The six small molecules screened in this study can be used as the lead compound that in NS1 CPSF30 binding pocket about drug designing in the future studies, and provide an important reference on the new influenza virus drug discovery.
5. Acknowledgements
This project was supported by Research Center for Computer Simulating and Information Processing of Biomacromolecules of Shenyang (Shenyang Science and Technology Bureau Public Service Platform Construction Project No. F10-188-8-00).
REFERENCES
- A. H. Reid, J. K. Taubenberger and T. G. Fanning, “The 1918 Spanish influenza: Integrating History and Biology,” Microbes and Infection, Vol. 3, No. 1, 2001, pp. 81-87. doi:10.1016/S1286-4579(00)01351-4
- H. Nicholls, “Pandemic Influenza: The Inside Story,” PLoS Biology, Vol. 4, No. 2, 2006, p. e50.
- WHO, “Pandemic (H1N1) 2009—Update 112,” 2010. http://www.who.int/csr/don/2010_08_06/en/
- B. G. Hale, R. E. Randall, J. Ortin and D. Jackson, “The Multifunctional NS1 Protein of Influenza A Viruses,” Journal of General Virology, Vol. 89, No. 10, 2008, pp. 2359-2376. doi:10.1099/vir.0.2008/004606-0
- R. M. Krug, W. Yuan, D. L. Noah and A. G. Latham, “Intracellular Warfare between Human Influenza Viruses and Human Cells: The Roles of the Viral NS1 Protein,” Virology, Vol. 309, No. 2, 2003, pp. 181-189. doi:10.1016/S0042-6822(03)00119-3
- J. Y. Min and R. M. Krug, “The Primary Function of RNA Binding by the Influenza A Virus NS1 Protein in Infected Cells: Inhibiting the 2 - 5 Oligo (A) Synthetase/ RNase L Pathway,” Proceedings of the National Academic of Sciences of the United States of America, Vol. 103, No. 8, 2006, pp. 7100-7105.
- Y. K. Shin, Q. Liu, S. K. Tikoo, L. A. Babiuk and Y. Zhou, “Influenza A Virus NS1 Protein Activates the Phosphatidylinositol 3-Kinase (PI3K)/Akt Pathway by Direct Interaction with the P85 Subunit of PI3K,” Journal of General Virology, Vol. 88, No. 1, 2007, pp. 13-18. doi:10.1099/vir.0.82419-0
- J. Y. Min, S. Li, G. C. Sen and R. M. Krug, “A Site on the Influenza A Virus NS1 Protein Mediates Both Inhibition of PKR Activation and Temporal Regulation of Viral RNA Synthesis,” Virology, Vol. 363, No. 1, 2007, pp. 236-243. doi:10.1016/j.virol.2007.01.038
- O. G. Engelhardt and E. Fodor, “Functional Association between Viral and Cellular Transcription during Influenza Virus Infection,” Reviews in Medical Virology, Vol. 16, No. 5, 2006, pp. 329-345. doi:10.1002/rmv.512
- D. L. Noah, K. Y. Twu and R. M. Krug, “Cellular Antiviral Responses against Influenza A Virus Are Countered at the Post Transcriptional Level by the Viral NS1A Protein via Its Binding to a Cellular Protein Required for the 3’End Processing of Cellular Pre-mRNAS,” Virology, Vol. 307, No. 2, 2003, pp. 386-395. doi:10.1016/S0042-6822(02)00127-7
- K. Y. Twu, D. L. Noah, P. Rao, R. L. Kuo and R. M. Krug, “The CPSF30 Binding Site on the NS1A Protein of Influenza A Virus Is a Potential Antiviral Target,” Journal of Virology, Vol. 80, No. 8, 2006, pp. 3957-3965. doi:10.1128/JVI.80.8.3957-3965.2006
- K. Das, et al., “Structural Basis for Suppression of a Host Antiviral Response by Influenza A Virus,” Proceedings of the National Academy of Sciences, Vol. 105, No. 35, 2008, pp. 13093-13098. doi:10.1073/pnas.0805213105
- D. Basu, et al., “Novel Influenza Virus NS1 Antagonists Block Replication and Restore Innate Immune Function,” Journal of Virology, Vol. 83, No. 4, 2008, pp. 1881-1891. doi:10.1128/JVI.01805-08
- M. P. Walkiewicz, D. Basu, J. J. Jablonski, H. M. Geysen and D. A. Engel, “Novel Inhibitor of Influenza NonStructural Protein 1 Blocks Multi-Cycle Replication in an RNase L-Dependent Manner,” Journal of General Virology, Vol. 92, No. 1, 2011, pp. 60-70. doi:10.1099/vir.0.025015-0
- R. Guha, et al., “The Blue Obelisk Interoperability in Chemical Informatics,” Journal of Chemical Information and Modeling, Vol. 46, No. 3, 2006, pp. 991-998. doi:10.1021/ci050400b
- T. Cheng, et al., “Computation of Octanol-Water Partition Coefficients by Guiding an Additive Model with Knowledge,” Journal of Chemical Information and Modeling, Vol. 47, No. 6, 2007, pp. 2140-2148. doi:10.1021/ci700257y
- C. A. Lipinski, “Drug-Like Properties and the Causes of Poor Solubility and Poor Permeability,” Journal of Pharmacological and Toxicological Methods, Vol. 44, No. 1, 2000, pp. 235-249. doi:10.1016/S1056-8719(00)00107-6
- H. Ai, et al., “Discovery of Novel Influenza Inhibitors Targeting the Interaction of dsRNA with the NS1 Protein by Structure-Based Virtual Screening,” International Journal of Bioinformatics Research and Applications, Vol. 6, No. 5, 2010, pp. 449-460. doi:10.1504/IJBRA.2010.037985
- G. M. Morris, et al., “AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility,” Journal of Computational Chemistry, Vol. 30, No. 16, 2009, pp. 2785-2791. doi:10.1002/jcc.21256
- H. Park, J. Lee and S. Lee, “Critical Assessment of the Automated AutoDock as a New Docking Tool for Virtual Screening,” Proteins: Structure, Function, and Bioinformatics, Vol. 65, No. 3, 2006, pp. 549-554. doi:10.1002/prot.21183
- M. Larkin, et al., “Clustal W and Clustal X Version 2.0,” Bioinformatics, Vol. 23, No. 21, 2007, pp. 2947-2948. doi:10.1093/bioinformatics/btm404
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, “LIGPLOT: A Program to Generate Schematic Diagrams of Protein-Ligand Interactions,” Protein Engineering, Vol. 8, No. 2, 1995, p. 127. doi:10.1093/protein/8.2.127
- P. Rice, I. Longden and A. Bleasby, “EMBOSS: The European Molecular Biology Open Software Suite,” Trends in Genetics, Vol. 16, No. 6, 2000, pp. 276-277. doi:10.1016/S0168-9525(00)02024-2
- W. L. DeLano, “The PyMOL Molecular Graphics System,” DeLano Scientific LLC, Palo Alto, 2002. http://www.pymol.org
NOTES
*Corresponding author.