Open Journal of Veterinary Medicine
Vol.3 No.2(2013), Article ID:32687,9 pages DOI:10.4236/ojvm.2013.32016
Feline Hypertrophic Cardiomyopathy Associated with the p.A31P Mutation in cMyBP-C Is Caused by Production of Mutated cMyBP-C with Reduced Binding to Actin
1Department of Clinical Biochemistry and Immunology, Statens Serum Institut, Copenhagen, Denmark
2Department of Small Animals Diseases, Faculty of Life Science, University of Copenhagen, Copenhagen, Denmark
3MRC Centre for Molecular and Cellular Biology, University of Stellenbosch, Cape Town, South Africa
4Institute of Biochemistry and Molecular Biology, University of Southern Denmark, Odense, Denmark
5Institute of Cardiovascular Science, The Heart Hospital, University College London, London, UK
Email: *mic@ssi.dk
Copyright © 2013 Mia T. N. Godiksen et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received February 7, 2013; revised March 7, 2013; accepted April 7, 2013
Keywords: Feline; HCM; cMyBP-C; Mutation; Actin; Y2H; MSMS
ABSTRACT
Hypertrophic cardiomyopathy (HCM) is a myocardial disorder, with complications including heart failure, thromboemboli and sudden death. Human and feline HCM (fHCM) are clinically comparable, thus fHCM may serve as a spontaneous animal model. fHCM in Maine Coon (MC) cats is associated with the p.A31P mutation in the cMyBP-C protein. The mutation is located in the cMyBP-C C0-domain which is known to interact with actin. The presence and levels of the wild type and mutated protein in heart tissue from mutant and wild type MC cats were examined by SDS-PAGE and mass spectrometry (MS). Quantitative yeast-2-hybrid (Y2H) protein-protein interaction analysis was used to assess the effect of the mutation on C0C1/actin interaction. The NMR-based structure of the C0 domain was used to calculate the energetic consequence of replacing alanine with a proline residue. In the homozygous MC cat, the mutated cMyBP-C protein was present, and cMyBPC-C levels were not reduced compared to that of the wild type cat. However, the interaction of actin with mutant cMyBP-C C0C1 was reduced compared to that of wild type. This may be because the substitution of the alanine with proline in position 31 was energetically highly unfavorable and resulted in only one hydrogen bond within the anti-parallel beta-strand compared to two hydrogen-bonds for alanine, possibly destabilizing the structure of the actin-interacting domain. The p.A31P mutation is present in cardiac tissue and the most likely pathogenic mechanism is interference with contractility by reducing binding of the C0C1 domain of cMyBP-C to actin.
1. Introduction
Hypertrophic cardiomyopathy (HCM) is a primary structural disorder of the myocardium; characterized by left ventricular hypertrophy and clinical complications ranging from no symptoms to sustained palpitation, heart failure and sudden cardiac death [1]. In adult humans, HCM has a prevalence of 0.2% [1], the prevalence in cats is unknown, however, it may be as high as 9% - 26.3% in some predisposed breeds including the Maine Coon (MC) breed [2,3]. Feline HCM (fHCM) is the most commonly diagnosed cardiac disease in cats and has been described as an excellent spontaneous animal model for human HCM, as it mimics the clinical characteristics of human disease e.g. risk of heart failure, cardiac arrhythmia and sudden death [4-6].
Within the MC breed, fHCM is frequently associated with a p.A31P substitution in the sarcomeric protein cardiac myosin binding protein-c (cMyBP-C), encoded by the feline MYBPC3 gene [6,7]. When first reported, the mutation was described in a research colony of cats where both homozygous and heterozygous mutationcarrier cats expressed fHCM, suggesting a dominant pattern of inheritance [6]. However, cross-sectional studies in large cohorts of MC cats reveal that the mutated allele has a high frequency (~20%) and suggests that p.A31P associated fHCM is a disease with a very low penetrance in young and middle-aged heterozygous cats [3,7,8].
One of the most frequently involved genes in human HCM is MYBPC3, which is responsible for ~30% of all cases. To date, over 240 MYBPC3 mutations have been associated with human HCM [9,10] these are usually private mutations, although distinct founder mutations have been identified in cohorts from the Netherlands and Finland [11,12]. One such founder mutation, cMyBP-C p.R502W, is the most common HCM-causing mutation in patients of European descent, and is associated with 2.4% of all cases [13]. Although mutations in the MYBPC3 gene are most often associated with autosomal dominant inheritance, recessive inheritance of MYBPC3-associated HCM has also been reported [14-16]. Patients carrying homozygous mutations in MYBPC3 often exhibit an earlier onset and more severe phenotype than heterozygous mutation carriers [14-16].
The cMyBP-C protein plays a regulatory role in the sarcomere of cardiomyocytes and is involved in regulation of cardiac contractility through its binding to myosin as well as to actin, the major component of the thin filament of the sarcomere [17]. The cMyBP-C protein also contributes to sarcomere assembly and stability, without being an essential sarcomere protein; its role is thus rather that of a modulator of sarcomere function [18]. The cMyBP-C protein consists of 11 domains (C0, C1, C2… through to C10); the N-terminus comprises the C0-C2 domains and the C-terminus comprises C8-C10. The feline cMyBP-C p.A31P mutation is located in the cMyBP-C C0 domain, which occurs only in the cardiac isoform [19]. Besides the C0 domain, the cMyBP-C has two other cardiac-specific features; a nine amino acid long insertion (LAGGGRRIS) in the motif between C1 and C2, and a 28-amino acid loop in the C5 domain (NCBI GenBank X84075.1) as reviewed by Flashman et al. in 2004 [20,21].
The majority of human HCM associated mutations in the MYBPC3 gene are located in the region encoding the central domain and the C-terminus of the protein (Biobase Biological Databases:
http://www.biobase-international.com/) [9,22-24]. In the C0 domain, two amino acid substitution mutations; p.G5R and p.T59A and a nonsense mutation p.E76x (x = stop codon), one splice mutation IVS2-2A > G, and finally three small deletions introducing stop codons at residues 59, 74 and 93 have been reported [9,22-24]. Haploinsufficiency is often associated with human cMyBP-C HCM; of the mutations identified in the C0 domain, IVS2-2A > G has been reported to cause haploinsufficiency [9], and the mutations which result in premature stop codons are expected to result in haploinsufficiency. Functional studies of the missense mutations have not yet been reported.
HCM caused by dominant-negative MYBPC3 mutations has also been reported [25] however, little is presently known about the pathogenic mechanism of mutated cMyBP-C in the diseased heart. In particular, the information on the functional consequences of mutations in the N-terminus is inadequate.
In this study, mass spectrometry (MS) analysis was used to compare the presence of cMyBP-C in heart tissue from a MC cat that was a homozygous carrier of the cMyBP-C p.A31P mutation with that of a homozygous wild type MC cat. Furthermore, we examined the effect of the p.A31P amino acid substitution on the interaction between the cMyBP-C C0C1 domain and actin using quantitative b-galactosidase assays in a yeast-two-hybrid (Y2H) system and by in silico molecular modeling.
2. Methods
2.1. Animals
Samples from cardiac muscle tissue were obtained from two MC cats. Cat A was a 36-month-old male homozygous for the p.A31P mutation [3], with a mild HCM phenotype. Cat B was a 16 months old, mutation negative queen, with no clinical signs of HCM. Both cats were examined by 2-D and M-mode echocardiography as previously described [3] just prior to euthanasia. Cat A had a mild asymmetric hypertrophy of the left ventricle consistent with mild HCM. The 2-D measurements of the left ventricle free wall in diastole (LVFWd) were 5.6 mm and the interventricular septum in diastole (IVSd) was 4.1 mm. Cat B had normal cardiac dimensions and function with IVSd and LVFWd less than 5 mm. For the used feline HCM diagnostic criterias we refer to recently publiched data [3]. Owners gave informed consent and the study was approved by the ethics committee at the Department of Small Animal Clinical Sciences, University of Copenhagen.
The procedure for euthanasia involved sedating the cats with a 1 ml intramuscular injection of Zoletil (zolazepam/tiletamin). Following sedation, an intravenous catheter was placed in the cephalic vein of the right front leg and 5 ml of pentobarbital (200 mg/ml) was injected intravenously. The hearts were removed immediately following euthanasia and were stored at −80˚C until use.
2.2. Protein Analysis
Tissue samples (~500 mg) were taken from the left ventricular free wall of the frozen hearts and treated as described previously [26,27] with only few modifications. Briefly, the samples were weighed, placed in a tube on ice and immediately added ice cold sample buffer (8 M urea (InVitrogen, Taastrup, Denmark), 2 M thiourea, T7875 (Sigma, Brondby, Denmark), 50 mM Trizma base, T1503 (Sigma), 75 mM dithiothreitol (DTT) (Sigma), 3% w/v SDS (Bie&Berntsen), and 0.004% w/v bromophenol blue, pH 6.8), at a ratio of 30 µl sample buffer to 1 mg tissue sample. The samples were homogenised using a UltraTurrax 25 Basic (Ika, Straufen, Germany) for 5 - 10 seconds and heated 2 min at 100˚C using a heat block, to be placed on ice for at least 5 min, with subsequently centrifugation at 12.000 rpm (Ole Dich centrifuge, Ole Dich, Hvidovre, Denmark) for 2 min, at 4˚C. In order to enable mass-based separation of sarcomeric proteins, the supernatant containing the extracted, denatured and reduced sarcomeric proteins, was analyzed by SDS-PAGE using Ready Gel Tris-HCl precast 7.5% polyacrylamide gels (BioRad, Copenhagen, Denmark) and Tris/Glycine/SDSbuffer (10XTGS, cat. 161-0772, BioRad, Copenhagen, Denmark) (Figure 1). The coomassie-stained bands were subjected to densitometric scanning on a BioSpectrumAC (UVP Inc., Upland, CA), and the scanned areas were quantified by use of the VisionWorksLS software. The cMyBP-C band was normalized to the bands of myomesin, α-actinin 2 and troponin T, to verify differences in specific loaded protein, Figure 1. Protein bands
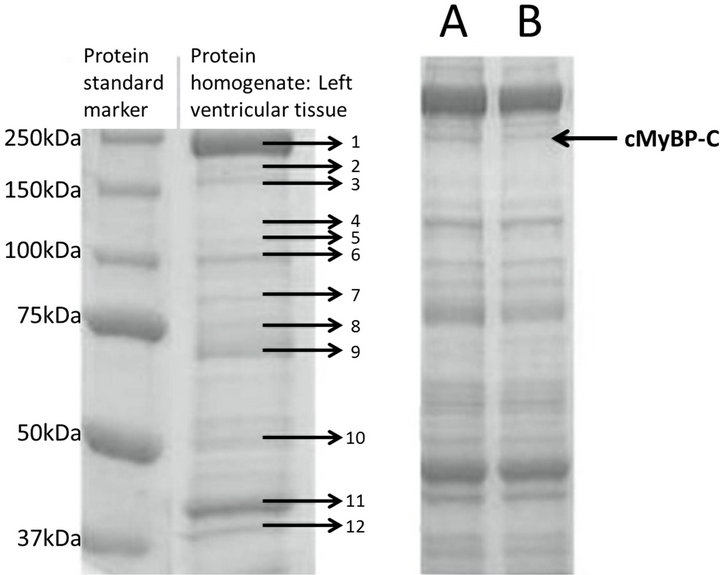
Figure 1. SDS-PAGE analysis of proteins extracted from feline heart tissue. Left-hand gel picture: Lane 1: Standard marker, lane 2: protein extract from feline left ventricle protein homogenate. Feline proteins identified from mass spectrometric analysis: 1) myosin heavy chain, 2) protein M (similar to myomesin), 3) cMyBP-C, 4) vinculin, 5) nicotinamide nucleotide (NAD(P)) trans hydrogenase, 6) α-actinin 2, 7) aconitase 2, 8 + 9) albumin, 10) ATP synthase H+ transporting F1 complex α plus β subunit, 11) actin and 12) troponin T. Gel picture to the right: SDS gel of the tissue homogenate of feline cardiac muscles. The arrow indicates the protein band representing the cMyBP-C. Right-hand gel picture: Cat A was homozygous for the p.A31P mutation, whereas cat B was a cMyBP-C wild type. Protein bands two, six and 12 were used as internal standards to evaluate the relative levels of cMyBP-C (band 3 indicated by arrow).
were cut from a gel reduced by DTT and digested over night with trypsin (Promega, WI, USA) at 37˚C, as described in [28]. Samples were purified on POROS 50R2 micro-columns and loaded on a steel target using 5 µg/µl α-cyano-4-hydroxycinnamic acid matrix [29]. For the identification of proteins, MS and MSMS, separating proteins based on mass-charge (m/z) ratio, were performed on either a MALDI Q-TOF (Micromass, Waters, MA, USA) or a 4700 Proteomics Analyzer (Applied Biosystems, CA, USA) and the data searched against the SwissProt database using the Mascot search engine (Matrix Science, UK). To verify the proposed mutation of cMyBP-C, MS was obtained on a Bruker Ultraflex (Bruker Daltonics, Germany) and MSMS on the 4700 Proteomics Analyzer. MSMS data was interpreted using GPMAW (Lighthouse, Denmark).
2.3. Mutagenesis
The region of MYBPC3 (NM_000256) encoding the C0C1 domain of the cMyBP-C protein was PCR-amplified from a full-length human MYBPC3 cDNA clone (a kind gift from Prof Hugh Watkins, University of Oxford, UK) and cloned in-frame with the GAL4 DNA-binding domain (BD) sequence in pGBKT7 (pGBKT7-C0C1) (Clontech, Mountain View, CA, USA). To assess what influence the p.A31P mutation had on the ability of cMyBP-C-C0C1 to interact with cardiac actin; PCRbased site-directed mutagenesis [30] was used to generate a fragment that coded for the replacement of alanine at position 31 with a proline (p.A31P) within the C0C1 region. Briefly, a set of complementary mutagenesis primers for the mutation, (Table 1), were used in conjunction with pGBKT7-specific outer primers, pGBKT7- F and pGBKT7-R, to separately generate a PCR fragment for the mutation from the pGBKT7-C0C1 construct. For each of the PCR reactions, 100ng of pGBKT7-C0C1 cDNA was used as template in a 50 µl PCR standard reaction. Following purification, 4 µl of each PCR-fragment was added to a second PCR mix and subjected to a further 15 cycles of thermal cycling. These PCR fragments were cloned into pGBKT7 to generate the mutant construct pGBKT7-C0C1-31P. All constructs were used together with a clone encoding the cDNA of cardiac actin (pACT2-ACTC) (a kind gift from Dr. Amsha Ramburan, University of Stellenbosch, SA) in Y2H prey vector pACT2, in quantitative yeast-based interaction assays.
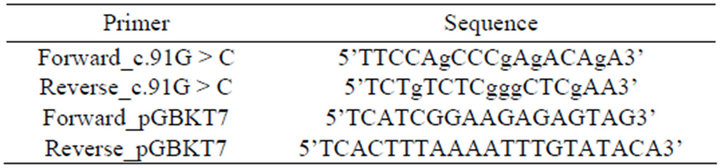
Table 1. Oligonucleotide primer sequences used to generate mutant cMyBP-C C0C1 construct.
2.4. Interaction Studies
The pGBKT7-C0C1-31A and the mutant construct pGBKT7-C0C1-31P were transformed separately into S. cerevisiae strain AH109, and prey plasmid pACT2- ACTC was transformed into strain Y187, by the lithium acetate procedure. The non-recombined pGBKT7 plasmid was used as a negative control. Two independent quantitative β-galactosidase assays were performed with diploid crosses of these baits and preys, each with triplicate samples, and data pooled after normalization to the β-galactosidase production of the negative control, as previously described [31]. One unit of β-galactosidase was defined as the amount of enzyme that hydrolyzes 1 nmol of ONPG to O-nitrophenol and D-galactose per minute per cell. Significant differences between test and control β-galactosidase production was determined by ANOVA followed by a post-hoc Bonferroni multiple comparison test, where a P-value < 0.05 indicated significance.
2.5. Homology Studies
Homology studies of the C0C1 domain were investigated using ClustalW multiple alignment analysis. The first 262 amino acids from cMyBP-C comprise the human C0C1 cMyBP-C domain and the first 262 amino acids of cMyBP-C sequences from different species were aligned: Mus musculus (O70468.1), Rattus norvegicus (NP_001099960.1), Homo sapiens (NP_000247.2), Pan troglodytes (XP_508410.2), Canis lupus familiaris (NP_001041571.1), Felis catus (ensfcap00000002329), Bos Taurus (NP_001070004.1), equus caballus (XP_001491151.2), Gallus gallus (Q90688.3), Xenopus laevis (NP_001082167.1) and Danio rerio (NP_001037814.2). The feline cMyBP-C amino acid sequence was not available at the database at the National Center for Biotechnology Information and was instead obtained from the Ensembl Genome Browser http://www.ensembl.org/index.html.
2.6. Molecular Visualization and Energy Calculations
The structure of the C0 domain of cMyBP-C was obtained from the NMR-based pdb-file 2K1M. This file contains 20 NMR structures and one energetically possible was chosen using the GROMOS forcefield (www.igc.ethz.ch/gromos) in DeepView
(www.spdbv.org). The structure was visualized using Yasara by Yasara Biosciences and the secondary structured was noted. The Yasara program was also used to visualize the C0 domain with the alanine residue substituted by transand cis-proline. The GROMOS force field was used to calculate the changes in total energy by introducing the proline into the secondary and tertiary structure of the C0 domain of cMyBP-C.
3. Results
3.1. Identification of Mutated cMyBP-C in Cardiac Tissue
Mapping of the tryptic peptides from the cardiac cMyBPC bands by MS confirmed the presence of proline at position 31 of the cMyBP-C protein (Figure 2) in the homozygous p.A31P cat (A), but not in the homozygous WT cat (B). A low abundance peak at m/z 1894 was also seen in the spectrum from cat B; however, MSMS confirmed that this did not correspond to the 18 - 35 peptide with a proline residue at position 31. The distribution of proteins extracted from feline cardiac tissue can be seen on the SDS PAGE gel in Figure 1. Clearly distinguishable were myosin heavy chain, protein M (myomesin), actin, α-actinin 2, troponin T and cMyBP-C. After normalizing of the cMyBP-C protein (Figure 1—band three) to the bands of myomesin, α-actinin 2 and troponin T (Figure 1—bands two, six and 12, respectively) no differences in the protein level of cMyBP-C were seen between the mutant and the wild type cMyBP-C form, thus the p.A31P mutation does not appear to result in haploinsufficiency of cMyBP-C in the heart.
3.2. Interaction with Actin
The Y2H assay was used to investigate the interaction between the C0C1 region of cMyBP-C and actin. The β-galactosidase production was higher in yeast colonies producing actin and wild type cMyBP-C C0C1-31A (normalized to negative control: 1.68 units, SE 0.28) than in yeast colonies producing actin and the mutant cMyBPC C0C1 p.31P (normalized to negative control: 0.24 units, SE 0.08). Comparison of β-galactosidase production of C0C1-31A versus C0C1-31P revealed a significant (P = 0.001) reduction of 85% in the production of β-galactosidase, suggesting that carrying a proline at position 31 inhibits the binding of cMyBP-C to cardiac actin (Figure 3).
3.3. Bioinformatics
The multiple alignments of the first ~100 amino acids of cMyBP-C sequences from different species represent the C0 domain of the protein. The alignment showed that the alanine in position 31 of cMyBP-C is conserved in all the examined species (Figure 4), and thus is likely important for protein structure of function.
3.4. Molecular Visualization and Energy Calculations
The visualization of the C0 domain of cMyBP-C from
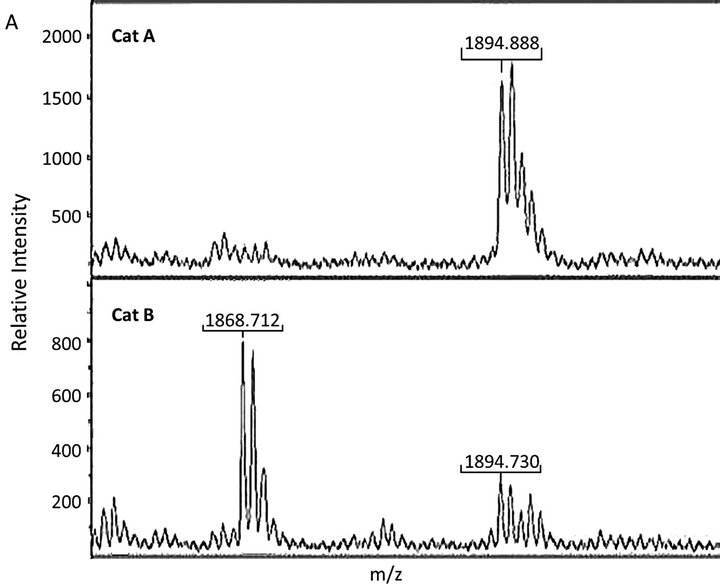
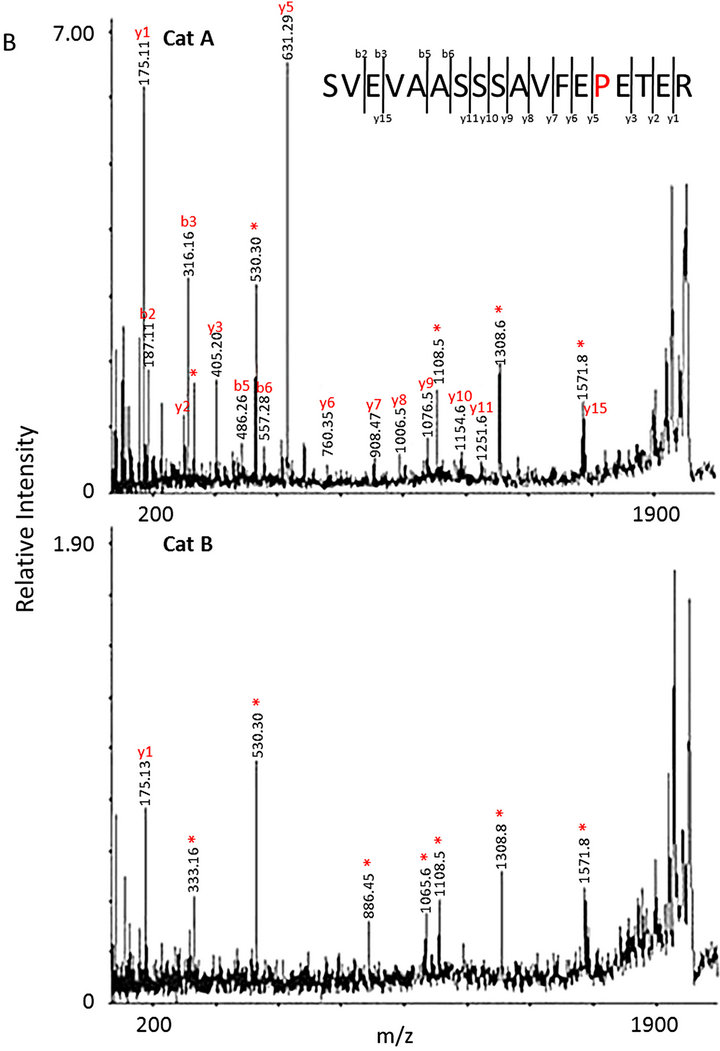
Figure 2. MS analysis of the cMyBP-c band isolated from the SDS-PAGE gel in Figure 1. (A) Peak 1894 (m/z) corresponded to the peptide 18-35 with a proline residue at position 31 identified in cat A. Peak 1868 (m/z) corresponded to the peptide 18-35 with an alanine residue at position 31 from cat B; (B) MSMS spectra of m/z 1894 (m/z) from cat A and cat B. In the top spectrum yand b-fragmentations that can be assigned to peptide 18 - 35 with a proline at position 31 are indicated. *indicates peaks that cannot readily be explained by fragmentation of peptide 18 - 35. The bottom spectrum only contains peaks corresponding to the unexplained ones from cat A showing the presence of an unidentified peptide.
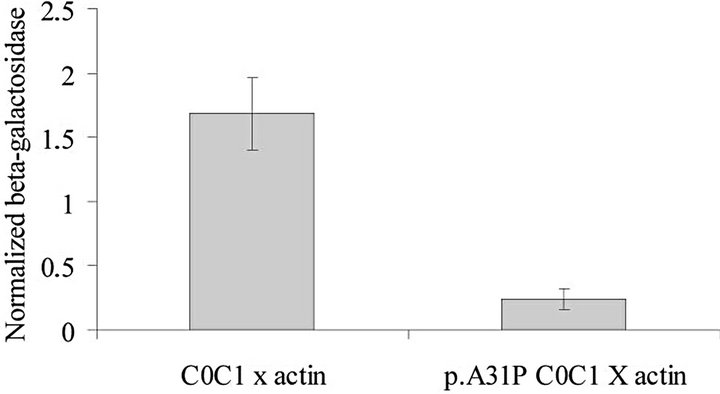
Figure 3. The results of the Y2H assay. Values represent mean ± SE of the mean for three independent assays, each performed with triplicate samples. β-galactosidase production was higher (P < 0.05) than control values in yeasts producing actin and domain C0C1 but not in yeast producing actin and domain C0C1-A31P. The β-galactosidase production of C0C1 versus C0C1-A31P was significantly higher (P = 0.001) suggesting that the p.A31P variant results in reduced binding of C0C1 to actin.
the NMR-structures in the pdb-file 2K1M showed the classical immunoglobulin I-set structure with 9 betastrands assembled into two beta-sheets (Figures 5(a) and (b)) [32,33]. Substitution of the alanine with trans-proline in residue 31 (30 in the finished protein sequence) was highly energetically unfavorable, the total energy of the C0 domain increased from −1847 kJ/mole to +431 kJ/mole following energy minimization, irrespective of which of three rotamers were chosen. As seen in Figure 6(b), the trans-proline extends above the plane of the beta-strand causing steric hindrance, and the energy calculations suggested that it interferes with the tryptophan residue in codon 42, and only one H-bond is possible with the anti-parallel beta-strand. This compared to two H-bonds with alanine.
The cis-proline substitution (Figure 6(c)) was less sterically hindered and the energy calculations following energy minimization resulted in a total energy of −729 kJ/mole, but still only resulted in only one H-bond with the anti-parallel beta-strand.
4. Discussion
In this study, we demonstrated that the feline cMyBP-C variant p.A31P is present in heart tissue from a homozygous MC cat with mild HCM, and that cMyBP-C levels are not reduced as a result of this mutation; hence its pathoetiology is not likely to be due to haploinsufficiency, but more likely involves a dominant-negative effect. Furthermore, we support this finding by showing that the known interaction between actin and domain C0-C1 of cMyBP-C is reduced in the mutant C0C1-31P, compared to that of the wild-type cMyBP-C C0C1-31A domain.
Our visualization of the structure of the C0 domain in cMyBP-C (Figures 5 and 6) suggests that the introduction of a proline, as occurs in the p.A31P cMyBP-C mu-
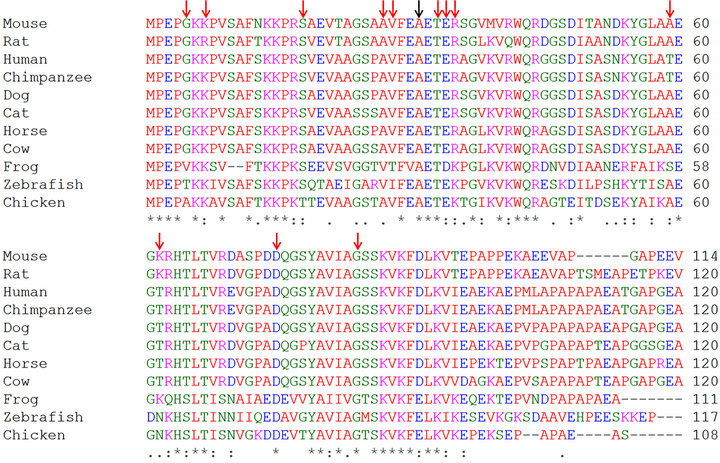
Figure 4. Alignment of the cMyBP-C C0C1 domain amino acid sequence with the sequences from a range of other species. Known missense mutations in human are marked by red arrows and the fHCM mutation is marked by a black arrow.
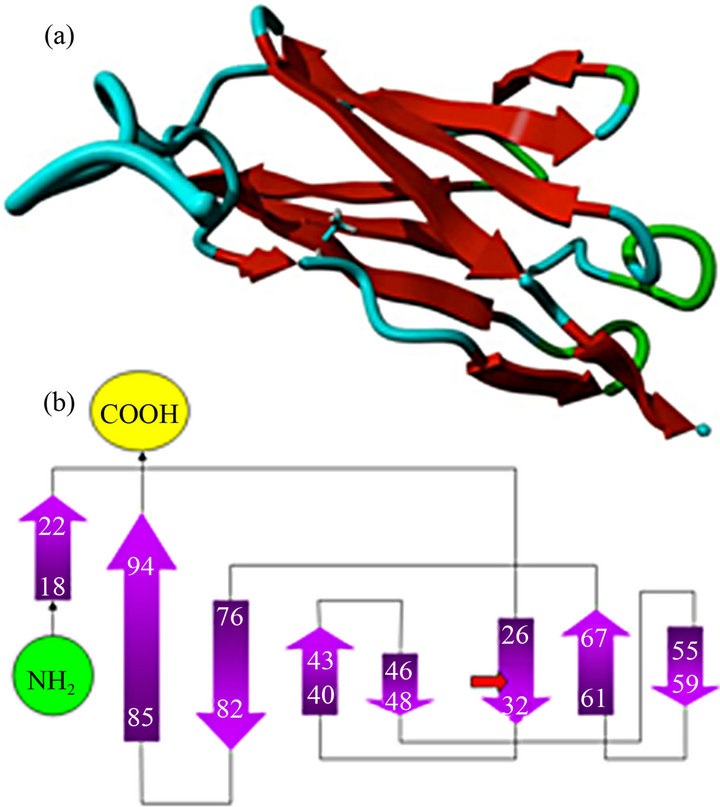
Figure 5. Molecular structure of the C0 domain based on the 2K1M PDB file. (a) C0 structure; (b) Schematic description of the localization of the nine beta strands and the location of the p.A31P mutation.
tant, may be highly energetically unfavorable. The sequence identity between the feline and human cMyBP-C C0 domain is > 95%, consequently the use of the human structure is considered acceptable considering the unavailability of the feline structure. It seems to be a general rule that the introduction of prolines into the inner
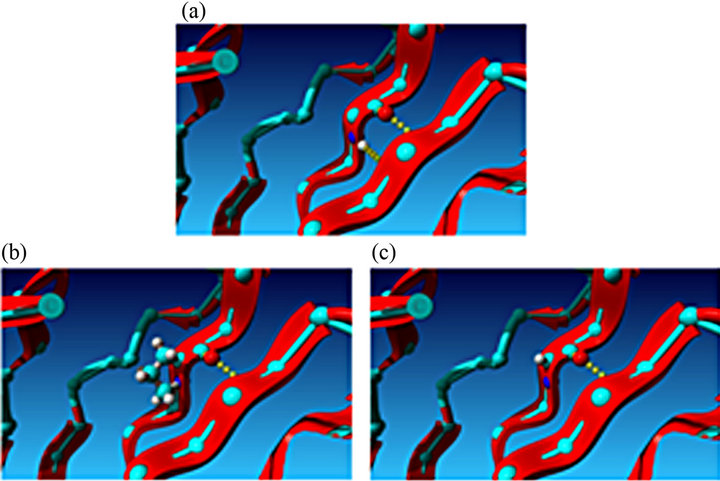
Figure 6. Schematic visualization of the effect of substituting the alanine residue with a proline residue. The proline interrupts the hydrogen bond between two beta strands. (a) Close-up of the alanine residue in the beta strand; (b) Close-up of the C0 domain with the alanine substituted by trans-proline. (c) Close-up of the C0 domain with the alanine substituted by cis-proline.
parts of buried β-strands and β-sheets are not possible without disruption of the structure [34], one reason being that a proline cannot participate in more than one H-bond [34]. Furthermore, prolines are known to contribute to protein folding, which may disrupt tertiary domain structures [35]. Introduction of a proline into a beta sheet strand has previously been reported as pathogenic; a notable example being the HFE p.Q283P mutation in hereditary hemochromatosis patients [36]. Molecular modeling of the pQ283P mutation predicts a destabilizing effect of the mutation on the tertiary structure of the HFE protein [36]. Hence, the feline A31P cMyBP-C mutation is likely to result in an unstable, conformationally distorted, C0 domain with reduced interaction with actin.
Cardiac contraction is regulated by the binding of cMyBP-C to actin and myosin, which are the major components of thin and thick filaments, respectively. The C-terminus of cMyBP-C binds light meromyosin (LMM) and titin and is believed to stabilise the sarcomere during assembly. The N-terminus binds reversibly to actin and myosin S2 [37-39], as determined by phosphorylation of a region (the M-motif) between the C1 and C2 domains [39]. Unphosphorylated cMyBP-C binds to actin and myosin S2; upon phosphorylation, interactions with both myosin S2 and actin are reduced, which allow myosin heads of thick filaments to interact with actin of thin filament [39,40].
Additional actin binding sites have been identified in the cMyBP-C N-terminus; these include regions of the C0 and C1 domains [38-40]. The C0 domain contains an actin binding site which has a similar actin affinity as that of skeletal MyBP-C in myofibrils [38]. Using a quantitative Y2H direct protein-protein interaction assay, we have determined that the binding between C0C1 and actin is significantly reduced in assays using a mutated C0C1 (p.31P) (P = 0.001) (Figure 3) compared to those containing normal wild type C0C1 (p.31A). Shaffer et al. (2009) showed that actin binding sites in C1 are not affected by M-motif phosphorylation and are thus not part of the reversible regulation of contraction [40]; this is most likely also the case for the actin binding site in the C0 domain.
The fact that the C0 domain is cardiac-specific indicates that the role of the C0 domain is not essential for sarcomere contraction. Therefore we suggest that the C0 domain might add an extra “tightness” to the sarcomere structure and may be important to sustain sarcomeric organization during the repeated strain of cardiac contraction. The loss of this support, caused by the homozygous presence of the p.A31P mutation may then result in compensatory hypertrophy seen as fHCM in MC cats [3].
This study is, to our knowledge, the first to present a functional effect of a mutation in the C0 domain of cMyBP-C protein and an association with HCM pathogenesis. Known human C0 domain mutations have only been associated with HCM based on their presence in HCM patients and absence in control groups. Two cMyBPC C0 domain missense mutations in human HCM patients have been described, p.G5R in compound heterozygosity with p.R502W is described as a founder mutation found in European patients [13,24], and p.T59A cMyBP-C was found in an elderly woman [22]. The latter mutation is very unlikely to be disease causing as alanine is the most common amino acid at residue 59 in mammals (Figure 4), and a threonine/alanine substitution is a very conservative change. This underscores that it is very difficult to verify if a potential disease-causing mutation segregates with the disease in single individuals and small families, and thus functional analysis of possible disease-causing mutations is important.
The fact that the cMyBP-C p.A31P associated fHCM exhibits a very low penetrance in cats which are heterozygous carriers of the mutation indicates that interference with the cMyBP-C C0 actin interaction is perhaps less pathogenic than mutations in the rest of the gene. Thus, both alleles are necessary to provoke fHCM in young and middle-aged cats, whereas single dose gene effects may be seen in mutations involving the rest of the gene [3,8].
In summary, we have shown that the p.A31P mutation of feline cMyBP-C results in expression of cMyBP-C in which the alanine residue in position 31 has been substituted by proline. The N-terminal of this protein has been shown to exhibit a markedly reduced interaction with actin, which is explained by the structural and energetic effects of the introduced proline. Most likely, the reduced interaction between cMyBP-C and actin results in a sarcomere with reduced lateral stability, which conceivably may, over time, trigger hypertrophic responses in an effort to compensate for a reduced ability to cope with contractile strain.
5. Acknowledgements
The human MYBPC3 cDNA clone was a kind gift Prof. Hugh Watkins, University of Oxford and the human cardiac ACTIN cDNA clone was a kind gift from Dr. Amsha Ramburan, University of Stellenbosch, SA. Both are highly acknowledged. This study was supported by the Novo Nordisk foundation.
REFERENCES
- J. A. Towbin, “Hypertrophic Cardiomyopathy,” Pacing and Clinical Electrophysiology, Vol. 32, No. S2, 2009, pp. S23-S31. doi:10.1111/j.1540-8159.2009.02381.x
- S. Gundler, A. Tidholm and J. Haggstrom, “Prevalence of Myocardial Hypertrophy in a Population of Asymptomatic Swedish Maine Coon Cats,” Acta Veterinaria Scandinavica, Vol. 50, 2008, p. 22. doi:10.1186/1751-0147-50-22
- M. T. N. Godiksen, S. Granström, J. Koch and M. Christiansen, “Hypertrophic Cardiomyopathy in Young Maine Coon Cats Caused by the p.A31P cMyBP-C Mutation— The Clinical Significance of Having the Mutation,” Acta Veterinaria Scandinavica, Vol. 53, No. 7, 2011, pp. 2-11.
- P. R. Fox, S. K. Liu and B. J. Maron, “Echocardiographic Assessment of Spontaneously Occurring Feline Hypertrophic Cardiomyopathy. An Animal Model of Human Disease,” Circulation, Vol. 92, No. 9, 1995, pp. 2645- 2651. doi:10.1161/01.CIR.92.9.2645
- M. D. Kittleson, K. M. Meurs, M. J. Munro, J. A. Kittleson, S. K. Liu, P. D. Pion and J. A. Towbin, “Familial Hypertrophic Cardiomyopathy in Maine Coon Cats: An Animal Model of Human Disease,” Circulation, Vol. 99, No. 31, 1999, pp. 72-80.
- K. M. Meurs, X. Sanchez, R. M. David, N. E. Bowles, J. A. Towbin, P. J. Reiser, J. A. Kittleson, M. J. Munro, K. Dryburgh, K. A. MacDonald and M. D. Kittleson, “A Cardiac Myosin Binding Protein C Mutation in the Maine Coon Cat with Familial Hypertrophic Cardiomyopathy,” Human Molecular Genetics, Vol. 14, No. 23, 2005, pp. 3587-3593. doi:10.1093/hmg/ddi386
- R. Fries, A. M. Heaney and K. M. Meurs, “Prevalence of the Myosin-Binding Protein C Mutation in Maine Coon Cats,” Journal of Veterinary Internal Medicine, Vol. 22, No. 4, 2008, pp. 893-896. doi:10.1111/j.1939-1676.2008.0113.x
- G. Wess, C. Schinner, K. Weber, H. Kuchenhoff and K. Hartmann, “Association of A31P and A74T Polymorphisms in the Myosin Binding Protein C3 Gene and Hypertrophic Cardiomyopathy in Maine Coon and Other Breed Cats,” Journal of Veterinary Internal Medicine, Vol. 24, No. 3, 2010, pp. 527-532. doi:10.1111/j.1939-1676.2010.0514.x
- P. S. Andersen, O. Havndrup, H. Bundgaard, L. A. Larsen, J. Vuust, K. A. Pedersen, K. Kjeldsen and M. Christiansen, “Genetic and Phenotypic Characterization of Mutations in Myosin-Binding Protein C (MYBPC3) in 81 Families with Familial Hypertrophic Cardiomyopathy: Total or Partial Haploinsufficiency,” European Journal of Human Genetics, Vol. 12, No. 8, 2004, pp. 673-677. doi:10.1038/sj.ejhg.5201190
- A. J. Marian, “Genetic Determinants of Cardiac Hypertrophy,” Current Opinion in Cardiology, Vol. 23, No. 3, 2008, pp. 199-205. doi:10.1097/HCO.0b013e3282fc27d9
- P. Jaaskelainen, J. Kuusisto, R. Miettinen, P. Karkkainen, S. Karkkainen, S. Heikkinen, P. Peltola, J. Pihlajamaki, I. Vauhkonen and M. Laakso, “Mutations in the Cardiac Myosin-Binding Protein C Gene Are the Predominant Cause of Familial Hypertrophic Cardiomyopathy in Eastern Finland,” Journal of Molecular Medicine, Vol. 80, No. 7, 2002, pp. 412-422. doi:10.1007/s00109-002-0323-9
- M. Alders, R. Jongbloed, W. Deelen, A. van den Wijngaard, P. Doevendans, F. Ten Cate, V. Regitz-Zagrosek, H. P. Vosberg, I. van Langen, A. Wilde, D. Dooijes and M. Mannens, “The 2373insG Mutation in the MYBPC3 Gene Is a Founder Mutation, Which Accounts for Nearly One-Fourth of the HCM Cases in the Netherlands,” European Heart Journal, Vol. 24, No. 20, 2003, pp. 1848-1853. doi:10.1016/S0195-668X(03)00466-4
- A. J. Saltzman, D. Mancini-Dinardo, C. Li., W. K. Chung, C. Y. Ho, S. Hurst, J. Wynn, M. Care, R. M. Hamilton, G. W. Seidman, J. Gorham, B. McDonough, E. Sparks, J. G. Seidman, C. E. Seidman and R. L. Rehm, “The Cardiac Myosin Binding Protein C Arg502Trp Mutation. A Common Cause of Hypertrophic Cardiomyopathy,” Circulation Research, Vol. 108, 2010, pp. 743-750.
- L. Nanni, M. Pieroni, C. Chimenti, B. Simionati, R. Zimbello, A. Maseri, A. Frustaci and G. Lanfranchi, “Hypertrophic Cardiomyopathy: Two Homozygous Cases with ‘Typical’ Hypertrophic Cardiomyopathy and Three New Mutations in Cases with Progression to Dilated Cardiomyopathy,” Biochemical and Biophysical Research Communications, Vol. 309, No. 2, 2003, pp. 391-398. doi:10.1016/j.bbrc.2003.08.014
- B. Xin, E. Puffenberger, J. Tumbush, J. R. Bockoven and H. Wang, “Homozygosity for a Novel Splice Site Mutation in the Cardiac Myosin-Binding Protein C Gene Causes Severe Neonatal Hypertrophic Cardiomyopathy,” American Journal of Medical Genetics Part A, Vol. 143A, No. 22, 2007, pp. 2662-2667. doi:10.1002/ajmg.a.31981
- K. Zahka, K. Kalidas, M. A. Simpson, H. Cross, B. B. Keller, C. Galambos, K. Gurtz, M. A. Patton and A. H. Crosby, “Homozygous Mutation of MYBPC3 Associated with Severe Infantile Hypertrophic Cardiomyopathy at High Frequency among the Amish,” Heart, Vol. 94, No. 10, 2008, pp. 1326-1330. doi:10.1136/hrt.2007.127241
- C. Moos, C. M. Mason, J. M. Besterman, I. N. Feng and J. H. Dubin, “The Binding of Skeletal Muscle C-Protein to F-Actin, and Its Relation to the Interaction of Actin with Myosin Subfragment-1,” Journal of Molecular Biology, Vol. 124, No. 4, 1978, pp. 571-586. doi:10.1016/0022-2836(78)90172-9
- S. P. Harris, C. R. Bartley, T. A. Hacker, K. S. McDonald, P. S. Douglas, M. L. Greaser, P. A. Powers and R. L. Moss, “Hypertrophic Cardiomyopathy in Cardiac Myosin Binding Protein-C Knockout Mice,” Circulation Research, Vol. 90, No. 5, 2002, pp. 594-601. doi:10.1161/01.RES.0000012222.70819.64
- L. Carrier, G. Bonne, E. Bahrend, B. Yu, P. Richard, F. Niel, B. Hainque, C. Cruaud, F. Gary, S. Labeit, J. B. Bouhour, O. Dubourg, M. Desnos, A. A. Hagege, R. J. Trent, M. Komajda, M. Fiszman and K. Schwartz, “Organization and Sequence of Human Cardiac Myosin Binding Protein C Gene (MYBPC3) and Identification of Mutations Predicted to Produce Truncated Proteins in Familial Hypertrophic Cardiomyopathy,” Circulation Research, Vol. 80, No. 3, 1997, pp. 427-434.
- M. Gautel, O. Zuffardi, A. Freiburg and S. Labeit, “Phosphorylation Switches Specific for the Cardiac Isoform of Myosin Binding Protein-C: A Modulator of Cardiac Contraction?” The EMBO Journal, Vol. 14, No. 9, 1995, pp. 1952-1960.
- E. Flashman, C. Redwood, J. Moolman-Smook and H. Watkins, “Cardiac Myosin Binding Protein C: Its Role in Physiology and Disease,” Circulation Research, Vol. 94, No. 10, 2004, pp. 1279-1289. doi:10.1161/01.RES.0000127175.21818.C2
- H. Niimura, K. K. Patton, W. J. McKenna, J. Soults, B. J. Maron, J. G. Seidman and C. E. Seidman, “Sarcomere Protein Gene Mutations in Hypertrophic Cardiomyopathy of the Elderly,” Circulation, Vol. 105, No. 4, 2002, pp. 446-451. doi:10.1161/hc0402.102990
- P. Richard, P. Charron, L. Carrier, C. Ledeuil, T. Cheav, C. Pichereau, A. Benaiche, R. Isnard, O. Dubourg, M. Burban, J. P. Gueffet, A. Millaire, M. Desnos, K. Schwartz, B. Hainque and M. Komajda, “Hypertrophic Cardiomyopathy: Distribution of Disease Genes, Spectrum of Mutations, and Implications for a Molecular Diagnosis Strategy,” Circulation, Vol. 107, No. 17, 2003, pp. 2227- 2232. doi:10.1161/01.CIR.0000066323.15244.54
- S. L. Van Driest, V. C. Vasile, S. R. Ommen, M. L. Will, J. A. Tajik, B. J. Gersh and J. M. Ackerman, “Myosin Binding Protein C Mutations and Compound Heterozygosity in Hypertrophic Cardiomyopathy,” Journal of the American College of Cardiology, Vol. 44, No. 9, 2004, pp. 1903-1910. doi:10.1016/j.jacc.2004.07.045
- J. L. Theis, J. M. Bos, J. D. Theis, D. V. Miller, J. A. Dearani, H. V. Schaff, B. J. Gersh, S. R. Ommen, R. L. Moss and M. J. Ackerman, “Expression Patterns of Cardiac Myofilament Proteins: Genomic and Protein Analysis of Surgical Myectomy Tissue from Patients with Obstructive Hypertrophic Cardiomyopathy,” Circulation: Heart Failure, Vol. 2, No. 4, 2009, pp. 325-333. doi:10.1161/CIRCHEARTFAILURE.108.789735
- E. R. Blough, E. R. Rennie, F. Zhang and P. J. Reiser, “Enhanced Electrophoretic Separation and Resolution of Myosin Heavy Chains in Mammalian and Avian Skeletal Muscles,” Analytical Biochemistry, Vol. 233, No. 1, 1996, pp. 31-35. doi:10.1006/abio.1996.0003
- P. J. Reiser and W. O. Kline, “Electrophoretic Separation and Quantitation of Cardiac Myosin Heavy Chain Isoforms in Eight Mammalian Species,” American Journal of Physiology, Vol. 274, No. 3, 1998, pp. H1048-H1053.
- A. Shevchenko, M. Wilm, O. Vorm and M. Mann, “Mass Spectrometric Sequencing of Proteins Silver-Stained Polyacrylamide Gels,” Analytical Chemistry, Vol. 68, No. 5, pp. 850-858. doi:10.1021/ac950914h
- J. Gobom, E. Nordhoff, E. Mirgorodskaya, R. Ekman and P. Roepstorff, “Sample Purification and Preparation Technique Based on Nano-Scale Reversed-Phase Columns for the Sensitive Analysis of Complex Peptide Mixtures by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry,” Journal of Mass Spectrometry, Vol. 34, No. 2, 1999, pp. 105-116. doi:10.1002/(SICI)1096-9888(199902)34:2<105::AID-JMS768>3.0.CO;2-4
- M. Hass, M. Westerkofsky, S. Muller, B. Becker-Ziaja, C. Busch and S. Gunther, “Mutational Analysis of the Lassa Virus Promoter,” Journal of Virology, Vol. 80, No. 24, 2006, pp. 12414-12419. doi:10.1128/JVI.01374-06
- E. Flashman, L. Korki, H. Watkins, C. Redwood and J. Moolman-Smook, “Support for a Trimeric Collar of Myosin Binding Protein C in Cardiac and Fast Skeletal Muscle, but Not in Slow Skeletal Muscle,” FEBS Letters, Vol. 582, No. 3, 2008, pp. 434-438. doi:10.1016/j.febslet.2008.01.004
- P. Bork, L. Holm and C. Sander, “The Immunoglobulin Fold. Structural Classification, Sequence Patterns and Common Core,” Journal of Molecular Biology, Vol. 242, No. 4, 1994, pp. 309-320. doi:10.1016/S0022-2836(84)71582-8
- D. K. Smith and H. Xue, “Sequence Profiles of Immunoglobulin and Immunoglobulin-Like Domains,” Journal of Molecular Biology, Vol. 274, No. 4, 1997, pp. 530-545. doi:10.1006/jmbi.1997.1432
- S. C. Li, N. K. Goto, K. A. Williams and C. M. Deber, “Alpha-Helical, but Not Beta-Sheet, Propensity of Proline Is Determined by Peptide Environment,” Proceedings of the National Academy of Sciences of the USA, Vol. 93, No. 13, 1996, pp. 6676-6681. doi:10.1073/pnas.93.13.6676
- K. W. Plaxco, C. Spitzfaden, I. D. Campbell and C. M. Dobson, “Rapid Refolding of a Proline-Rich All-Beta-Sheet Fibronectin Type III Module,” Proceedings of the National Academy of Sciences of the USA, Vol. 93, No. 20, 1996, pp. 10703-10706. doi:10.1073/pnas.93.20.10703
- G. G. Le, F. Y. Dupradeau, C. Mura, S. Jacolot, V. Scotet, G. Esnault, A. Y. Mercier, J. Rochette and C. Ferec, “Phenotypic Expression of the C282Y/Q283P Compound Heterozygosity in HFE and Molecular Modeling of the Q283P Mutation Effect,” Blood Cells Molecules and Diseases, Vol. 30, No. 3, 2003, pp. 231-237. doi:10.1016/S1079-9796(03)00036-6
- G. Kunst, K. R. Kress, M. Gruen, D. Uttenweiler, M. Gautel and R. H. Fink, “Myosin Binding Protein C, a Phosphorylation-Dependent Force Regulator in Muscle That Controls the Attachment of Myosin Heads by Its Interaction with Myosin S2,” Circulation Research, Vol. 86, No. 1, 2000, pp. 51-58. doi:10.1161/01.RES.86.1.51
- I. Kulikovskaya, G. McClellan, J. Flavigny, L. Carrier and S. Winegrad, “Effect of MyBP-C Binding to Actin on Contractility in Heart Muscle,” The Journal of General Physiology, Vol. 122, No. 6, 2003, pp. 761-774. doi:10.1085/jgp.200308941
- J. M. Squire, P. K. Luther and C. Knupp, “Structural Evidence for the Interaction of C-Protein (MyBP-C) with Actin and Sequence Identification of a Possible ActinBinding Domain,” Journal of Molecular Biology, Vol. 331, No. 3, 2003, pp. 713-724. doi:10.1016/S0022-2836(03)00781-2
- J. F. Shaffer, R. W. Kensler and S. P. Harris, “The Myosin-Binding Protein C Motif Binds to F-Actin in a Phosphorylation-Sensitive Manner,” The Journal of Biological Chemistry, Vol. 284, No. 18, 2009, pp. 12318-12327. doi:10.1074/jbc.M808850200
NOTES
*Corresponding author.