Computational Molecular Bioscience
Vol.2 No.4(2012), Article ID:25315,4 pages DOI:10.4236/cmb.2012.24012
A Brief Overview of a Few Popular and Important Protein Databases
Department of Biochemistry and Biophysics, University of Kalyani, Kalyani, India
Email: angshuman_bagchi@yahoo.com
Received September 5, 2012; revised October 18, 2012; accepted November 13, 2012
Keywords: Database; SCOP; CATH; UniProt; PDB
ABSTRACT
Database is a repository of information. In today’s world there are different types of databases available. In this present review the focus is on a few popular and most widely used biological databases that store protein sequence and structure information. The databases that are of utmost importance to do basic biological research work are PDB, SCOP, CATH and UniProt/SwissProt and GenBank. These databases have different utilities & they play important roles in different fields of biology and bioinformatics. PDB provides the structural information of proteins, protein-complexes and proteins complexed with other macromolecules. SCOP & CATH store various annotations of protein sequences and structures. UniProt is a central repository of protein sequences & functions created by joining the information contained in SwissProt, TrEMBL.
1. Introduction
A database is a repository or collection of information that are organized in such a way that it can easily be accessed, managed and updated. In other words, database may be defined as a collection of related tables that themselves contain lists of records. To ensure that each record is unique, each of the records is given a primary key, usually in the form of an identification number. There are basically two different kinds of databases. They are primary database and secondary database. A primary database consists of data derived experimentally such as sequences and structures of biological molecules. On the other hand, a secondary database contains data derived from the analyses and annotations of the data present in primary databases [1-10]. In today’s world the gathering and processing of data constitute a very important part of research. Specially, in the field of biology data collection plays a vital role. A comparatively new but very important and upcoming field of modern day biotechnological research is bioinformatics which specifically deals with data processing. With time various new biological databases have been evolved. All the databases have certain important features associated with them. Therefore in the present review the focus is on the five most important protein sequence and structure databases viz., Protein Data Bank (PDB), SCOP (Structural Classification of Protein), CATH (Class Architecture Topology Homology), UniProt (Universal Protein Structure)/Swissprot and another database GenBank which contains both nucleotide and protein sequence information. One thing that worth mentioning here is there are certain other very important and useful biological databases which store information about proteins. But the databases that have been discussed here are the most important ones and used by majority of the biologists and biotechnologists. This review may serve as a good starting material for researches interested to use and process the biological information. The review would provide a first hand guide to those people who would like to start their bioinformatics research.
2. The Protein Data Bank (PDB) (www.rcsb.org/pdb)
The Protein Data Bank (or PDB) is a primary database. PDB is a repository of the 3-dimensional structural data of large biological macromolecules such as protein and nucleic acid or their complexes. PDB stores the data that are obtained by X-ray crystallography, NMR, electron microscopy. The PDB is the key resource for structural biotechnologists. Even scientists from other areas search the PDB to have idea about the structures of biological macromolecules. PDB database is updated weekly. As of November 1, 2011 the PDB contains the following components presented in Table 1. These data show that most of the data in PDB are determined by X-ray crystallography. About 12% of structures are determined by protein
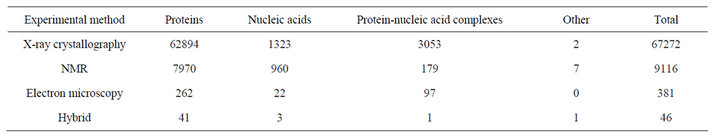
Table 1. Contents of PDB.
NMR. From X-ray diffraction method, the approximations of the coordinates of the atoms of the protein are obtained whereas NMR method estimates the distances between pairs of atoms of the protein. So for NMR method a distance geometry problem has to be solved to obtain final conformation of the protein. Very few protein structures are determined by electron microscopy [11-17].
3. PDB File Format
The file format that was used by the PDB was called the PDB file format. However, during the late nineties, the “macromolecular Crystallographic Information File” format, mmCIF, was introduced. The details of the file format have been presented in an XML version which is called PDBML. The structure files can be downloaded from the pdb web server in any of these formats. The individual pdb files can easily be downloaded into graphics packages using web addresses. By convention, the names of each pdb files start with a number followed by three letters like 1smt. This is also called the PDB ID. It has been observed that the PDB files contain numerous inconsistencies and errors. In some cases the pdb file format is violated. There are inconsistent residue numbering and missing values for experimental parameters. Many authors have pointed out the problems with the experimental data or its interpretation by the submitters [11-17].
4. Contents
The deposited data to PDB are considered as primary data. These primary data may include atomic coordinates, information related to the chemistry of the macromolecule, the small-molecule ligands, some data collection details, structure refinement, and some structural descriptors. A PDB entry may contain about 400 unique items of data.
5. Data Acquisition and Processing
There are three main steps in Data processing: data deposition, annotation and validation. Previously data were submitted to PDB via email. But now author can submit his/her data online via the PDB AutoDep Input Tool (ADIT; http://pdb.rutgers.edu/adit/) based on mmCIF developed by the RCSB. Within minutes of structure deposition using ADIT, a PDB identifier is sent to the author automatically. Although different tools are used for data processing, all of them use the same principles and algorithms. Then the entry is annotated using ADIT to identify errors or inconsistencies in the files. Validation procedure is used for assessing the quality of deposited atomic models (structure validation) and agreement of these models with the experimental data. For accuracy PDB files are checked in various aspects such as nomenclature, chemistry of the polymer and ligands, stereochemical validation etc. In present times structure factors and NMR constraint files are also deposited along with most of the data files. So now it becomes possible to calculate the agreement with experimental data using SFCheck [18]. The author reviews the processed files and sends back after any further revisions. Depending on the importance of these revisions, previous steps may be repeated. As soon as approval is received from the author it becomes ready for distribution.
6. Shortcomings
PDB may have some better representation such as for: very large macromolecules, disordered structures, X-ray structures refined with multiple models. Most difficult problem for the PDB is that the files are not uniform.
7. SCOP (Structural Classification of Protein) (http://scop.mrc-lmb.cam.ac.uk/scop/)
The Structural Classification of Proteins (SCOP) database is basically a database with manual classification of protein structural domains. The whole concept is based on similarities of the amino acid sequences and threedimensional structures of the proteins. The database was originally published in 1995 and it is usually updated at least once yearly by Alexei G. Murzin and his colleagues [19-22].
SCOP database uses the following protein structural hierarchy:
Class—It is the general structural architecture of the protein domains.
Fold—It represents similar arrangement of regular secondary structures but without evidence of evolutionary relatedness.
Superfamily—It represents whether the protein structures have sufficient structural and functional similarities to each other to infer a divergent evolutionary relationship but not necessarily a detectable sequence homology.
Family—Proteins belonging to the same family share some sequence similarity.
This classification of proteins in SCOP is more significantly based on the human expertise. It is generally accepted that SCOP gives a better justified classification. It is generally the human expertise that is important to decide whether some proteins are evolutionarily related and therefore should be placed in the same super family, or their similarity is only a result of structural constraints present in the proteins to classify them to the same fold.
SCOP has the following classes:
1) Proteins with mostly α-helical domains;
2) Proteins with mostly β-sheet domains;
3) Proteins with α/β domains which contain betaalpha-beta structural units or motifs that form mainly parallel β-sheets;
4) Proteins with mostly α + β domains consisting of independent α-helices and mainly antiparallel β-sheets;
5) Multi-domain proteins;
6) Membrane proteins and cell surface proteins and peptides;
7) Small proteins;
8) Coiled-coil proteins;
9) Low-resolution protein structures;
10) Peptides and fragments;
11) Proteins designed from non-natural sequence.
Currently the contents of SCOP database are as in the Table 2. This statistics is as per the latest SCOP update; SCOP: Structural Classification of Proteins. 1.75 release based on 38221 PDB Entries as of 23 Feb 2009.
8. CATH (Class Architecture Topology Homology) (http://www.cathdb.info/)
The CATH Protein Structure Classification method is a semi-automatic, hierarchical classification of protein domains. The database was first published in 1997 by Christine Orengo, Janet Thornton and their colleagues. CATH carries many broad features with its principal rival, SCOP. However there are also many areas in which the detailed classifications in the two databases differ greatly [22-25].
CATH clusters proteins at four major levels:
1) Class (C): Class is derived from secondary structure contents of proteins. It is assigned for more than 90% of protein structures automatically.
2) Architecture (A): Architecture describes the gross orientation of secondary structures, independent of connectivity in proteins.
3) Topology (T): Topology level of CATH clusters protein structures according to their topological connections & numbers of secondary structures.
4) Homologous Superfamily (H): Homologous superfamilies of CATH cluster the proteins with highly similar structures & functions. The assignments of protein structures to the last two categories i.e., the topology & homologous superfamilies, are made by sequence & structure comparisons [22-25].
In order to distinguish between SCOP and CATH the steps to classify proteins in SCOP and CATH are to be discussed.
In the first step of the classification in CATH, the proteins are separated into domains. It is difficult to produce an unequivocal definition of a domain. So in this is area CATH and SCOP differ. Then the domains are automatically sorted into classes and then they are clustered on the basis of sequence similarities. The topology level in CATH is then formed by structural comparisons of the
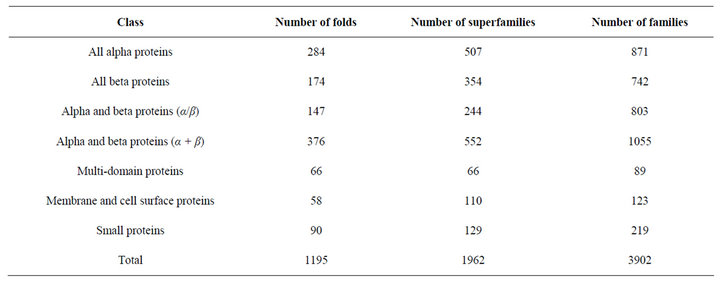
Table 2. Contents of SCOP.
homologous groups of proteins. Then the Architecture level is assigned manually in CATH [22-25].
In CATH the Class level classification is done on the basis of the following 4 criteria: The secondary structure contents of proteins, the secondary structure contacts of proteins, the secondary structure alternation score and the percentage of parallel strands in proteins [22-25]. The latest version of CATH (V 3.4) includes 104,238 PDB chains. CATH V 3.4 has a total of 1282 folds, 2549 superfamilies, 11330 sequence families, 152,920 domains. CATH V 3.4 has recently been linked to Gene3D 10.2 (released September 2011) 16,118,154 structural annotations for 14,963,305 protein sequences.
9. UniProt/Swiss-Prot (Universal Protein Resource) (http://www.uniprot.org/) [26-30]
UniProt is a protein sequence database. It is a very comprehensive and high-quality database which can be accessed via the World Wide Web (WWW) free of charge. The protein sequences have been derived from genome sequencing projects. This database also contains a large amount of information about the biological function of proteins curated from the research literature [25-27]. The Swiss-Prot, TrEMBL, and PIR protein databases are combined to form the Universal Protein Resource (UniProt). The UniProt acts as a central resource of protein sequences & functional annotations collaborating with three databases, each of which addresses a key need in protein bioinformatics. The components are:
1) UniProt Knowledgebase (UniProtKB);
2) UniProt Reference Clusters (UniRef);
3) UniProt Archive (UniParc).
The UniProt Knowledgebase (UniProtKB) is considered to be the central access point for extensively curated protein information which includes protein function, classification & cross reference. UniProtKB incorporates a range of data from other resources as well.
UniProtKB consists of two sections: UniProtKB/ Swiss-Prot and UniProtKB/TrEMBL.
10. UniProtKB/Swiss-Prot
It contains manually annotated records and combined information extracted from scientific literature and biocurator-evaluated computational analysis. For a particular protein UniProtKB/Swiss-Prot gives all known relevant information. It has become important because of its high quality annotation, direct links to specialized databases and minimal redundancy. It uses standardized nomenclature. In each entry the core data mainly contains amino acid sequence, description of protein, taxonomic data and citation information. If additional information of protein is available, the entries contain detailed annotation such as the function(s) of the protein, some enzyme-specific information like catalytic activity, cofactors, metabolic pathway, regulation mechanisms, molecular weight determined by mass spectrometry, secondary structure, quaternary structure, tissue-specific expression etc.
11. UniProtKB/Trembl
It is composed of automatically annotated records. It contains the translations of all coding sequences (CDS) present in the EMBL/GenBank/DDBJ databases. It also contains sequences from PDB and incorporates gene prediction including Ensembl, RefSeq and CCDS.
The UniProt Reference Clusters (UniRef) databases incorporate closely related sequences into a single record in order for speedy searches and recovery of the data. This database is a comprehensive & non-redundant database. The UniRef contains clustered sets of sequences from the UniProtKB & selected UniProt Archive records and thereby helps to obtain complete coverage of sequence space at several resolutions while hiding redundant sequences.
The UniProt Archive (UniParc) is also a comprehensive repository containing the history of all protein sequences. The protein sequences are retrieved from predominant, publicly accessible protein information resources. This database gathers all new & updated protein sequences. However, the UniParc contains only protein sequences & database cross-references. All other information must be retrieved from the source databases.
12. Unimes
The UniProt Metagenomic and Environmental Sequences (UniMES) database is a repository for metagenomic and environmental data. In recent times UniMES contains data from the Global Ocean Sampling Expedition (GOS). This may predict nearly 6 million proteins, primarily from oceanic microbes. From this dataset the predicted proteins are combined with automatic classification by InterPro and enhance original information with further analysis.
13. GenBank (http://www.ncbi.nlm.nih.gov/genbank/) [31]
The GenBank is a sequence database that stores nucleotide sequences and the proteins obtained from them by translations. This database is maintained by National Center for Biotechnology Information (NCBI). As of April 2011, the GenBank contains approximately 126,551,501,141 numbers of bases in 135,440,924 numbers of sequences. Each sequence submitted to GenBank is assigned a unique GenBank identifier or GenBank accession number.
14. Conclusion
The different databases discussed here provide different information. PDB gives both structural and sequence information of macromolecules whereas SCOP & CATH have structures based on their evolutionary relationships and folding classes. The structural classifications of proteins are generally obtained from SCOP and CATH. On the other hand, the UniProt/Swissprot database provides the sequence annotations of proteins along with links to the external databases like PDB. All these databases are increasing day by day. There are other databases and some new databases are coming. But the databases discussed here are considered to be the foundation stones of bioinformatics.
REFERENCES
- L. Liu and M. T. Özsu, “Encyclopedia of Database System,” Springer, Berlin, 2009.
- P. Beynon-Davies, “Database Systems,” 3rd Edition, Palgrave, Houndmills, Basingstoke, 2004.
- T. Connolly and B. Carolyn, “Database Systems,” Harlow, New York, 2002.
- C. J. Date, “An Introduction to Database Systems,” 8th Edition, Addison Wesley, Boston, 2003.
- D. M. Kroenke and D. J. Auer, “Database Concepts,” 3rd Edition, Prentice, New York, 2007.
- T. Teorey, S. Lightstone and T. Nadeau, “Database Modeling & Design: Logical Design,” 4th Edition, Morgan Kaufmann Press, Burlington, 2005.
- J. W. Tukey, “Exploratory Data Analysis,” Addison Wesley, Reading, 1977.
- L. Manovich, “Database as a Symbolic Form,” MIT Press, Cambridge, 2001.
- J. Galindo, “Handbook on Fuzzy Information Processing in Databases,” Information Science Reference (An Imprint of Idea Group Inc.), 2008. doi:10.4018/978-1-59904-853-6
- J. Gray and A. Reuter, “Transaction Processing: Concepts and Techniques,” Morgan Kaufmann Publishers, Burlington, 1992.
- H. M. Berman, “The Protein Data Bank: A Historical Perspective,” Acta Crystallographica Section A: Foundations of Crystallography, Vol. A64, 2008, pp. 88-95. doi:10.1107/S0108767307035623
- E. F. Meyer, “The First Years of the Protein Data Bank,” Protein Science, Vol. 6, No. 7, 1997, pp. 1591-1597. doi:10.1002/pro.5560060724
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, “The Protein Data Bank,” Nucleic Acids Research, Vol. 28, No. 1, 2000, pp. 235-242. doi:10.1093/nar/28.1.235
- J. Westbrook, N. Ito, H. Nakamura, K. Henrick and H. M. Berman, “PDBML: The Representation of Archival Macromolecular Structure Data in XML,” Bioinformatics, Vol. 21, No. 7, 2005, pp. 988-992. doi:10.1093/bioinformatics/bti082
- H. M. Berman, K. Henrick, H. Nakamura, J. Markley, P. E. Bourne and J. Westbrook, “Realism about PDB,” Nature Biotechnology, Vol. 25, 2007, pp. 845-846. doi:10.1038/nbt0807-845
- C. Schierz, L. N. Soldatova and R. D. King, “Overhauling the PDB,” Nature Biotechnology, Vol. 25, 2007, pp. 437-442. doi:10.1038/nbt0407-437
- F. C. Bernstein, T. F. Koetzle, G. J. B. Williams, E. F. Meyer Jr., M. D. Brice, J. R. Rodgers, O. Kennard, T. Shimanouchi and M. Tasumi, “The Protein Data Bank: A Computer-Based Archival File for Macromolecular Structures,” Journal of Molecular Biology, Vol. 112, No. 3, 1977, pp. 535-542. doi:10.1016/S0022-2836(77)80200-3
- A. Vaguine, J. Richelle and S. J. Wodak, “SFCHECK: A Unified Set of Procedure for Evaluating the Quality of Macromolecular Structure-Factor Data and Their Agreement with Atomic Model,” Acta Crystallographica, Vol. D55, 1999, pp. 191-205. doi:10.1107/S0907444998006684
- G. Murzin, S. E. Brenner, T. Hubbard and C. Chothia, “SCOP: A Structural Classification of Proteins Database for the Investigation of Sequences and Structures,” Journal of Molecular Biology, Vol. 247, No. 4, 1995, pp. 536- 540. doi:10.1016/S0022-2836(05)80134-2
- L. Lo Conte, S. E. Brenner, T. J. Hubbard, C. Chothia and A. G. Murzin, “SCOP Database in 2002: Refinements Accommodate Structural Genomics,” Nucleic Acids Research, Vol. 30, No. 1, 2002, pp. 264-267. doi:10.1093/nar/30.1.264
- Andreeva, D. Howorth, S. E. Brenner, T. J. Hubbard, C. Chothia and A. G. Murzin, “SCOP Database in 2004: Refinements Integrate Structure and Sequence Family Data,” Nucleic Acids Research, Vol. 32, Suppl. 1, 2004, pp. D226-D229. doi:10.1093/nar/gkh039
- R. Day, D. A. Beck, R. S. Armen and V. Daggett, “A Consensus View of Fold Space: Combining SCOP, CATH, and the Dali Domain Dictionary,” Protein Science, Vol. 12, No. 10, 2003, pp. 2150-2160. doi:10.1110/ps.0306803
- A. Orengo, A. D. Michie, S. Jones, D. T. Jones, M. B. Swindells and J. M. Thornton, “CATH—A Hierarchic Classification of Protein Domain Structures,” Structure, Vol. 5, No. 8, 1997, pp. 1093-1108. doi:10.1016/S0969-2126(97)00260-8
- Hadley and D. T. Jones, “A Systematic Comparison of Protein Structure Classifications: SCOP, CATH and FSSP,” Structure, Vol. 7, No. 9, 1999, pp. 1099-1112. doi:10.1016/S0969-2126(99)80177-4
- L. Cuff, I. Sillitoe, T. Lewis, A. B. Clegg, R. Rentzsch, N. Furnham, M. Pellegrini-Calace, D. Jones, J. Thornton and C. A. Orengo, “Extending CATH: Increasing Coverage of the Protein Structure Universe and Linking Structure with Function,” Nucleic Acids Research, Vol. 39, Suppl. 1, 2011, pp. D420-D426. doi:10.1093/nar/gkq1001
- R. Apweiler, A. Bairoch, C. H. Wu, W. C. Barker, B. Boeckmann, S. Ferro, E. Gasteige and H. Huang, “UniProt: The Universal Protein Knowledgebase,” Nucleic Acids Research, Vol. 32, Suppl. 1, 2004, pp. 1115-1119. doi:10.1093/nar/gkh131
- O’Donovan, M. J. Martin, A. Gattiker, E. Gasteiger, A. Bairoch and R. Apweiler, “High-Quality Protein Knowledge Resource: SWISS-PROT and TrEMBL,” Briefings in Bioinformatics, Vol. 3, 2002, pp. 275-284.
- C. Uniprot, “The Universal Protein Resource (UniProt),” Nucleic Acids Research, Vol. 36, Suppl. 1, 2007, pp. D190-D195. doi:10.1093/nar/gkm895
- R. Leinonen, F. G. Diez, D. Binns, W. Fleischmann, R. Lopez and R. Apweiler, “UniProt Archive,” Bioinformatics, Vol. 20, No. 17, 2004, pp. 3236-3237. doi:10.1093/bioinformatics/bth191
- B. E. Suzek, H. Huang, P. McGarvey, R. Mazumder and C. H. Wu, “UniRef: Comprehensive and Non-Redundant UniProt Reference Clusters,” Bioinformatics, Vol. 23, No. 10, 2007, pp. 1282-1288. doi:10.1093/bioinformatics/btm098
- A. Benson, I. Karsch-Mizrachi, D. J. Lipman, J. Ostell and E. W. Sayers, “GenBank,” Nucleic Acids Research, Vol. 39, Suppl. 1, 2011, pp. D32-D37. doi:10.1093/nar/gkq1079