American Journal of Analytical Chemistry
Vol.07 No.08(2016), Article ID:69429,6 pages
10.4236/ajac.2016.78056
Unsaponifiable Matter in Carnuba (Cera carnuba) Wax, a Modification of the USP/NF and FCC Methods
Yusuf Yildiz*, Manjista Dasgupta
Complete Analysis Laboratories, Analytical Chemistry Research Department, Parsippany, NJ, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 10 June 2016; accepted 30 July 2016; published 3 August 2016
ABSTRACT
Carnuba wax consists chiefly of myricyl cerotate (MW 817.4) and small quantities of free cerotic acid (C26H52O2, Mw 396.7) and myricyl alcohol (C30H62O, mp 90˚C). Of the two common extraction solvents, ethyl ether or petroleum ether, Lewkowitsch prefers the former. Concerning separation of phases, he advocates addition of small amounts of alcohol or caustic, and he also states that formation of a flocculant layer between the aqueous layer and the solvent does not interfere with the correct estimation of the unsaponifiable matter. These statements were not corroborated in the hands of this chemist. The “Unsaponifiable Matter” in oils or and fats, which consist mainly of hydrocarbons, sterols and aliphatic alcohols of high molecular mass that are not saponifiable by alkali hydroxides but are soluble in the ordinary fat solvents, and to products of saponification that are soluble in such solvents. Carnuba wax, a rather expensive wax, may be adulterated with less expensive paraffin by dishonest merchants. ASTM has a method for determining paraffinic material in carnuba wax. It uses heptanes at its boiling point to dissolve the wax, apply it to a silica gel column, and elute only the nonpolar (i.e. alkane) material. The method has the disadvantage of using a large volume of haptane, nor is it called for by either US Pharmacopeia/National Formulary (USP/NF) or Food Chemicals Codex (FCC). The test for unsaponifiable matter on pure carnuba wax will yield a result of 50.0% - 55.0%, while a higher result will betray the presence of paraffin adulterants.
Keywords:
Carnuba (Cera carnuba) Wax, Unsaponifiable Matter, Fatty Acids

1. Introduction
Carnuba wax (Cera carnuba) is purified wax obtained from the leaves of Copernicia cerifera Mart. (Palmae) [1] grown only in Brazil. The carnuba wax is often called the “queen of waxes”, also known as “palm wax” or Brazil wax.” [2] . Carnuba wax occurs as light yellow to light brown, hard and brittle masses or white to light yellow moderately coarse powder or flakes, possessing a slight characteristic bland odor, and free from rancidity. It is nontoxic and hypoallergenic. Specific gravity is about 0.99. Carnuba wax consists of fatty acid esters (80% - 85%), fatty alcohols (10% - 16%), acids (3% - 6%) and hydrocarbons (1% - 3%). It is around 20% esterified fatty diols, 10% methoxylated or hydroxylatedcinnamic acid, and 6% hydroxylated fatty acids. Carnuba wax has a very high melting point of 82˚C - 86˚C (180˚F - 187˚F). It is tasteless and partially insoluble in water, in ethanol (95%) and in diethyl ether and xylenes [3] , soluble on heating in ethyl acetate, in warm chloroformand in warm toluene and in xylene [4] . It is slightly soluble in boiling alcohol [5] .
Carnuba wax is by scraping or cooking leaves from the carnuba palm (Copernicia cerifera Mart.). The carnuba leaves are cut, and the adult leaves are separated from the youngest. After the sun and or wind dries leaves, then the hitting occurs for complete removal of the powder. The process can be done manually or using a machine. The powder is then stored in cotton bags. The raw material can be forwarded to the industry in powder form or lumps. The solid form is obtained by boiling the powder in water, for later solidification and packaging [6] .
There is no formula for carnuba wax. Fatty acids are carboxylic acid with hydrocarbon chains of 4 to 36 carbons. They can be saturated and unsaturated. The esters of unsaturated hydroxylated fatty acids have about 12 carbon atoms in the acid chain. The usual names for the constituents, i.e., cerotic acid, melissylcerotate, canaubic acid, etc., are meaningless [7] . An older source gives the constituents as myricyl alcohol, myricyl cerotate, a small amount of cerotic acid, and 55% of unsaponifiable waxes [8] .
The simple lipids which are constructed from fatty acids are triacylglycerols or triglycerides. Glycerides are lipid esters. Fats (triglycerides) upon alkali hydrolysis (either with KOH or NaOH) yield glycerol and potassium or sodium salts of fatty acids (soap).
In this study, a test for unsaponifiable matter has been successfully determined in carnuba wax. This is in contradistinction to the US Pharmacopeia/National Formulary (USP/NF) and Food Chemicals Code (FCC) methods, which for carnuba wax fails by given results far below the specific range. The comparison of two tests is mentioned in Table 1, as well as for an overview of the pertinent chemistry [3] .
The modified method for determination of unsaponifiable matter in carnuba wax is here noted, and emphatically stated, that the FCC and USP/NF methods, as written, fail miserably in the separation of the unsaponifiable matter from the reaction mixture after saponification [3] .
2. Materials and Method
2.1. Chemicals
Carnuba (Cera carnuba) wax, ethyl alcohol (Pharmco-Aaper, 36.5% - 38.0%, HPLC Grade), heptane (spectrum 99 + %, HPLC grade), Deionized water.
2.2. Apparatus
Condenser with 24/40 ground glass joint, 250 mL and 500 mL conical flask with 24/40 ground glass joint. Magnetic stir bar, ribbed, dimensions 35 mm × 9 mm. Corning hotplate stirrer. Pasteur pipet and 1 mL pipet bulb. Two 600 mL beakers (one for preparation of 1:1 EtOH:H2O, one for water bath). Glass beads boiling aid. Oven 100˚C. Analytical balance. Stopper for 24/40 flask. 250 mL beaker (for gravimetry).
2.3. Solutions Were Prepared as Follows
1) Methyl Orange Indicator Solution: 10 mg methyl orange is dissolved in 100 g of water to obtain 0.01% w/w.
2) 0.5 N HCl Volumetric Solution: Prepared by diluting 40 mL of concentrated HCl to 1 L with deionized water. Standardized vs Na2CO3.
3) Masked Phenolphthalein Indicator Solution: Dissolve 1.6 g phenolphthalein and 2.7 g methylene blue in

Table 1. Overview of the comparison of methods [3] [5] [9] . In each method, 5.0 g of carnuba wax was taken.
500 mL alcohol, adjust pH with alcoholic alkali solution so that the greenish blue color is faintly tinged with purple. The color change, when going from acid to alkali, is from green to purple [9] .
4) 0.5 N Sodium hydroxide ethanolic Volumetric Solution: Prepared by dissolving 20 g sodium hydroxide in a minimal amount of water, cooling to room temperature, and diluting to 1L ethyl alcohol. Standardized vs. Potassium Hydrogen Phthalate (KHP).
2.4. Procedure
1) 2.5 g sodium hydroxide was dissolved in minimal water (about 10 mL) on cold water bath. Add 50 mL Ethanol, mix well.
2) Weight 5.0 g carnuba wax into 250 mL conical flask with 24/40 ground glass joint for condenser.
3) Put ribbed stir bar and add alcoholic sodium hydroxide prepared in 1).
4) Saponify by refluxing 1 hour with sitirring using corning hotplate until clear brown color liquid observed. Heat setting 6 and stir setting 6.
5) Remove from heat and add heptanes 60 mL.
6) Set hotplate setting to 3 ½ and wait for temperature to equilibrate. Put to reflux, stir setting 5, heat setting 3 ½ gradually rise to 4 ½.
7) Stir and reflux until homogeneous clear solution with no solids. Be careful not to stir sofast. Avoid forming froth. Modulate stir setting to 7 or 8 and back to 5, to mix upper layer with lower layer. Repeat this a dozen times.
8) Remove from heat, when 2 layers form, carefully drawoff top layer to 500 mL flask having 24/40 ground glass joint, by using tared 250 mL beaker, glass beads, and 1 mL pipet bulb.
9) Add 60 mL heptanes and perform the 2nd extraction as per 6)-8), also 3rd - 6thextractions.
10) Prepare 500 mL of 1:1 Et-OH: H2O and 2 mL of methyl orange indicator and enough hydrochloric acid to turn indicator red. Add to 500 mL flask to bring level to 2/3 full, put ribbed stir bar, put to heat/stir with reflux.
11) After single clear phase obtained, remove from heat, and with stirring add 0.5 N hydrochloric acid untilred color appears. Stopping the stirring to let phases separate shows the color more clearly. Once indicator has become red, add another 1 - 2 mL of 0.5 N hydrochloric acid.
12) Stir and heat until a single clear phase exists, and be sure it is still red. Remove from heat, stop stirring, and allow phases to separate. Draw off top clear heptanes layer into tared beaker.
13) Add H2O: Ethanol to raise the interface level nearer the narrow part of the flask and repeat 12).
14) Repeat 13) until the top heptanes layer is about 2 cm is height, in the narrow part of the flask.
15) Add about 20 mL heptanes and repeat 12).
16) Repeat 15) two times.
17) Position the beaker containing the heptane extract in a boiling water bath using clamps to stabilize it. The boiling water is in a 600 mL beaker, including glass beads.
18) After heptanes has been driven off; put the beaker with the pipet (no bulb) in a 100˚C oven overnight.
19) Cool, weigh the beaker (with pipet, no bulb, and glass bead should be there still). Subtracttare to obtain weight of extracted material.
20) Dry the residue at 105˚C until successive weighing differ by not more than 1 mg. calculate the percentage of saponification matter in the portion of oil or fat taken by the formula [5] :
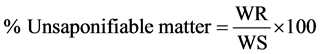
where;
WR = weight in g of the residue (unsaponifiable matter).
WS = weight of sample in g, carnuba wax taken for the test.
Dissolve the residue in 20 mL of alcohol, previously neutralized to the phenolphthalein endpoint, add phenolphthalein Test Solution (TS), and titrate with 0.1 N alcoholic sodium hydroxide Volumetric Solution (VS) to the first appearance of a pink color that persist for not less than 30 seconds. If the volume of 0.1 N alcoholic sodium hydroxide required is greater than 0.2 mL, the separation of the layers was incomplete; the residue weighed cannot be considered as “unsaponifiable matter,” and the test must be repeated [4] .
3. Result and Discussion
The unsaponifiable matter was separated as such, and to this end the fat must first be saponified. It was convenient to combine the determination of the unsaponifiable matter with that of the saponification value; it must, however, be borne in mind, that in view of the very small quantity of unsaponifiable matter, it has been necessary to take at least 5 g for the test [10] . Expected unsaponifiable matter for carnuba wax is 50.0% - 55.0%. The percent unsaponifiable has been found 52.4% or sample A, and 52.2% for the sample B (as duplicate) (Table 2), % RPD is 0.38%, and result meets the requirements.
The Food Chemicals Codex (FCC-9) method was tried first. A white mass was formed between the bottom aqueous/ethanol layer and top petroleum ether layer, which Lewkowitsch calls “flocculant layer”. Taking only the clear colorless petroleum ether washings, the result was negligible, less than 1% unsaponifiable matter. Taking the flocculant layer, with extensive drying, the result was greater than 100%. Lunge [10] method was tried next: 25 mL ethanolic sodium hydroxide (2 g sodium hydroxide dissolved in water, put 25 mL ethanol) in porcelain basin on water bath. Boil down to dryness. Add 50 mL ethyl ether. Shake thoroughly. Add alcohol to speed separation. Separate, drain aqueous layer to another separatory funnel for further extraction with ethyl ether. Collect ether layers, wash with water, and proceed as usual. Result: 11.4% unsaponifiable matter. Comments: top (ethyl ether) layers were clear and yellow. The yield, it was thought, would be improved by saponifying with refluxing instead of ceramic basin on a water bath. Such hope was not realized, and the method was abandoned.
Next, the US Pharmacopeia/National Formulary USP/NF 38 method was tried. Gentle swirling and rotating of separatory flask did not impart a yellow color to the ethyl ether layer, being interpreted that no unsaponifiable matter was extracted by gentle swirling. Upon vigorous shaking, a flocculant layer was formed, and testing was halted. Result: 15%.
Table 2. Calculations of unsaponifiable matter.
Table 3. The method given is this study may be shematacized as follows [3] .
Notes: 1) Correction as perFood Chemicals Codex (FCC), fatty acid subtracted from weight of extracted unsaponifiable matter. FCC uses Mw of 283, but for carnuba wax use 396.7 Mw for the cerotic acid; 2) If any hydrochloric acid is carried into heptanes, it would inflate of sodium hydroxide consumed, giving a falsely large correction and a falsely low unsaponifiable matter result. This does not seem to be the problem, as results were too high; 3) pKa of RCOOH is about 4.8 and methyl orange transition range is 3.5 - 4.5. Acidifying to methyl orange and adding excess ensures over 90% protonation of RCOOH for correction by subsequent titration with sodium hydroxide pH of wash was found to be 2.22.
Lewkowitsch [11] states: the method above (Lunge’s) was not suitable for determining the unsaponifiable matter in beeswax, carnuba wax, and other solid waxes. Instead, neutralize the soap solution, after adding phenolphthalein, with acetic acid, and precipitate with either barium chloride or lead acetate. The precipitate is then washed, dried, mixed with sand in a mortar, and extracted in a Soxhlet apparatus with petroleum ether boiling below 80˚C. Comments: it required addition of Celite filter aid to wash the precipitate. So much dried material was obtained that each wax sample required two extraction thimbles. Result: 36.75%.
It was then thought to try to break down flocculant layer by use of emulsion breakers. The following were applied to already-formed flocculant layer: split 100 cationic emulsion breaker, split 420 emulsion breaker, and split 430 emulsion breaker. They all failed to break down the flocculant layer.
It is to be noted that not even boiling heptane, when applied to an already-formed flocculant layer, could break down the flocculant layer. From this, it may be concluded that the fatty acid salts (i.e. “wax soaps”) are insoluble in this solvent.
In the search for an answer, recourse was made to a book of emulsions [12] , which stated that the three needed ingredients for the formation of the stable emulsion were two immiscible phases and an emulsifying agent―a molecule being of mixed non-polar-polar type, bearing hydrophobic and hydrophilic portions, for example, the salt of the fatty acid. Then, vigorous shaking is all that is needed to form a stable emulsion.
There must be a solvent, for this study that will more effectively extract unsaponifiable matter from the aqueous/ ethanolic layer without need for vigorous shaking.
The penultimate method tried, which also failed, was to use superheated petroleum ether in a makeshift pressure vessel with gentle stirring. The pressure vessel failed and run was aborted.
The method that finally worked was to go to the next higher member of the homologous series of alkanes, from petroleum ether (which is a mixture of hexane, cyclohexane, and other low boiling components) to heptane, which boiled at 98.6˚C. The suggested heptane extraction method has been shematicized in Table 3. This, it was thought, and shown by experience to be true, world allows gentle stirring of the two phases for efficient extraction of unsaponifiable matter without the formation of a flocculant layer. This is the method described in this study. The two phases, in the case of petroleum ether as solvent, can not be gently stirred, because at the boiling point of the solvent, the aqueous-alcoholic saponification mixture is solid. The solid mixture melts at a temperature between the boiling pointsif petroleum ether and heptane.
Acknowledgements
The authors wish to thank Ilgin Yildiz for the critical review. We are also thankful to Dr. Raghavendra Sahai, President at Complete Analysis Laboratory to give us the opportunity to complete this research work.
Cite this paper
Yusuf Yildiz,Manjista Dasgupta, (2016) Unsaponifiable Matter in Carnuba (Cera carnuba) Wax, a Modification of the USP/NF and FCC Methods. American Journal of Analytical Chemistry,07,611-616. doi: 10.4236/ajac.2016.78056
References
- 1. Japonese Pharmacopeia (JP), Official Monographs, Carnuba Wax. 2008.
- 2. Wolfmeier, U., Schmidth. Heinrichs, F.L., et al. (2000) Waxes. Ullmann’s Encyclopedia of Industrial Chemistry.
- 3. Edens, G. and Yildiz. Y. (2013) SOP for Unsaponifiable Matter in Carnuba Wax. Complete Analysis Laboratories, NJ, USA, 21 February 2013.
- 4. European Pharmacopeia (EP), 01/2008:0597.
- 5. United State Pharmacopeia/National Formulary (USP/NF) 38. Official Monographs. Carnuba Wax.
- 6. Natural Wax, Distrito Industrial, Maracanau Ceare-Brasil 61939-130.
- 7. Merck Index, 12th Edition.
- 8. Holde & Mueller “Hydrocarbon Oils & Saponifiable Fats Waxes”.
- 9. Food Chemical Codex (FCC), 5th Edition. Institute of Medicine of the National Academies Press, Washington DC, 1 January 2004.
- 10. George Lunge, Ed., Technical Methods of Chemical Analysis, Vol. III, Part I, 1914, 125.
- 11. Lewkowitsch, J. (1904) Chemical Technology and Analysis of Oils, Fats, and Waxes, Vol. II, 874, 294-295.
- 12. Sutheim, G.M. (1946) Introduction to Emulsions. 37 and Chapter 3.
NOTES

*Corresponding author.