Surgical Science
Vol. 3 No. 12 (2012) , Article ID: 25245 , 4 pages DOI:10.4236/ss.2012.312111
Giant Mediastinal Neurofibroma in a Child with Neurofibromatosis Type I
1Department of Gastroenterological Surgery I, Hokkaido University Graduate School of Medicine, Sapporo, Japan
2Department of Pediatrics, Hokkaido University Graduate School of Medicine, Sapporo, Japan
3Department of Surgical Pathology, Hokkaido University Hospital, Sapporo, Japan
Email: okadata@med.hokudai.ac.jp
Received August 14, 2012; revised September 15, 2012; accepted September 26, 2012
Keywords: Mediastinal Tumor; Neurofibromatosis Type I; Neurofibroma, Open Biopsy
ABSTRACT
Aim: There are a variety of malignant tumors related to neurofibromatosis type 1 (NF1). This report describes a rare pediatric NF1 case with an unresectable giant mediastinal tumor. Case: A 6-year-old girl with wheezing was admitted to our institution for the further evaluation of a right mediastinal mass on plain chest radiography. On examination, there were multiple café au lait spots mainly on the trunk, and a well-defined, immobile, painless mass was palpable on her neck. The mediastinal lesion was detected as nonuniform mass surrounding the aortic arch, pulmonary artery, and right main bronchus on the contrast-enhanced CT and MRI. Open biopsy was useful to rule out malignancy and revealed neurofibroma, and contributed to follow up and treatment. Discussion: Open biopsy was useful to rule out malignancy, such as malignant peripheral nerve sheath tumor, revealed neurofibroma, and also contributed to follow up and treatment. The authors report successful management by open biopsy and discuss several clinical points regarding mediastinal neurofibroma for NF1.
1. Introduction
Neurofibromatosis type I (NF1) is a genetic disorder. Mutation of the NF1 gene results in a loss of functional protein, neurofibromin, which is expressed in many tissues, and causes a wide spectrum of clinical findings such as NF1-associated tumors including optic pathway glioma and Malignant Nerve Sheath Tumor (MNST). However, the overall risk of mediastinal tumors is only 2.7% in NF1 patients [1], which are benign of neurofibromas, gliomas, schwannomas, and MPNSTs. Here in, we present a pediatric NF1 case with an unresectable giant mediastinal tumor, which was diagnosed as a neurofibroma. There are some reports that NF1 is rarely associated with resectable mediastinal neurofibroma. This report describes a pediatric NF1 patient with a huge, unresectable mediastinal neurofibroma, which was diagnosed as a neurofibroma on open tumor biopsy. The literature on cases of NF1 with mediastinal neurofibroma is reviewed herein, and the clinical problems associated with this condition are discussed.
2. Case Report
A 6-year-old girl with wheezing was admitted to our institution for the further evaluation of a right mediastinal mass on plain chest radiography. Her mother had previously been diagnosed with NF1. She had no history of perinatal or developmental problems except for the family history. There were no symptoms except for wheezing. She was well-nourished, 107 cm in height, and 16.9 kg in weight. On examination, there were multiple café au lait spots (larger than 5 mm) mainly on the trunk, and a welldefined, immobile, painless mass was palpable on her neck. Her breath sounds were weak at the right upper back. Due to the café au lait spots and her family history, she was diagnosed with NF1.
Laboratory evaluations were normal, with a white blood cell count of 4.2 × 10 × 3/μl and C-reactive protein <0.24 mg/dl. The results of tumor makers were normal, with alfa-fetoprotein of 1.2 mg/ml, human chorionic gonadotropin-β < 0.5 ng/ml, CA19-9 of 11.4 U/ml, CA125 of 6.9 U/ml, urine-vanillyl mandelic acid of 4.6 μg/mgCr, and urine homovanillic acid of 9.6 μg/mgCr. These data showed no abnormal findings except for neuron-specific enolase of 11.29 ng/ml.
On plain chest radiography (Figure 1), there was a poorly defined mass shadow at the right upper mediastinum and the right bronchus deviated to the left. Enhanced Computed Tomography (CT) (Figure 2(A)) revealed a 3.5 × 4.0 × 10.0 cm well-demarcated, nonuniform mass surrounding the aortic arch, pulmonary artery, and right main bronchus, spreading along the internal carotid artery up to near the temporomandibular joint. A plain CT scan of the brain was conducted to rule out NF1-related brain tumor, showing no tumor. Chest magnetic resonance imaging (Figure 2(B)) identified the mass, which was of hypo and hyper intensity on T1-weighted images and hyper intensity on T2-weighted images, showed a clear border with the vertebral canal suggesting no invasion. Therefore, she was diagnosed with a NF1 with mediastinal tumor. Based on those findings, it was impossible to resect the entire mediastinal tumor. Therefore, open tumor biopsy was decided upon for emergent pathological diagnosis and treatment, because she showed slight respiratory symptoms caused by the tumor. Open biopsy was performed via a neck incision. Macroscopically, the tumor was soft, appeared white in color, and was capsulated. Pathological study (Hematoxylin-Eosin stain) (Figure 3) revealed cell proliferation containing
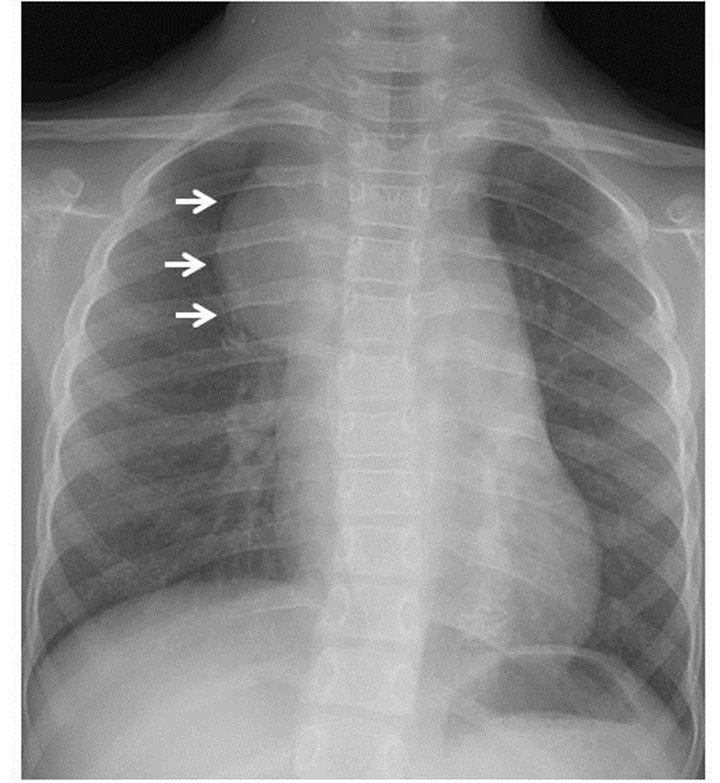
Figure 1. Chest radiography. The arrows indicate a poorly defined mass in the right mediastinum.
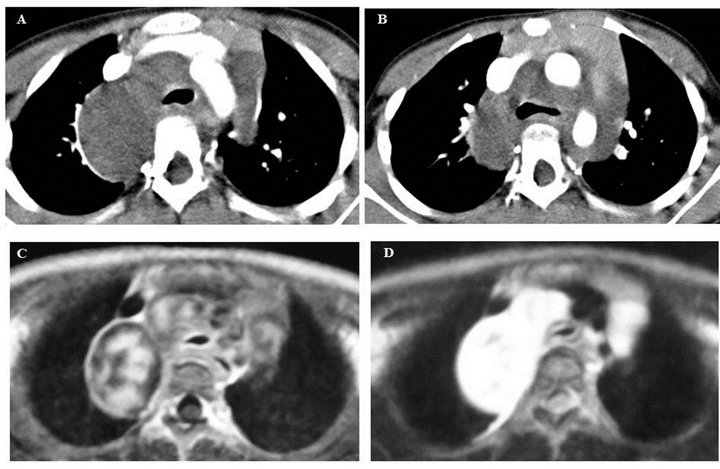
Figures 2. Mediastinal tumor images. (A), (B) Neck and chest CT scan show a tumor surrounded by the aortic arch, pulmonary artery, and right main bronchus. It spreads along the internal caroted artery up to near the temporomandibular joint.
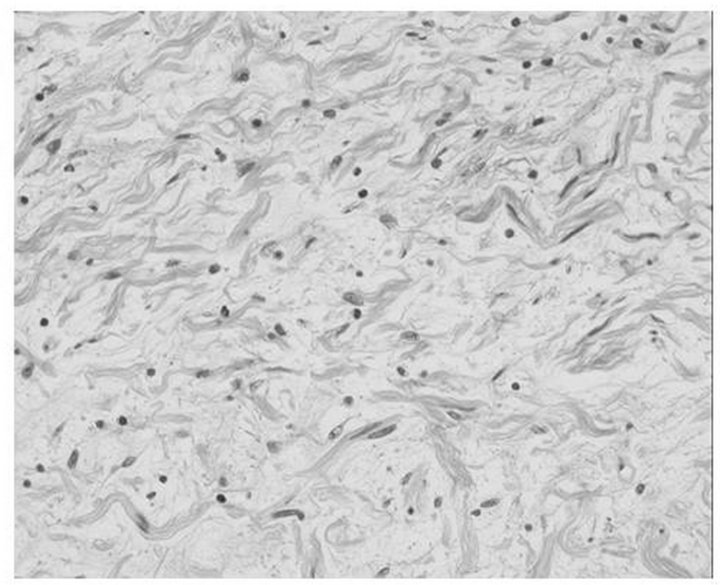
Figure 3. Histopathological manifestation (HE stain). The proliferation of the cell with a poorly heterotypic, spindleshaped nucleus is revealed.
poorly heterotypic spindle shaped nuclei and no malignant findings. These findings confirmed the diagnosis of neurofibroma. Her respiratory symptom subsequently resolved and her course has been uneventful for 2 years.
3. Discussion
NF1 is an autosomal dominant genetic disorder (approximately one-half of cases are familial, and the reminder represent de novo mutations), with an incidence of approximately 1 in 2600 to 1 in 3000 individuals [2]. The NF1 gene is a tumor suppressor gene [3]. Mutation of the NF1 gene, mapped to chromosome 17 q 11.2, result in the loss of a functional protein, neurofibromin, via the inactivation of Ras protein, causing a wide spectrum of clinical findings. Patients with NF1 develop both benign and malignant tumors (NF1-associated tumors) as a wide variety of neuroectodermal, neuroendocrine, and mesenchymal tumors at an increased frequency throughout life [4].
The lifetime risk for NF1 patients developing benign and malignant tumors is higher than in the general population. NF1 related benign tumors are plexiform (15% to 53%), optic glioma (6%), and astrocytoma (1.5%). NF1- related malignant tumors are phenochromocytoma (1.5%) and Malignant Peripheral Nerve Sheath Tumor (MPNST) (7.5% to 15%). Based on these findings, NF1 patients have an increased risk of developing malignancies in general. The overall risk of developing malignancies in NF1 is reported to be 15% to 20%, with a median age of 7 years old [5]. Furthermore, pediatric and adolescent NF1 patients are at an increased risk of developing Central Nervous System (CNS) tumors and malignant connective or soft tissue neoplasms, such as MNST [6,7]. MNST are one of the most aggressive factors associated with NF1, and lead to a poor prognosis [3]. CNS tumors include optic pathway gliomas (15% of NF1 children younger than 6 years old [8]), astrocytomas, brain stem gliomas [9,10]. These are the leading causes of death in NF1 patients. In this case, NF1-related tumors except for mediastinal tumor were not observed on radiological images.
Neurofibroma accounts for 4% of all mediastinal tumors. Neurofibroma with NF1 is associated in less than 1% of all cases [11]. However, it is estimated that 4.5% of NF1 patients develop mediastinal tumors [3]. In our case, it might have been too invasive for her to undergo extirpation of the mediastinal tumor, because of the huge, enrolled aortic arch, pulmonary artery, and right main bronchus. Open tumor biopsy was conducted to establish treatment, which identified neurofibroma. Mediastinal neurofibroma has been reported to arise from the intercostal nerves and sympathetic trunk rather than the vagal and phrenic nerves [11]. In our case, the origin of the mediastinal neurofibroma was thought to be the vagal nerve due to its distribution on radiological images.
Benign neurofibroma is reported to occur in 20% to 50% of NF1 patients [1,12-14], most of whom are less than 30 years old of age, particularly between 0 and 19. Neurofibroma might undergo malignant transformation to MNST. The lifetime risk of developing MNST in patients with NF1 is estimated to be 5% to 8% [5,7,12]. MNST usually develops from pre-existing nodular or diffuse plexiform neurofibroma and present as a prgressively enlarging tumor, often accompanied by pain and neurological symptoms. Unfortunately, delays in the diagnosis of MNST in NF1 are frequent [3]. Therefore, pain, neurological deficit, and the enlargement of preexisting neurofibroma must be investigated by surgical biopsy to rule out malignant transformation [3]. On CT scan, mediastinal tumors appear round and well-circumscribed, and include a cystic and internal calcified part along the distribution of the major mediastinal nerves.
It is generally difficult to determine NF1-related malignant tumor based on imaging studies. However, recently, the findings of enhanced CT, enhanced MRI, and Positron Emission Tomography (PET) are useful for the diagnosis of NF1-related malignant tumor [15,16].
The treatment of NF1-related mediastinal neurofibroma in general remains a surgical challenge. Surgery is usually performed when there are accompanying symptoms and in lesions exhibiting rapid growth [17]. If it is symptomatic and completely resectable surgically, this should be recommended to excise the tumor without damaging other organs and confirm the histopathological characteristics, facilitating a diagnosis. However, when the tumor is too large to fully resect, as in the present case, open tumor biopsy should be performed to rule out malignancy, such as MNST, and help determine an appropriate course of treatment, like in the present case. Medical treatment using antiangiogenic drugs and farnesyl transferase inhibitors is being evaluated [18]. Furthermore, the unpredictable natural history of neurofibromas requires more prospective and neuroimaging studies to determine patients with the highest risk of MNST development [3]. Pain, neurological deficit, and enlargement of the mediastinal tumor in NF1 are suggestive of malignant transformation. In these situations, open tumor biopsy should be conducted to confirm the diagnosis.
Once NF1-related neurofibromas undergo malignant transformation, the 5-year-event free and overall survival rates of patients are 19% and 28%, respectively [19]. NF1 patients with neurofibroma who underwent incomplete resection with a grossly or microscopically positive margin showed a 22% survival rate, whereas those who underwent a complete gross resection or had unknown margins showed a 34% survival rate [20]. In our case, the tumor was too large and located close to major vessels and nerves, and so its complete excision was very difficult without damaging a surrounding organ. In the specific case of dumbbell-shaped mediastinal tumors with spinal cord compressive symptoms, urgent decompression of the spinal cord is needed. Patients who already have symptoms of spinal cord compression might suffer from severe disabilities, even though decompressions are performed correctly.
4. Conclusion
In conclusion, if symptomatic and the mediastinal tumor with NF1 can be completely removed surgically with no harm to surrounding organs, this should be recommended, facilitating a histopathological diagnosis. However, when the mediastinal tumor is too large to resect completely, as in our case, open tumor biopsy should be conducted to rule out malignancy.
REFERENCES
- R. D. Brasfield and T. K. das von Gupta, “Recklinghausen’s Disease: A Clinicopathological Study,” Annals of Surgery, Vol. 175, No. 1, 1972, pp. 86-104. doi:10.1097/00000658-197201000-00015
- M. Lammert, J. M. Friedman, L. Kluwe and V. F. Mautner, “Prevalence of Neurofibromatosis 1 in German Children at Elementary School Enrollment,” Archives of Dermatology, Vol. 141, No. 1, 2005, pp. 71-74. doi:10.1001/archderm.141.1.71
- L. Valeyrie-Allanore, N. Ismaili, S. Bastuji-Garin, J. Zeller, J. Wechsler, J. Revuz and P. Wolkenstein, “Symptoms Associated with Malignancy of Peripheral Nerve Sheath Tumours: A Retrospective Study of 69 Patients with Neurofibromatosis 1,” British Journal of Dermatology, Vol. 153, No. 1, 2005, pp. 79-82. doi:10.1111/j.1365-2133.2005.06558.x
- M. M. Feldkamp, D. H. Gutmann and A. Guha, “Neurofibromatosis Type 1: Piecing the Puzzle Together,” Canadian Journal of Neurological Sciences, Vol. 25, No. 3, 1998, pp. 181-191.
- J. M. McGaughran, D. I. Harris, D. Donnai, D. Teare, R. MacLeoa, R. Westerbeek, H. Kingston, M. Super, R. Harris and D. Q. Evans, “A Clinical Study of Type 1 Neurofibromatosis in North West England,” Journal of Medical Genetics, Vol. 36, No. 3, 1999, pp. 197-203.
- S. A. Rasmussen, Q. Yang and J. M. Friedman, “Mortality in Neurofibromatosis 1: An Analysis Using US Death Certificates,” The American Journal of Human Genetics, Vol. 68, No. 5, 2001, pp. 1110-1111. doi:10.1086/320121
- M. Zoller, B. Rembeck, H. O. Akesson and L. Angervall, “Life Expectancy, Mortality and Prognostic Factors in Neurofibromatosis Type 1: A Twelve-Year Follow-Up of an Epidemiological Study in Goteborg, Sweden,” Acta Dermato-Venereologica, Vol. 75, No. 2, 1995, pp. 136- 140.
- R. A. Lewis, L. P. Gerson, K. A. Axelson, V. M. Riccardi and R. P. von Whitford, “Recklinghausen Neurofibromatosis. II: Incidence of Optic Gliomata,” Ophthalmology, Vol. 91, No. 8, 1984, pp. 929-935.
- A. Creange, J. Zeller, S. Rostaing-Rigattieri, P. Brugières, J. D. Degos, J. Revuz and P. Wolkenstein, “Neurological Complications of Neurofibromatosis Type 1 in Adulthood,” Brain, Vol. 122, No. 3, 1999, pp. 473-481. doi:10.1093/brain/122.3.473
- J. S. Guillamo, A. Creange, C. Kalifa, J. Grill, D. Rodriquez, F. Doz, S. Barbarot, M. Zerah, M. Sanson, S. Bastuji-Garia and P. Wolkenstein, “Prognostic Factors of CNS Tumours in Neurofibromatosis 1 (NF1): A Retrospective Study of 104 Patients,” Brain, Vol. 126, No. 1, 2003, pp. 152-160.
- A. R. Wychulis, W. S. Payne, O. T. Clagett and L. B. Wodner, “Surgical Treatment of Mediastinal Tumors: A 40- Year Experience,” The Journal of Thoracic and Cardiovascular Surgery, Vol. 62, No. 3, 1971, pp. 379-392.
- B. S. Ducatman, B. W. Scheithauer, D. G. Piepgras, H. M. Reiman and D. M. Ilstrup, “Malignant Peripheral Nerve Sheath Tumors. A Clinicopathologic Study of 120 Cases,” Cancer, Vol. 57, No. 10, 1986, pp. 2006-2021. doi:10.1002/1097-0142(19860515)57:10<2006::AID-CNCR2820571022>3.0.CO;2-6
- R. E. Ferner and D. H. Gutmann, “International Consensus Statement on Malignant Peripheral Nerve Sheath Tumors in Neurofibromatosis,” Cancer Research, Vol. 62, No. 5, 2002, pp. 1573-1577.
- J. M. Meis, F. M. Enzinger, K. L. Martz and J. A. Neal, “Malignant Peripheral Nerve Sheath Tumors (Malignant Schwannomas) in Children,” The American Journal of Surgical Pathology, Vol. 16, No. 7, 1992, pp. 694-707. doi:10.1097/00000478-199207000-00008
- J. S. Suh, P. Abenoza, H. R. Galloway, L. I. Everson and H. J. Griffiths, “Peripheral (Extracranial) Nerve Tumors: Correlation of MR Imaging and Histologic Findings,” Radiology, Vol. 183, No. 2, 1992, pp. 341-346.
- D. G. Varma, A. Moulopoulos, A. S. Sara, N. Leeds, R. Kumar and E. E. Kim, “MR Imaging of Extracranial Nerve Sheath Tumors,” Journal of Computer Assisted Tomography, Vol. 16, No. 3, 1992, pp. 448-453. doi:10.1097/00004728-199205000-00020
- T. T. Zacharia, D. Jaramillo, T. Y. Poussaint, B. Korf, “MR Imaging of Abdominopelvic Involvement in Neurofibromatosis Type 1: A Review of 43 Patients,” Pediatric Radiology, Vol. 35, No. 3, 2005, pp. 317-322. doi:10.1007/s00247-004-1352-z
- R. J. Packer, D. H. Gutmann, A. Rubenstein, D. Viskochil, R. A. Zimmerman and G. Vezina, “Plexiform Neurofibromas in NF1: Toward Biologic-Based Therapy,” Neurology, Vol. 58, No. 10, 2002, pp. 1461-1470.
- A. Ferrari, G. Bisogno, A. Macaluso, M. Casanova, P. D’Angelo, P. Pierani, I. Zanetti, R. Alaggio, G. Cecchetto and M. Carti, “Soft-Tissue Sarcomas in Children and Adolescents with Neurofibromatosis Type 1,” Cancer, Vol. 109, No. 7, 2007, pp. 1406-1412.
- H. Neville, C. Corpron, M. L. Blakely and R. Andrassy, “Pediatric Neurofibrosarcoma,” Journal of Pediatric Surgery, Vol. 38, No. 3, 2003, pp. 343-346.