Computational Molecular Bioscience
Vol.2 No.3(2012), Article ID:22490,5 pages DOI:10.4236/cmb.2012.23007
Molecular Dynamics Simulations of DOPC Lipid Bilayers: The Effect of Lennard-Jones Parameters of Hydrocarbon Chains
1Leadership Computing Facility, Argonne National Laboratory, Argonne, USA
2Department of Internal Medicine, University of Cincinnati College of Medicine, Cincinnati, USA 3Division of Human Genetics, Department of Pediatrics, Cincinnati Children’s Hospital Medical Center, Cincinnati, USA
Email: xiaoyang.qi@uc.edu, *aliu@alcf.anl.gov
Received June 19, 2012; revised July 25, 2012; accepted August 10, 2012
Keywords: Molecular Dynamics; Simulations; DOPC; Lipid Bilayers
ABSTRACT
The current Chemistry at Harvard Molecular Mechanics (CHARMM) force field cannot accurately describe the properties of unsaturated phospholipid membranes. In this paper, a series of simulations was performed in which the LennardJones (L-J) parameters of lipid acyl chains of dioleoylphosphatidylcholine (DOPC) were systematically adjusted. The results showed that adjustment of the L-J parameters in lipid acyl chains can significantly improve the current CHARMM force field. It was found that the L-J parameters have different influences on the order parameters of the top half and bottom half of the chain, separated by the cis double bond. The order parameters of the top half and the bottom half of the chain are related to the area/lipid and the length of the chain, respectively.
1. Introduction
The functioning of living biological systems is directly dependent on the structure of biological membranes [1] and [2]. The main structural component of biological membranes is a liquid-crystalline lipid double layer. Depending on the properties of their interactions with the bilayer, proteins or peptides can either insert into or attach to the bilayer, which in turn can activate or prevent the biological functions of proteins or peptides. As a result of rapid advances in the capabilities of computational hardware and simulation software, biological membrane systems have been studied extensively using molecular dynamics (MD) simulation methodology [3-12].
Significant progress has been made in the development of software and force fields. CHARMM (Chemistry at Harvard Molecular Mechanics) and GROMOS (University of Gronizn Atom. Simulations of lipid bilayers at various hydrations have been performed in different ensembles, such as NPAT (constant normal pressure and lateral surface area of membranes and constant temperature) [8,13,14], NPγT (constant normal pressure and lateral surface tension of membranes and constant temperature) [15-19] and NPT (constant number of molecules, constant pressure, and constant temperature) ensembles [5,20-23]. Data from the literature show that GROMOS simulations yielded good values of area/lipid but shorter d-spacing in comparison with experimental data, e.g. 71.1 Å2/35.08 Å [5] and 70.55 Å2/35.8 Å [24] versus the experimental data of 72.5 Å2/36.9 Å for the dioleoylphosphatidylcholine (DOPC) bilayer [25], 59.2 Å2/49.7 Å [3] versus 59.3 Å2/49.1 Å [26,27] at 66% RH. On the other hand, CHARMM simulations produced smaller values of area/lipid and wider spacing distances in the NPT ensemble, e.g., 56.6 Å2/50.4 Å [6] versus the experimental data of 59.3 Å2/49.1 Å at 66% RH [26,27], which suggests that the temperature of the bilayer is lower and the hydrocarbon chains of the lipid are less flexible than observed experimentally. Since the area/lipid and d-spacing are of fundamental importance for the structure properties, a more realistic and effective all-atom force field that can more Accurately reproduce these two characteristic properties is highly desirable.
This paper discusses simulations of a DOPC bilayer that were performed with modified van der Waals interaction parameters to show that adjustment of the Lennard-Jones (L-J) parameters of the carbon atoms in the hydrocarbon chains alone can significantly improve the current all-atom force field for lipid bilayers.
2. Methods
MD simulations of DOPC bilayer systems were carried out in the NPT ensemble. The system is fully hydrated (128 DOPC molecules and 6483 tip3p water molecules, a total of 37113 atoms). The CHARMM MD program [28] version 28 with the CHARMM27 force-field parameters (toppar_c31b1.tar.gz, http://mackerell.umaryland. edu/CHARMM_ff_params.html) was used for the initial system setup and for some analyses. The NAMD (Nanoscale Molecular Dynamics) MD program [29] version 2.6 was used with the same force-field parameters given above for the equilibrium and production runs. The temperature was maintained at 303 K by means of Langevin dynamics using a collision frequency of 5/ps. A fully flexible cell constrained to an orthorhombic system at a constant pressure of 1 atm was applied by means of the Nosé- Hoover Langevin Piston method [21] and [30] as implemented in the NAMD program. The van der Waals interactions were switched smoothly to zero over the region 10 - 12 Ǻ, and the PME method was applied for the calculation of electronic interactions. A neighbor list was kept to 13.5 Ǻ and updated every 10 steps. The time step was 2 fs. The DOPC molecule was built using the insightII program (InsightII version 2001) [31] and optimized in bulk. A library of DOPC molecules was generated by randomly rotating the DOPC molecule. The bilayer system was constructed according to the protocol from Roux et al. [4] [11]. In order to fully relax the polar head groups and hydrocarbon chains of the lipids, the initial system was heated up to 500 K from 303 K in a period of 80 ps. The system stayed at 500 K for 200 ps and was then cooled down to 303 K within 80 ps. The subsequent equilibrium and production runs, with different parameters for the carbon atoms in the hydrocarbon chains, were carried out for 8 - 20 ns. Data averages and error estimations were taken from the last 4 ns trajectories. Uncertainties were estimated by the block averages.
3. Results and Discussion
The periodic simulation box contained 128 DOPC lipids (64 per monolayer) and 6483 water molecules in a full hydration of the bilayer (≈50.6 water molecules per lipid).
The system was first simulated with the CHARMM c31b2 force-field parameters at 303 K in the NPT ensemble and then simulated with modified L-J parameters for the carbon atoms in the hydrocarbon chains at which experimental values of the DOPC bilayer are available. The simulation yielded A = 69.15 ± 1.27 Ǻ2 for area/lipid and Dp-p = 37.47 ± 0.37 Ǻ for phosphate-phosphate distance, respectively. The area/lipid was calculated by the area of the simulation box divided by the number of lipids per monolayer.
The area/lipid value is about 4.6% smaller than the experimental value of 72.5 [25], and the d-spacing is about 1.5% wider than the experimental value of 36.9 [25]. In order to investigate the influence of the hydrocarbon chains on the bilayer structure, a series of simulations was performed with the change of the L-J parameter ε or parameter Rmin/2 for the carbon atoms in the chains. There are six energy and size L-J parameters for carbons in three different groups in the chains, namely, the methylene group (-CH2-), the alkene group (-CH= ) and the methyl group (-CH3), respectively. The simulation values of the area/lipid and the distance between phosphate atoms on the opposite monolayers are presented in Table 1.
Comparison of the Sys#2 with the Sys#1 in Table 1 shows that the reduction of the parameter ε for the carbon of the methylene group (-CH2-) in the chains significantly increased the area/lipid of the bilayer. With a decrease of 50% of ε alone, the area/lipid increased to the experimental value. At the same time, the chains expanded, which is in contradiction to the intuitive expectation of a decrease because more subsequent systems, in which the parameter ε for the carbon of the methylene group was kept the same as in Sys#2. The L-J parameter Rmin/2 free space is available for the chains to move.
Sys#2 was used as a reference system and Rmin/2 was reduced by 10% in Sys#3. The reduction of Rmin/2 for the carbon of the methylene group increased the value of the area/lipid, whereas the length of the chains changed littlein comparison with the values from Sys#2. The ε
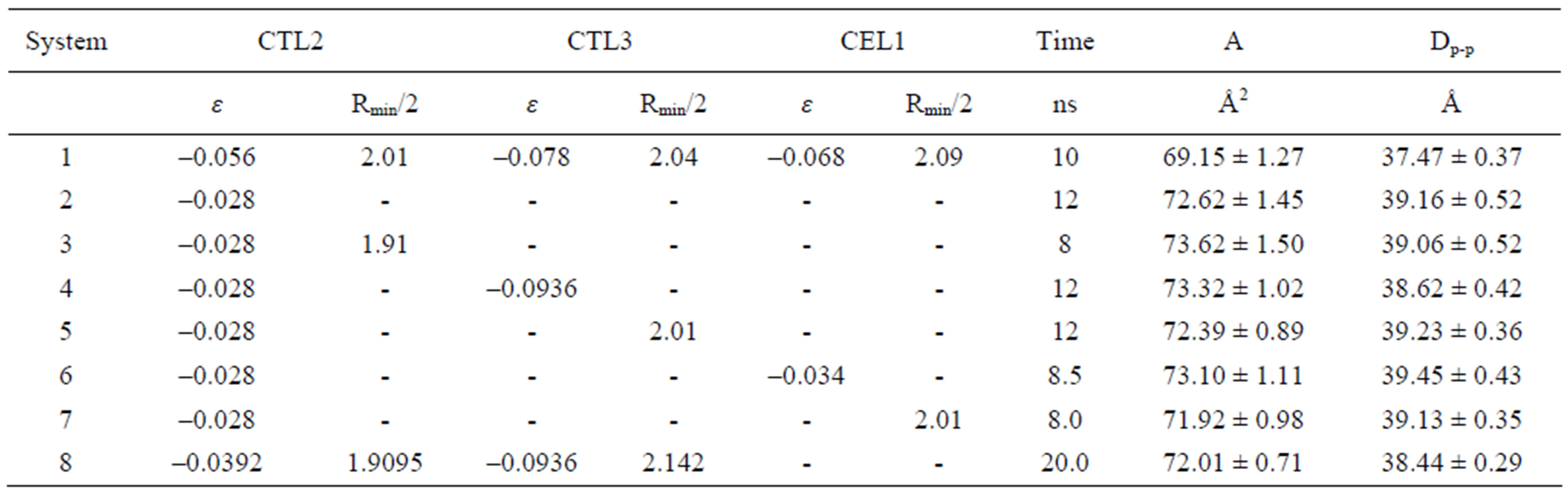
Table 1. Simulation values of DOPC bilayers with modified L-J parameters for hydrocarbon chains.
parameter for the carbon of the methyl group (-CH3) increased 20% in Sys#4. This change produced a larger value of area/lipid and a shorter length of chains compared with Sys#2. The decrease of Rmin/2 of the carbon in the methyl group reduced the value of the area/lipid and increased the chain length slightly in Sys#5 compared with Sys#2. In Sys#6 the ε parameter for the alkene group (-CH=) was reduced 50%. This increased both the values of the area/lipid and the p-p distance compared with Sys#2. The reduction of Rmin/2 of the carbon in the alkene group reduced the area/lipid and produced very small changes in the chain length. The corresponding lipid scd order parameters are shown in Figure 1.
It is interesting to note that the L-J parameters do not affect the flexibilities of the hydrocarbon chains uniformly. Figure 1 shows that the modified parameters made the first half of the hydrocarbon chains (C2-C8) more flexible (scd smaller) but the second half of the hydrocarbon chains (C9-C14) more rigid (scd higher) in comparison with the original parameters in charmm c31b2. From the figure we can conclude that the surface area or area/lipid changes were mainly contributed by the properties of the first half of the hydrocarbon chains. The second half of the hydrocarbon chains contributed to the extension of the chains.
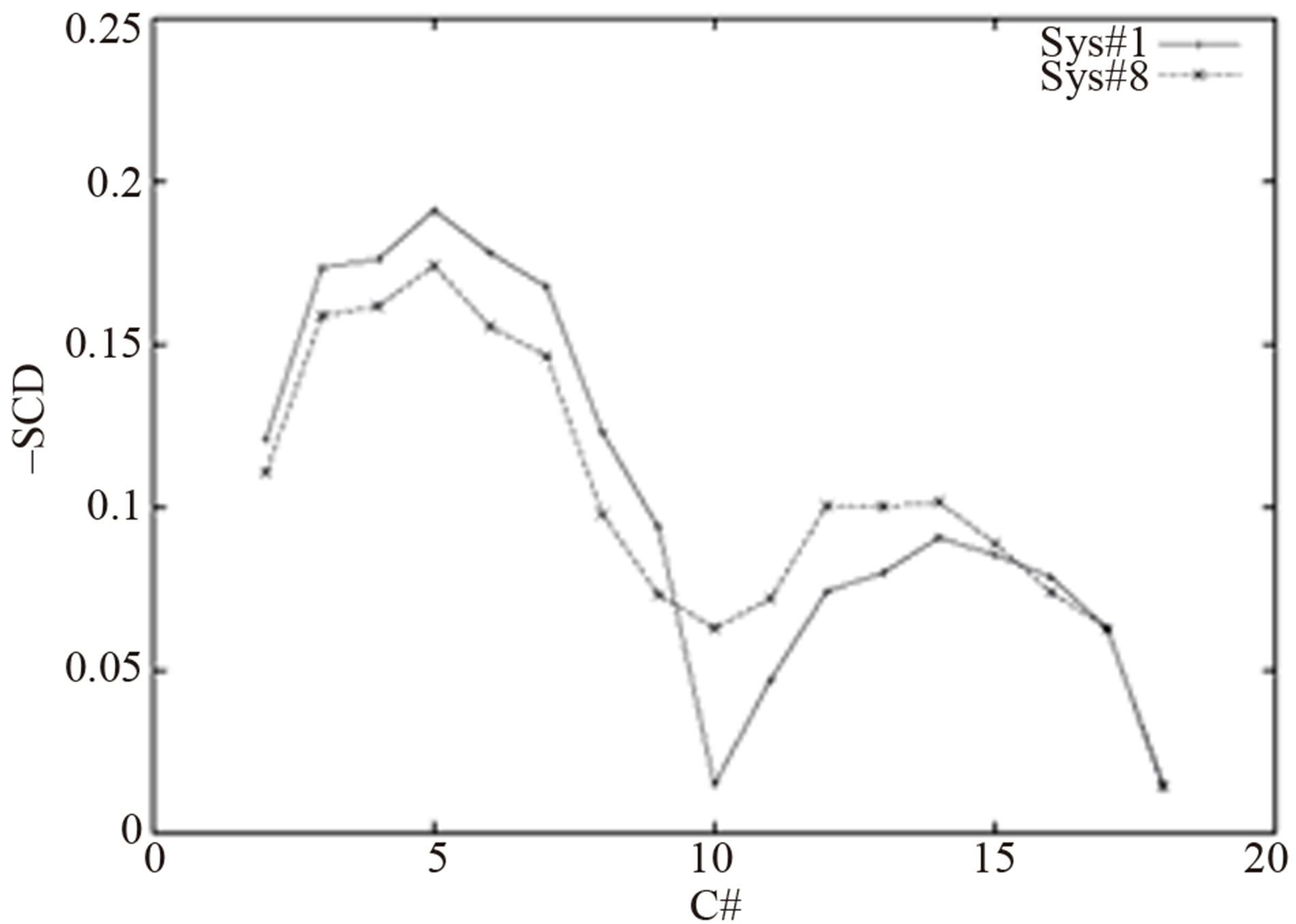
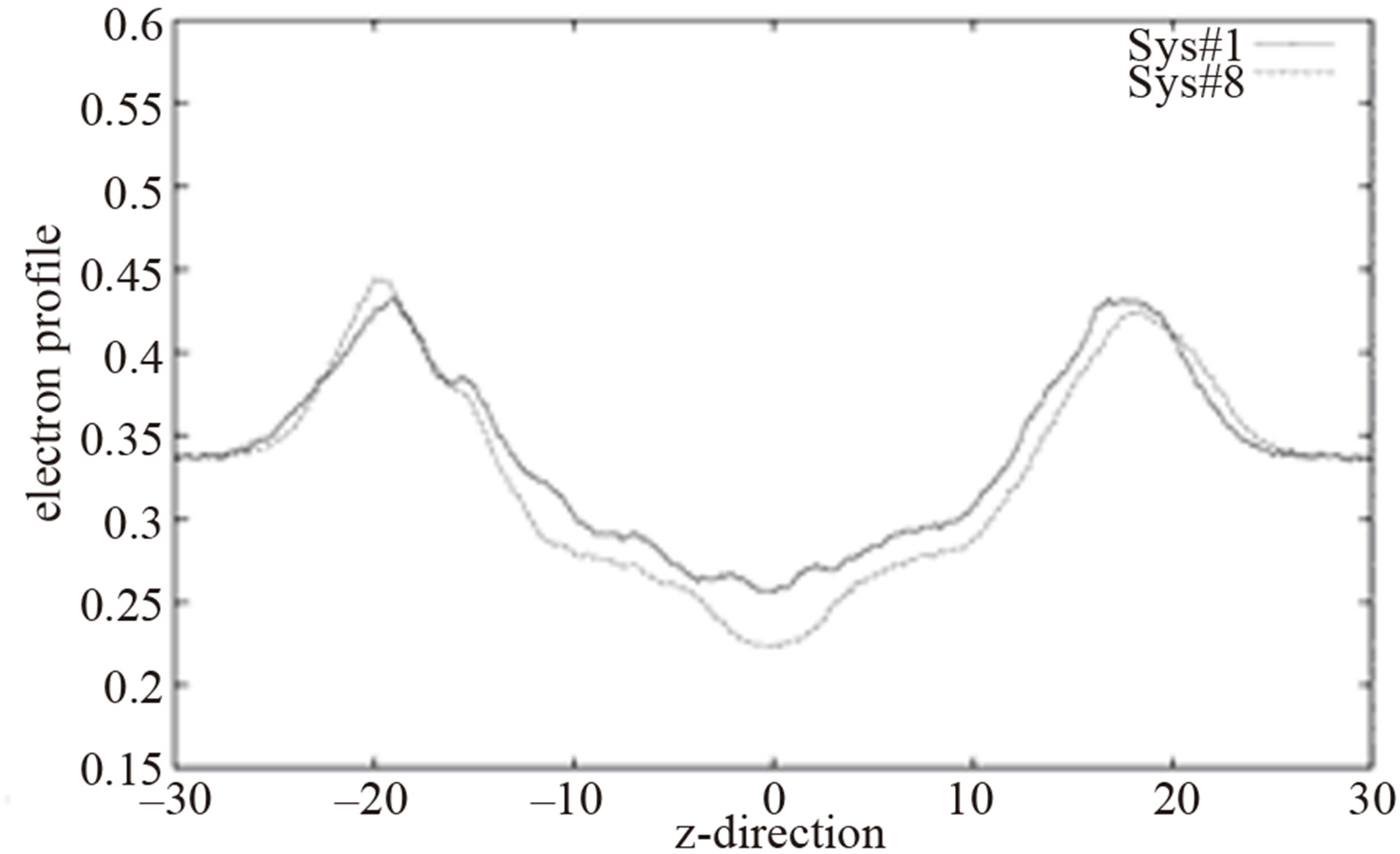
A long simulation was done for a system in which four L-J parameters were changed in order to obtain a shorter chain length (Sys#8 in Table 1). Figures 2 and 3 show the order parameters and the electron profiles of the modified system and the system with the original CHARM force field, respectively. Figure 3 shows that the system with modified parameters has wider and deeper electron density profiles than the one with the original parameters. Wider profile and less electron density in the hydrocarbon core range corresponds to a larger Dp-p value. When the L-J parameters of the carbon atoms in the hydrocarbon chains are adjusted, the fundamental structure properties, such as area/lipid and d-spacing, can be described more accurately. Certainly many combinations of the
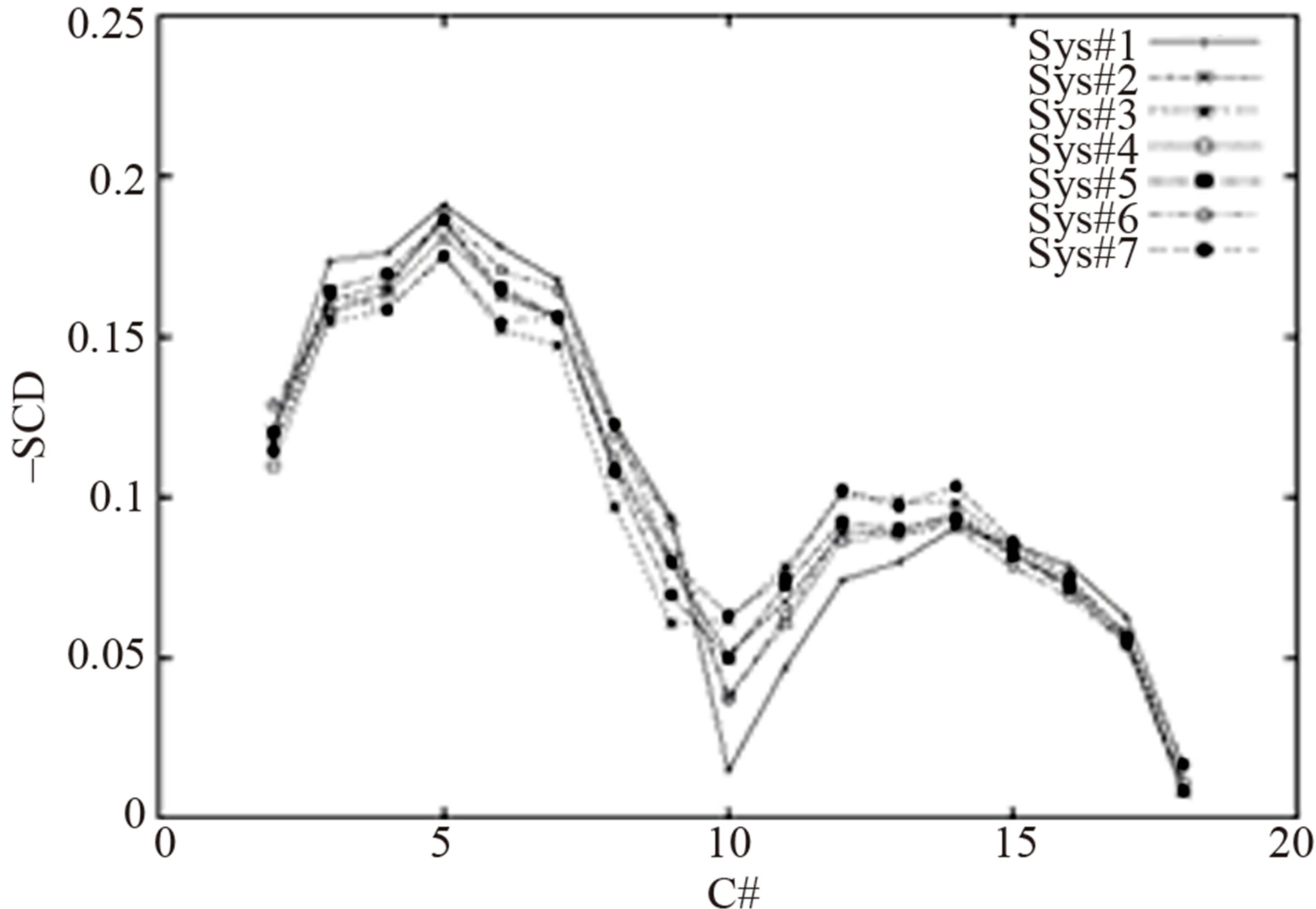
Figure 1. Order parameters of the hydrocarbon chains.
parameters could be found to reproduce the experimental value within the experimental uncertainties. The advantage to having an effective potential based on the modification of the L-J parameters is that it is simple and the properties depending on long-range interactions will not be affected. It is arguable whether the surface pressure exists in a bilayer system or whether the introduction of the surface pressure parameter γ is due to the imperfecttion of the effective potentials. The reproduction of the experimental structure properties by modification of the L-J parameters indicates that the surface pressure effect might be included in these parameters if there are any surface pressures in these systems.
4. Conclusion
The effect of the Lennard-Jones (L-J) parameters of DOPC lipid acyl chains on the area/lipid and length of chains has been investigated by MD simulations. It was found that the L-J parameters have different influences on the order parameters of the top half and bottom half of the chain, separated by the cis double bond. The order parameters of the top half of the chain are directly related to the area/lipid, whereas the order parameters of the
bottom half of the chain are directly related to the length of the chain. It is possible to adjust a few L-J parameters on the acyl chains to obtain more accurate structure properties of lipid bilayers.
5. Acknowledgements
We thank Sandy Grabowski for manuscript editing.
REFERENCES
- P. K. J. Kinnunen, A. Kõiv, J. Y. A. Lehtonen, M. Rytömaa and P. Mustonen, “Lipid Dynamics and Peripheral Interactions of Proteins with Membrane Surfaces,” Chemistry and Physics of Lipids, Vol. 73, No. 1-2, 1994, pp. 181- 207. doi:10.1016/0009-3084(94)90181-3
- M. Kobayashi, R. K. Nutharasan, J. W. Feng, M. F. Robert and J. W. Lomasney, “Identification of Hydrophobic Interactions between Proteins and Lipids: Free Fatty Acids Activate Phospholipase C δ1 via Allosterism,” Biochemistry, Vol. 43, No. 23, 2004, pp. 7522-7533. doi:10.1021/bi035966c
- R. W. Benz, F. Castro-Román, D. J. Tobias and S. H. White, “Experimental Validation of Molecular Dynamics Simulations of Lipid Bilayers: A New Approach,” Biophysical Journal, Vol. 88, No. 2, 2005, pp. 805-817. doi:10.1529/biophysj.104.046821
- S. Berneche, M. Nina and B. Roux, “Molecular Dynamics Simulation of Melittin in a Dimyristoylphosphatidylcholine Bilayer Membrane,” Biophysical Journal, Vol. 75, No. 4, 1998, pp. 1603-1618. doi:10.1016/S0006-3495(98)77604-0
- S. Y. Bhide and M. L. Berkowitz, “Structure and Dynamics of Water at the Interface with Phospholipid Bilayers,” The Journal of Chemical Physics, Vol. 123, No. 22, 2005, pp. 224702.1-224702.16. doi:10.1063/1.2132277
- F. Castro-Román, R. W. Benz, S. H. White and D. J. Tobia, “Investigation of Finite System-Size Effects in Molecular Dynamics Simulations of Lipid Bilayers,” The Journal of Chemical Physics B, Vol. 110, No. 47, 2006, pp. 24157-24164. doi:10.1021/jp064746g
- S. E. Feller, R. M. Venable and R. W. Pastor, “Computer Simulation of a DPPC Phospholipid Bilayer: Structural Changes as a Function of Molecular Surface Area,” Langmuir, Vol. 13, No. 24, 1997, pp. 6555-6561. doi:10.1021/la970746j
- S. E. Feller, D. Yin, R. W. Pastor and J. A. D. MacKerell, “Molecular Dynamics Simulation of Unsaturated Lipid Bilayers at Low Hydration: Parameterization and Comparison with Diffraction Studies,” Biophysical Journal, Vol. 73, No.5, 1997, pp. 2269-2279. doi:10.1016/S0006-3495(97)78259-6
- L. R. Forrest and M. S. P. Sansom, “Membrane Simulations: Bigger and Better?” Current Opinion in Structural Biology, Vol. 10, No. 2, 2000, pp. 174-181. doi:10.1016/S0959-440X(00)00066-X
- P. Mukhopadhyay, L. Monticelli and D. P. Tieleman, “Molecular Dynamics Simulation of a Palmitoyl-Oleoyl Phosphatidylserine Bilayer with Na+ Counterions and NaCl,” Biophysical Journal, Vol. 86, No. 3, 2004, pp. 1601-1609. doi:10.1016/S0006-3495(04)74227-7
- B. Roux and T. B. Woolf, “Molecular Dynamics of Pf1 Coat Protein in a Phospholipid Bilayer,” In: K. M. Merz Jr. and B. Roux, Eds., Biological Membranes: A Molecular Perspective from Computation and Experiment, Birkhauser Press, Boston, 1996, pp. 555-587.
- D. J. Tobias, K. Tu and M. L. Klein, “Atomic-Scale Molecular Dynamics Simulations of Lipid Membranes,” Current Opinion in Colloid & Interface Science, Vol. 2, No. 1, 1997, pp. 15-26. doi:10.1016/S1359-0294(97)80004-0
- S. W. Chiu, E. Jakobsson, S. Subramaniam and H. L. Scott, “Combined Monte Carlo and Molecular Dynamics Simulation of Fully Hydrated Dioleyl and Palmitoyloleyl Phosphatidylcholine Lipid Bilayers,” Biophysical Journal, Vol. 77, No. 5, 1999, pp. 2462-2469. doi:10.1016/S0006-3495(99)77082-7
- R. J. Mashl, H. L. Scott, S. Subramaniam and E. Jokobsson, “Molecular Simulation of Dioleoylphosphatidylcholine Lipid Bilayers at Differing Levels of Hydration,” Biophysical Journal, Vol. 81, 2001, pp. 3005-3015. doi:10.1016/S0006-3495(01)75941-3
- E. A. Dolan, R. M. Venable, R. W. Pastor and B. R. Brooks, “Simulations of Membranes and Other Interfacial Systems Using P21 and Pc Periodic Boundary Conditions,” Biophysical Journal, Vol. 82, 2002, pp. 2317-2325. doi:10.1016/S0006-3495(02)75577-X
- S. E. Feller, Y. Zhang and R. W. Pastor, “Computer Simulation of Liquid/Liquid Interfaces. II. Surface Tension—Area Dependence of a Bilayer and Monolayer,” Journal of Chemical Physics, Vol. 103, No. 23, 1995, pp. 10267-10276. doi:10.1063/1.469928
- F. Jähnig, “What Is the Surface Tension of a Lipid Bilayer Membrane?” Biophysical Journal, Vol. 71, No. 3, 1996, pp. 1348-1349. doi:10.1016/S0006-3495(96)79336-0
- B. Roux, “Commentary: Surface Tension of Biomembranes,” Biophysical Journal, Vol. 71, No. 3, 1996, pp. 1346-1347. doi:10.1016/S0006-3495(96)79335-9
- R. Sankararamakrishman and H. Weinstein, “Surface Tension Parameterization in Molecular Dynamics Simulations of a Phospholipid-Bilayer Membrane: Calibration and Effects,” The Journal of Chemical Physics B, Vol. 108, No. 31, 2004, pp. 11802-11811. doi:10.1021/jp048969n
- O. Berger, O. Edholm and F. Jähnig, “Molecular Dynamics Simulations of a Fluid Bilayer of Dipalmitoylphosphatidylcholine at Full Hydration, Constant Pressure, and Constant Temperature,” Biophysical Journal, Vol. 72, No. 5, 1997, pp. 2002-2013. doi:10.1016/S0006-3495(97)78845-3
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, “Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method,” The Journal of Chemical Physics, Vol. 103, No. 11, 1995, pp. 4613-4621. doi:10.1063/1.470648
- K. Tu, D. J. Tobias and M. L. Klein, “Constant Pressure and Temperature Molecular Dynamics Simulation of a Fully Hydrated Liquid Crystal Phase Dipalmitoylphosphatidylcholine Bilayer,” Biophysical Journal, Vol. 69, No. 6, 1995, pp. 2558-2562. doi:10.1016/S0006-3495(95)80126-8
- K. Tu, D. J. Tobias and M. L. Klein, “Constant Pressure and Temperature Molecular Dynamics Simulations of Crystals of the Lecithin Fragments: Glycerylphosphorylcholine and Dilauroylglycerol,” The Journal of Chemical Physics, Vol. 99, No. 24, 1995, pp. 10035-10042. doi:10.1021/j100024a053
- A. A. Polyansky, P. E. Volynsky, D. E. Nolde, A. S. Arseniev and R. G. Efremov, “Role of Lipid Charge in Organization of Water/Lipid Bilayer Interface: Insights via Computer Simulations,” The Journal of Chemical Physics B, Vol. 109, 2005, pp. 15052-15059.
- H. I. Petrache, S. Tristram-Nagle, K. Gawrisch, D. Harris, V. A. Parsegian and J. F. Nagle, “Structure and Fluctuations of Charged Phosphatidylserine Bilayers in the Absence of Salt,” Biophysical Journal, Vol. 86, No. 3, 2004, pp. 1574-1586. doi:10.1016/S0006-3495(04)74225-3
- M. C. Wiener and S. H. White, “Structure of a Fluid Dioleoylphosphatidylcholine Bilayer Determined by Joint Refinement of X-Ray and Neutron Diffraction Data. II. Distribution and Packing of Terminal Methyl Groups,” Biophysical Journal, Vol. 61, No. 2, 1992, pp. 428-433. doi:10.1016/S0006-3495(92)81848-9
- R. E. Jacobs and S. H. White, “The Nature of the Hydrophobic Binding of Small Peptides at the Bilayer Interface: Implications for the Insertion of Transbilayer Helices,” Biochemistry, Vol. 28, No. 8, 1989, pp. 3421-3437. doi:10.1021/bi00434a042
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, “CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations,” Journal of Computational Chemistry, Vol. 4, No. 2, 1983, pp. 187-217. doi:10.1002/jcc.540040211
- L. Kalé, R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan and K. Schulten, “NAMD2: Greater Scalability for Parallel Molecular Dynamics,” Journal of Computational Physics, Vol. 151, No. 1, 1999, pp. 283-312. doi:10.1006/jcph.1999.6201
- G. J. Martyna, D. J. Tobias and M. L. Klein, “Constant Pressure Molecular Dynamics Algorithms,” Journal of Chemical Physics, Vol. 101, No. 5, 1994, pp. 4177-4189. doi:10.1063/1.467468
- InsightII (San Diego, California, USA) http://www.accelrys.com.
NOTES
*Corresponding author.