American Journal of Analytical Chemistry
Vol.07 No.03(2016), Article ID:64342,8 pages
10.4236/ajac.2016.73021
Thermal Decomposition and a Kinetic Study of Poly(Para-Substituted Styrene)s
Ayşegül Şenocak, Cemil Alkan, Ahmet Karadağ*
Chemistry Department, Art and Science Faculty, Gaziosmanpaşa University, Tokat, Turkey

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 8 January 2016; accepted 6 March 2016; published 9 March 2016
ABSTRACT
The thermal decompositions of polystyrene (PS), poly(p-methyl styrene) (PMS), poly(p-bromo styrene) (PBrS), and poly(p-chloro styrene) (PClS) were investigated through thermogravimetric analysis (TGA). For this aim, Flynn-Wall-Ozawa method was applied to derivative thermogravimetric (DTG) curves. Continuous distribution kinetics was employed with a stoichiometric kernel to determine the rate coefficients for decomposition reactions. TGA data for the polymers were investigated by nonlinear fitting procedures that yielded activation energies and frequency factors for the combined chemical reactions. The reaction order values of PS derivatives are just about 1 in the nonisothermal decomposition process. Ea values for PS, PMS, and PClS increase with % conversion individually as they decrease in the order of PS/PMS/PClS which is consistent with the molecular weight increase. On the other hand, PBrS has the highest activation energy. Also its activation energy decreases with the % conversion. Thus it is suggested that PBrS degrades with somehow different mechanism.
Keywords:
Polystyrenes, Thermogravimetric Analysis, Kinetic Analysis

1. Introduction
Overview and Background
Polystyrene (PS) is a thermoplastic material having so many using areas like home appliances, construction, packaging, and other industrial applications. It is cheap and easily processable. However, it has restricted mechanical properties and is very flammable. Due to its advantages, different kinds of derivatives have been produced. To enhance PS’s competition capacity in engineering resin applications, to protect its properties like thermal stability is crucial. In this work, poly(para-substituted styrene)s have been investigated using nonisothermal degradation kinetics.
Usually, polystyrene, exposed to thermal degradation, softens and melts at 160˚C. Up to 275˚C, volatility of fused polymer with high molecular weight is lowered and this molten version of the polymer keeps its stability. However, polymer fragments with lower molecular weight have higher volatility form and the decomposition is completed after the temperature reaches to 470˚C [1] . In addition, molecular weight rapidly decreases in the beginning of the degradation and the rate of this decline slows down at later stages [2] . This case applies to not only polystyrene but also some of its substituted derivatives. Literature suggests that styrene is the major decomposition product in the complete depolymerization taking place below 500˚C. After this temperature, further fragmentation carries out and the oligomers turn into gaseous species having lower molecular weight [3] .
The decomposition of polystyrene is a radical chain process composed of sequential 1) initiation, 2) propagation, and 3) termination steps [4] . C-C bond strength determines the thermal stability of polystyrene. Random scission or breakage of “weak links” can cause initiation reactions followed by C-C bond chain-end cleavage producing radicals. In the second step of the degradation, hydrogen-abstraction and unzipping reactions occur. The reaction of free radicals produced and polystyrene results in short chain radicals. Termination generally happens as a result of the recombination of two radical chains. There are many examples for thermal degradation of different PS materials [5] [6] .
For kinetic interpretations in this study, poly(p-substituted styrene)s’ thermal degradation data were collected in nitrogen atmosphere. With intent to understand the origin of the equations to determine the kinetic parameters, a brief derivation is included below. For a simple degradation step, the rate of conversion is described as follows:
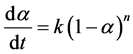 (1)
(1)
In this equation, a is the degree of chemical conversion, n is the reaction order, k is the reaction rate, and d/dt is the time derivative. A relation between the reaction rate and temperature is usually assumed:
 (2)
(2)
Here, Z is the preexponential constant, R is the ideal gas constant, Ea is the activation energy, and T is the temperature in Kelvin. This is known as the Arrhenius relationship. Equation (3) is attained by combining Equations (1) and (2):
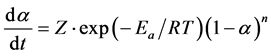 (3)
(3)
In order to obtain a precise solution, one of three variables in Equation (3) either may be held as a constant or may be related to another variable when the reaction process is carried out. In this way, the reaction process can be defined. For years, a differential or an integral version of Equation (3) has been applied by researchers to calculate the kinetic parameters consisting of Ea, Z, and n. While some of the techniques rely on the functional version of conversion, f(a), in the literature [7] - [12] , activation energy values have also been indicated to be estimated without any preceding knowledge of the functional form.
There is a linear relationship between temperature and time as following:
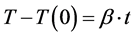 (4)
(4)
and
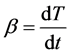 (5)
(5)
where T is the current temperature, T(0) is the initial temperature, t is time, and b is the scanning rate. Combining Equation (3) with Equation (5) yields:
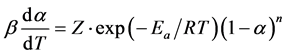 (6)
(6)
This equation shows the theoretical shape of the TGA curve that is determined in the Kinetics Software. Logarithmic form of the equation is
 (7)
(7)
A multilinear regression is performed by using ln(bda/dT), −l/RT, and ln(1−a) which have been appraised from TGA curve, and solving for Z, Ea, and n.
Kinetic parameters of degradation are determined upon the observations of Ozawa [13] and Flynn and Wall [14] . Their statements involve an approximation that has been discussed by Doyle [15] and by Flynn and Wall [14] , among others. This approximation comprises the so-called “exponential integral” and methods lowering error to a negligible level and by this way improving the results. Zsako and Zsako have suggested a closed form approximation for the exponential integral [16] . Although the determination of the activation energy is straightforward, one cannot carry out further kinetic computations without further data or assumptions as to the form of the kinetic equation. It has often been observed that the initial portion of a TG curve can usually be well-fitted by a first-order reaction equation. Since the interest in lifetime or stability studies is most often concerned with the initial portion (low conversion level), it has become a procedural practice to assume first-order kinetics and compute the rate constants, pre-exponential factors, etc. on this assumption. When the value of n = 1 is used (first order kinetics), the method is essentially the same as that used by ASTM Standart Test Method E 1641.
In this work, the mathematical treatment followed in calculating the parameters are called as Standard method. A head start of this method is to obtain the activation energy exactly independently of any assumptions regarding the relation between the reaction rate and the percent reacted. The quality of the statistical fit can be judged from the 95% confidence limits and from observing the lnβ vs 1/T plot. In the ideal case, the data points fall on parallel straight lines, the slope of which is used to calculate the activation energy.
2. Experimental
Apparatus
Low heating rate degradation experiments were conducted in a Perkin Elmer PYRIS Diamond TG/DTA (DSC) apparatus in a dynamic nitrogen atmosphere which was calibrated with calcium oxalate. All TGA experiments were carried out with a linear heating rate of 5˚C - 25˚C/min up to 600˚C under nitrogen atmosphere. Kinetic parameters of degradation were determined by the Scanning Kinetics Software.
Polystyrene with Mw = 230,000 g/mol (430,102), poly(p-methylstyrene) with Mw = 72000 (182,273), poly(p-chlorostyrene) with Mw = 75,000 (434,124), and poly(p-bromostyrene) with Mw = 65,000 (181,366) beads were all supplied from Aldrich company and used without any further process. TG analysis was carried out on approximately 10 mg samples.
3. Result and Discussion
3.1. Effect of Heating Rate on p-Substituted PSs
Nonisothermal TGA of the samples was carried out at 5, 10, 15, 20, and 25 K/min. Thermogravimetric curves for PS, PMS, PClS, and PBrS are represented in Figure 1 and Figure 2. The kinetic analysis of the degradation of samples was performed by utilizing the thermograms obtained from the TGA. Increase in the heating rate results in a shift toward higher temperatures in the thermograms (Figure 1 and Figure 2). The reason for this behavior is that the sample needs shorter time to catch a given temperature for a faster heating rate.
Same behavior with thermograms was observed for the peak temperatures. An empirical relation between the peak volatilization temperature (Tpv in ˚C) and the heating rate (R in ˚C/min) was reported by Liu et al. [17] .
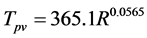 (8)
(8)
The temperature at which the decomposition rate of the sample is maximum is called as peak volatilization temperature. Tpv values for the PS and the para-substituted derivatives at different heating rates were calculated by using this relationship in the current study, and compared with the TGA experimental data. Experimental values are quite close with that of Liu et al.’s model (Table 1).
ln(k) versus 1/T plots for poly(para-substituted styrene)s are shown in Figure 3. The lines obtained at different conversion rates are observed to be roughly parallel to one another and the correlation coefficient of the li-

Figure 1. TGA scans of poly(p-substituted styrene)s at different heating rates in nitrogen.

Figure 2. DTG curves of poly(p-substituted styrene)s at different heating rates in nitrogen.

Figure 3. Kinetic analysis using Ozawa’s method for poly(p-substituted styrene)s degradation in nitrogen environment.

Table 1. Comparison of experimental poly(p-substituted styrene)s peak volatilization decomposition temperatures (Tpv) measured and expected values using Liu’s model in nitrogen at different heating rates.
nearity for the calculation of each activation energy are larger than 0.98. Also there is no systematic divergence from linearity. Therefore it is suggested that the method fitted well for the nonisothermal degradation of poly(p- substituted styrene)s and rate order of degradation is 1 in all poly(p-substituted styrene)s.
3.2. Effect of Substituent Group on Activation Energy and lnZ Values for the Decomposition of p-Substituted PSs
The activation energies and lnZ values of PS, PMS, PClS, and PBrS at the percent conversion levels of 3.0, 5.0, 8.0, 12.0, 18.0, and 26.0 are tabulated in Table 2. Activation energy is observed to increase with the degree of conversion for PS, PMS, and PClS, which should be attributed to the nature of random scission degradation and the differences in deviation from stationary reaction state at the different heating rates [18] .
The electronegativity values of methyl and hydrogen which are both bonded to the same group are close to each other. The bonding character of methyl group in PMS is most likely to bonding character of hydrogen in PS. The only difference between the two polymers is the weight of the repeating unit in the backbone. The repeating segment in PMS is slightly heavier in comparison with PS. Therefore the activation energies of PMS are slightly lower than the activation energies of PS. In the case of PClS, the weight of the repeating elements with Cl atom is much higher than the repeating units of PS and PMS and so the activation energy of the PClS is much lower. However PBrS exhibits completely different attitude. Its activation energy is considerably higher than PS and it decreases with the weight loss value. Therefore it can be suggested that the degradation mechanism of PBrS is different.
In order to understand the dependency of activation energies to the substituent in poly(p-substituted styrene)s, their activation energy values were graphed with respect to molecular weight of the repeating unit in Figure 4.
Table 2. Kinetic analysis results for poly(p-substituted styrene)s.

Figure 4. Activation energies of poly(p-substituted styrene)s at different weight loss percentages.
The outcome of Figure 4 is as follows;
1) The activation energies of degradation of PS and PMS are keeping up with its values upon different weight loss values as the activation energies of PClS and PBrS are considerably affected by the weight loss ratio.
2) The activation energies of PS, PMS, and PClS increases with the weight loss ratio, as the activation energy value of PBrS decreases.
3) The activation energy of the substituted styrenes decreases with the molecular weight of the repeating units, PBrS shows an extraordinary behavior. Its activation energy values are higher than the others up to high values of the weight losses.
Upon handling individually, PS degrades with chain scission mostly. The bonding character of methyl group in PMS is very similar to bonding character of H in PS. The electronegativity values of methyl and H which are both bonded to the same group are close to each other. The only difference between the two polymers is the weight of the repeating unit in the backbone. The weight of repeating segment of PMS is slightly higher than that of PS. Therefore the activation energies of PMS are slightly lower than PS. In the case of PClS, the weight of the repeating elements with Cl atom is much higher than the repeating units of PS and PMS and the activation energy of the PClS is much lower.
The bonds between the substituent and the polystyrene backbone are C-H in PS, C-C in PMS, C-Cl in PClS, and C-Br in PBrS. The trend in the electronegativity values of the substituent of polystyrenes is as H(2.20) < C(2.55) < Br(2.96) < Cl(3.16) and, the bonding character of Br is much close to H and C than Cl. As a result, it is seen from the Figure 4 that the activation energy values of the degradation of PBrS is more close to the PS and PMS for any of the weight loss rates. On the other hand the character of Br substituent makes the chemical bonds between repeating groups stronger, leading to a high activation energy values.
4. Conclusion
Thermal decomposition kinetics of poly(p-substituted styrene)s was examined in nitrogen atmosphere. Five different heating rates were applied to the samples and as heating rate increased, the degradation shifted to higher temperatures accompanied with an increase in the rate of weight loss. From the nonisothermal degradation data, kinetic parameters were calculated using Flynn-Wall-Ozawa method and the values of the reaction order of poly(p-substituted styrene)s were deduced as 1 with the correlation factor higher than 0.98. Activation energies of thermal degradations are increasing individually in PS, PMS, PClS with weight loss as it is decreasing in PbrS with weight loss. In general, the decrease in the activation energies of poly(p-substituted styrene)s with PS/PMS/ PClS trend is consistent with the increase in repeating unit molecular weight except for PBrS. Therefore PBrS is suggested to decompose with somehow different mechanism.
Cite this paper
AyşegülŞenocak,CemilAlkan,AhmetKaradağ, (2016) Thermal Decomposition and a Kinetic Study of Poly(Para-Substituted Styrene)s. American Journal of Analytical Chemistry,07,246-253. doi: 10.4236/ajac.2016.73021
References
- 1. Mehta, S., Biederman, S. and Shivkumar, S. (1995) Thermal Degradation of Foamed Polystyrene. Journal of Materials Science, 30, 2944-2949.
http://dx.doi.org/10.1007/BF00349667 - 2. Westerhout, R.W.J., Waanders, J., Kuipers, J.A.M. and Swaaij, W.P.M. (1997) Kinetics of the Low-Temperature Pyrolysis of Polyethene, Polypropene, and Polystyrene Modeling, Experimental Determination, and Comparison with Literature Models and Data. Industrial & Engineering Chemistry Research, 36, 1955-1964.
http://dx.doi.org/10.1021/ie960501m - 3. Poutsma, M.L. (2009) Further Considerations of the Sources of the Volatiles from Pyrolysis of Polystyrene. Polymer Degradation and Stability, 94, 2055-2064.
http://dx.doi.org/10.1016/j.polymdegradstab.2009.07.011 - 4. Carniti, P., Beltrame, P.L., Armada, M., Gervasini, A. and Audisio, G. (1991) Polystyrene Thermodegradation. 2. Kinetics of Formation of Volatile Products. Industrial & Engineering Chemistry Research, 30, 1624-1629.
http://dx.doi.org/10.1021/ie00055a032 - 5. Jun, H., Oh, S.C., Lee, H.P. and Kim, H.T. (2006) A Kinetic Analysis of the Thermal-Oxidative Decomposition of Expandable Polystyrene. Korean Journal of Chemical Engineering, 23, 761-766.
http://dx.doi.org/10.1007/BF02705924 - 6. Peterson, J.D., Vyazonkin, S. and Wight, C.A. (2001) Kinetics of the Thermal and Thermo-Oxidative Degradation of Polystyrene, Polyethylene and Poly(propylene). Macromolecular Chemistry and Physics, 202, 775-784.
http://dx.doi.org/10.1002/1521-3935(20010301)202:6<775::AID-MACP775>3.0.CO;2-G - 7. Sanchez-Jimenez, P.E., Perez-Makueda, L.A., Perejon, A. and Criado, J.M. (2009) Combined Kinetic Analysis of Thermal Degradation of Polymeric Materials under Any Thermal Pathway. Polymer Degradation and Stability, 94, 2079-2085.
http://dx.doi.org/10.1016/j.polymdegradstab.2009.07.006 - 8. Madras, G., Chung, G.Y., Smith, J.M. and McCoy, B.J. (1997) Molecular Weight Effect on the Dynamics of Polystyrene Degradation. Industrial & Engineering Chemistry Research, 36, 2019-2024.
http://dx.doi.org/10.1021/ie9607513 - 9. Murakata, T., Saito, M., Sato, H., Hirai, T. and Sato, S. (1998) Selective Pyrolysis of Polystyrene to That with a Desired Low Polymeric Degree. Journal of Applied Polymer Science, 70, 2299-3305.
http://dx.doi.org/10.1002/(SICI)1097-4628(19981212)70:11<2299::AID-APP24>3.0.CO;2-W - 10. Marcilla, A. and Beltran, M. (1995) Kinetic Study of the Thermal Decomposition of Polystyrene and Polyethylene-Vinyl Acetate Graft Copolymers by Thermogravimetric Analysis. Polymer Degradation and Stability, 50, 117-124.
http://dx.doi.org/10.1016/0141-3910(95)00138-C - 11. Dauengauer, S.A., Utkina, O.G., Popova, G.S. and Sazanov, Y.N. (1987) Investigation of Thermal Degradation of Polystyrene with the Aid of Thermal Analysis. Journal of Thermal Analysis and Calorimetry, 32, 311-314.
http://dx.doi.org/10.1007/BF01914571 - 12. Vyazovkin, S., Drancal, I., Fan, X. and Advincula, R. (2004) Kinetics of the Thermal and Thermo-Oxidative Degradation of a Polystyrene-Clay Nanocomposite. Macromolecular Rapid Communications, 25, 498-503.
http://dx.doi.org/10.1002/marc.200300214 - 13. Ozawa, T. (1965) A New Method of Analyzing Thermogravimetric Data. Bulletin of the Chemical Society of Japan, 38, 1881-1886.
http://dx.doi.org/10.1246/bcsj.38.1881 - 14. Flynn, J.H. and Wall, L.A. (1966) A Quick, Direct Method for the Determination of Activation Energy from Thermogravimetric Data. Polymer Letters, 4, 323-328.
http://dx.doi.org/10.1002/pol.1966.110040504 - 15. Doyle, C.D. (1961) Kinetic Analysis of Thermogravimetric Data. Journal of Applied Polymer Science, 5, 285-292.
http://dx.doi.org/10.1002/app.1961.070051506 - 16. Zsako, J. and Zsako, J.J. (1980) Kinetic Analysis of Thermogravimetric Data. Journal of Thermal Analysis and Calorimetry, 19, 333-345.
http://dx.doi.org/10.1007/BF01915809 - 17. Liu, Y., Bakhtiyarov, S.I. and Overfelt, R.A. (2001) Decomposed Eps Gases Pressure Measurements in Iron Lost Foam Casting Process. Proceedings of the 2001 ASME International Mechanical Engineering Congress and Exposition, New York, 11-16 November 2001, 9-12.
- 18. Gao, Z.M., Kaneko, T., Amasaki, I. and Nakada, M. (2003) A Kinetic Study of Thermal Degradation of Polypropylene. Polymer Degradation and Stability, 80, 269-274.
http://dx.doi.org/10.1016/S0141-3910(02)00407-X
NOTES

*Corresponding author.