American Journal of Analytical Chemistry
Vol. 3 No. 4 (2012) , Article ID: 18426 , 7 pages DOI:10.4236/ajac.2012.34039
Preparation of Fame by Microwave Irradiation Using Boron Trifluoride as a Catalyst
1Department of Chemical Processes, Federal Technological University of Parana, Toledo, Brazil
2Department of Chemistry, State University of Maringa, Maringa, Brazil
Email: *g_bonafe@yahoo.com.br
Received January 10, 2012; revised February 28, 2012; accepted March 9, 2012
Keywords: Vegetable Oil; Boron Trifluoride; Transesterification; Surface Response Methodology; FAME
ABSTRACT
Fatty acid methyl ester (FAME) preparation is an important step to determine fatty acid composition of oils and fats. Transesterification with boron trifluoride (BF3) in methanol has been widely used; however, the transesterification of triacylglycerols usually involves a long heating time, whereas much shorter reaction times may be sufficient when microwave irradiation is used. The purpose of this paper was to optimize FAME preparation with BF3 by microwave irradiation. A three-factor central composite routable (CCRD) design was used to study the effect of the heating time (A), irradiation power (B), and volume of sodium hydroxide (C) on FAME concentration. A second-order polynomial model was employed to generate a surface response. Optimum conditions obtained for FAME preparation were time of 60 s, irradiation power of 220 W, and base volume of 1.5 mL. Compared to conventional methods, the advantages of this method are the quick result and the accuracy.
1. Introduction
In the last decades, many of the functions of lipids in human metabolism have been elucidated, affording a better understanding of their relationships to health and disease. As a result, much emphasis has been placed on the increase in the intake of n – 3 fatty acids and the decrease in the intake of saturated fatty acids [1] and trans fatty acids. This has also led to a growing concern about the intake of lipids, and consequently, about the fatty acid composition of foods.
Chromatographic techniques, especially gas chromatography (GC) and high performance liquid chromatography (HPLC), have been widely used for the analysis of food lipids. In the analysis of fatty acids, GC plays a unique role due to its excellent ability to resolve complex mixtures that involve both geometric and positional isomers [2]. Thus, GC allows for obtaining more detailed information about the composition of fatty acids. The use of GC to analyze fatty acids usually involves the preparation of fatty acid methyl esters (FAME). Transesterification makes the fatty acid more volatile, a condition that is necessary in many determinations, due to the limit temperature to which the column stationary phase can be subjected to [3].
In routine analysis, time is critical, and gas chromatographic methods are usually time consuming; thus, the introduction of improvements in each step of the process helps to shorten the analysis time. Microwave irradiation has proven to be faster than thermal heating for the synthesis of methyl esters from triglycerides in the presence of heterogeneous catalysts [4].
Derivatization of fatty acids generally falls into two categories: acid and base catalysis. The reagents most commonly used in the transesterification of acylglycerols by basic catalysis are sodium (NaOH) or potassium (KOH) hydroxide in methanol and sodium methoxide (NaOCH3) in methanol. Transesterification with these reagents may be carried out at room temperature in a very short time. However, a disadvantage is that they do not convert free fatty acids to FAME, which limits their application to highly acid oils. Moreover, use of sodium or potassium hydroxide in methanol has a potential risk of saponification during transesterification. On the other hand, the primary advantage of acid catalysis is the general applicability, with both bound and free fatty acids (FFA) being converted concurrently to FAME. Among the various acid-catalyzed reagents (such as methanolic hydrochloric acid, sulfuric acid in methanol, and acetyl chloride in methanol), boron trifluoride in methanol has had wide application as a good reagent to convert both the acylglycerols and the FFA into methyl esters [5].
The Lewis acid, boron trifluoride, in the form of its coordination complex with methanol, is a powerful acidic catalyst for the transesterification of fatty acids. In the method proposed by Metcalfe and Schmitz [6], esterification of free fatty acids was completed in two minutes with 12% to 14% boron trifluoride in methanol under reflux. Morrison and Smith [7] determined the optimum conditions for transesterification of triacylglycerols (TAGs), sterol esters, monoacylglycerols, diacylglycerols, phosphocylglycerols, and sphingolipids. Under the conditions recommended (heating at 100˚C), transesterification occurs within 10 min for phosphocylglycerols and within 30 min for triacylglycerols.
The popularity of FAME preparation reaction with boron trifluoride in methanol is due, in part, to its acceptance by the American Oils Chemists’ Society [8], Association of Official Analytical Chemists [9], and International Union of Pure and Applied Chemistry [10]. However, boron trifluoride in methanol has a few disadvantages. The reagent is expensive and has a limited shelf life if not refrigerated. Moreover, its use may result in the production of artifacts or loss of polyunsaturated fatty acids. Fulk and Shorb [11] showed that the reaction time affects the formation of artifacts. Thus, reaction time must be reduced to avoid the formation of artifacts. Much shorter reaction times have been shown to suffice if conventional heating is replaced by microwave irradiation [12].
Microwave heating involves direct absorption of energy by functional groups that have a dipole rotation effect or ionic conduction and its release to the surrounding solution, leading to a rapid rise in temperature. Dipole rotation is an interaction in which polar molecules try to align themselves with the rapidly changing electric field of the microwave. The second way to transfer energy is ionic conduction, which results if there are free ions or ionic species present in the substance being heated.
The results of one-factor-at-a-time experiments do not reflect actual changes in the environment, as they ignore interactions between factors that are present simultaneously. When many factors and interactions affect desired responses, response surface methodology (RSM) is an effective optimization tool. Its use provides the relevant information in the shortest time with the least number of experiments. In addition to analyzing the effects of the independent variables, this experimental methodology generates a mathematical model which describes the chemical processes [13]. The central composite routable design (CCRD) is one of the most useful and desirable designs for fitting second order models. The design is considered routable because the variance of the predicted response Y is the same at all points X that are at the same distance from the design center. This means that the variance of predicted response is constant on concentric circles. A design with this property does not change the variance of the predicted response when the design is rotated around the center point [14]. Therefore, association of CCRD with RSM is very effective for reducing the number of combinations required without compromising the validity of the results in studies containing a large number of independent variables.
There is very limited literature examining the use of microwave energy for driving fatty acid transesterification reactions [15]. Thus, in this paper, the FAME preparation method proposed by Joseph and Ackman [16], which uses heating in a water bath, was modified for microwave heating. Optimization of the reaction conditions was made by CCRD and RSM, which evaluated the influence of heating time, irradiation power, and base volume, with the objective of obtaining the best conditions for this modification.
2. Materials and Methods
2.1. Materials
Refined soybean oil was purchased from a local supermarket. Tricosanoic acid methyl ester (99%), linoleic acid (99%), tripalmitin (99%), triestarin (99%), trilinolein (98%), and a FAME standard mixture (189-19) were purchased from Sigma (USA). All other chemicals used in the study were analytical grade. A microwave oven with an adjustable power level was used for microwave irradiation.
2.2. Transesterification
Soybean oil (25 ± 0.1 mg) was weighed in a test tube with a screw cap. Then, 250 µL of a 10 mg∙mL–1 solution of linoleic acid in chloroform was added, in order to obtain a final concentration of approximately 10% of free fatty acids in the sample. The solvent was evaporated under nitrogen flow. Next, 1.5 mL of a 0.50 mol∙L–1 solution of NaOH in methanol was added. The mixture was heated in a water bath at 100˚C for about 5 min and then cooled to room temperature (25˚C). A boron trifluoride solution (2.0 mL, 12% in methanol) was added, and the mixture was heated in a water bath at 100˚C for 30 min. The test tube containing the mixture was cooled in running water to room temperature before adding 1.0 mL of isooctane. The mixture was vigorously stirred for 30 s. Saturated sodium-chloride (5.0 mL) was added, and after phase separation by resting the mixture at –5˚C, the upper phase containing the methyl esters was collected and stored in a freezer (–18˚C) for subsequent chromatographic analysis.
2.3. Experimental Design
In the evaluation of the transesterification method, the water bath heating (30 min) was replaced with microwave oven heating. The variables analyzed were time (30 - 70 s), irradiation power (30% - 70%), and base volume (0.5 - 2.5 mL). The factor of central composite routable design resulted in 20 tests (Table 1), which were run in six replicates and randomly to minimize the effect of unexplained variability on the observed responses due to extraneous factors.
A quadratic polynomial regression model was assumed for predicting individual Y variables. The model proposed for response of Y was:
 (1)
(1)
where β0, βi, βii, and βij are intercept, linear, quadratic, and interaction regression coefficient terms, respectively, and Xi and Xj are independent variables [14], with j ≠ i. Response surface and contour plot were developed using the fitted quadratic polynomial equations obtained from response surface regression analysis.
2.4. Analysis of FAME
The chromatographic analysis was carried out by using a gas chromatograph CP3380 (Varian, USA) equipped with a flame ionization detector (FID) and a fused capillary column CP-Select CB-FAME (100 m × 0.25 mm i.d. and 0.25 µm film thickness, Varian, USA). The column was heated to 197˚C in 18 min and then the temperature was raised to 235˚C at 20˚C∙min–1 and held for 5 min. Injector and detector temperatures were 225˚C and 245˚C∙min–1, respectively. Samples (2 µL) were injected in triplicate. The gas flow rates used were 1.4 mL∙min–1 carrier gas (H2), 30 mL∙min–1 make-up gas (N2), and 30 and 300 mL∙min–1 flame gases (H2 and synthetic air, respectively). Sample splitting rate was 1:80. The peak areas were determined with Workstation 5.0 software (Varian, USA).
FAME were identified by comparison of sample FAME peak relative retention times with those of standards from Sigma (USA) and by equivalent chain length (ECL) values determined according to [17], with methyl esters of 16:0, 18:0, and 20:0 acids as reference compounds. ECL values were calculated for FAME of soybean oil and a standard mixture 189-19 (Sigma, USA), and were compared with the literature values [18-20].
Analysis for determination of ECL values was performed by using a gas chromatograph 14A (Shimadzu, Japan) equipped with a flame ionization detector (FID) and a fused capillary column Carbowax 20 M (50 m × 0.25 mm i.d., 0.20 m film thickness). The column temperature was maintained constant at 200˚C for 60 min. Injector and detector port temperatures were 220˚C and 245˚C, respectively. The gas flow rates used were 1.4 mL∙min–1 carrier gas (H2), 30 mL∙min–1 make-up gas (N2), and 30 and 300 mL∙min–1 flame gases (H2 and synthetic air, respectively). Sample splitting rate was 1:100. Peak areas were determined with an Integrator-Processor CG- 300 (Instrumentos Científicos CG). All the determinations of ECL values were carried out in six replicates.

Table 1. Coded and uncoded variables used for two level, three-factorial central composite rotatable design and response for FAMEs concentration.
2.5. Quantification of FAME
Quantification of FAME from soybean oil was made against methyl ester of tricosanoic acid (23:0Me) as an internal standard (IS). In a test tube, 25 ± 1 mg of soybean oil was weighted and 500 µL of 1.0 mg∙mL–1 solution of methyl tricosanoate in isooctane was added. The amount of IS added was established to maintain a maximum ratio of 200:1 between oil weight and IS weight. After the addition of the internal standard solution, the solvent was evaporated under nitrogen flow. The concentration of FAME obtained after transterification was calculated with Equation (2), as proposed by Cantellops, Reid, Eitenmiller, and Long [21]. The results were expressed in mg of FAME per gram of oil.
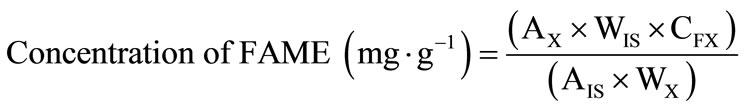 (2)
(2)
where AX is the peak area of FAME, WIS is the weight (mg) of internal standard (IS) added to the sample (in mg), CFX is the theoretical correction factor, WX is the sample weight (in mg), AIS the peak area of IS (23:0) added to the sample (in mg), and WX is the sample weight (in mg).
Because unsaturated FAME are subject to autoxidation, it is not possible to obtain and maintain high purity standards. Bannon, Craske, and Hilliker [22] have demonstrated that a theoretical correction factor (CFx) may be applied for unsaturated FAME after verifying that the empirical correction factors (ECFx) for saturated FAME are in accordance with CFx values.
ECFx was determined analyzing data of the 189-19 standard mixture (Sigma, USA) and fish oil in n-heptane, according to Martin, de Oliveira, Visentainer, Matsushita, and de Souza [23] and Aguiar et al. [24].
The limits of detection (LOD) and quantification (LOQ) were estimated by triplicate analysis of successively diluted methyl arachidate standard solution (1.0 mg∙mL–1) considering the signal-noise rate relative to the background signal as 3 and 10, respectively [25].
2.6. Recovery Assay
The method accuracy was evaluated by recovery assay. Initially, the linoleic acid concentration in soybean oil was determined, and known quantities of linoleic acid and IS were added to soybean oil. A standard mixture constituted of tripalmitin (TG 16:0), triestarin (TG 18:0), trilinolein (TG 18:3n – 3), and linoleic acid dissolved in chloroform was also prepared in concentrations of 0.310, 0.318, 0.323, and 1.11 mg∙mL–1, respectively. These triacylglycerols were selected because they are constituents of soybean oil. Before transesterification, tricosanoic acid methyl ester (IS) was added to soybean oil and followed by addition of triacylglycerols and a linoleic acid mixture. Solvent evaporation was carried out under nitrogen flow. Recovery assays were performed with three replicates.
2.7. Statistical Analysis
Factorial design experiments were performed in triplicate at all points except at the centre point (0, 0, 0) where eighteen replications were carried. Thus three sets of data were obtained, that were submitted to analysis of variance (ANOVA) and did not show significant difference between them. All experiments were carried out in a randomized order to minimize the effect of unexplained variability in the observed responses due to extraneous factors.
The software Statistica [26] was used for the multiple regression analysis, linear regression analysis, ANOVA, Student-t test, and factorial design. Significance level of 0.05 was used for all analysis. The software Scilab [27] was used in the canonical analysis.
3. Results and Discussion
The gas chromatographic method was remarkably stable and robust in the analysis of all sample FAME. Under the selected operating conditions, LOD and LOQ were estimated at 0.15 and 0.50 mg∙g–1, respectively. The CFx values for the saturated FAME were quite close to the experimental values. Thus, in the quantification of fatty acids, the CFx values for saturated and unsaturated fatty acid methyl esters were used.
With the results from experiments based on central composite design (Table 1), it was possible to obtain a highly significant quadratic model (p < 0.01) with a coefficient of determination (R2) of 0.952, according to Equation (3):
 (3)
(3)
where A, B, and C are coded variables related to the heating time, irradiation power, and volume of the sodium hydroxide solution, respectively.
The stationary point was determined by deriving the equation of the proposed model for all variables and making the derivatives equal to zero. The solution of the linear system gave the values of 0.471 for time, 0.388 for power, and 0.789 for the volume of the base solution. These values, in terms of uncoded variables, correspond to 59.4 seconds, 59.8% of the microwave oven maximum power, and 2.3 mL of NaOH solution.
The canonical analysis was conducted to characterize the stationary point of the response surface, determining if it corresponds to a maximum point, minimum point, or saddle point. The canonical form of the proposed quadratic model is as follows:
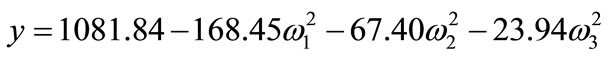 (4)
(4)
where ω1, ω2, and ω3 are the transformed independent variables and the coefficients of the variables are the eigenvalues. If all eigenvalues are positive, the stationary point corresponds to a minimum; if all of them are negative, the stationary point corresponds to a maximum; and if they have positive and negative values, it is a saddle point. Considering that all eigenvalues are negative and that the stationary point is within the region explored by the factorial design, it is possible to conclude that the stationary point corresponds to a maximum [14].
Taking into account the agreement with the method proposed by Joseph and Ackman [16], we chose to use the base volume of 1.5 mL. In this condition, the value of variable C in Equation (3) is zero, and the stationary point is determined through partial derivatives in relation to A and B, and by making the derivatives equal to zero. Solving the linear system obtained, we found the values of 0.518 for time and 0.516 for power. For the uncoded variables, the values for time and power were 60.4 s and 60.3% (220 W).
The analysis of the response surface (Figures 1 and 2) indicates that the stationary point is a maximum. In the contour plot, each contour represents contour lines with the same response value in a plane whose coordinates represent the levels of the independent variables. Thus, considering the different contour levels, it is possible to determine the levels of the independent variables in which the surface height value changes.
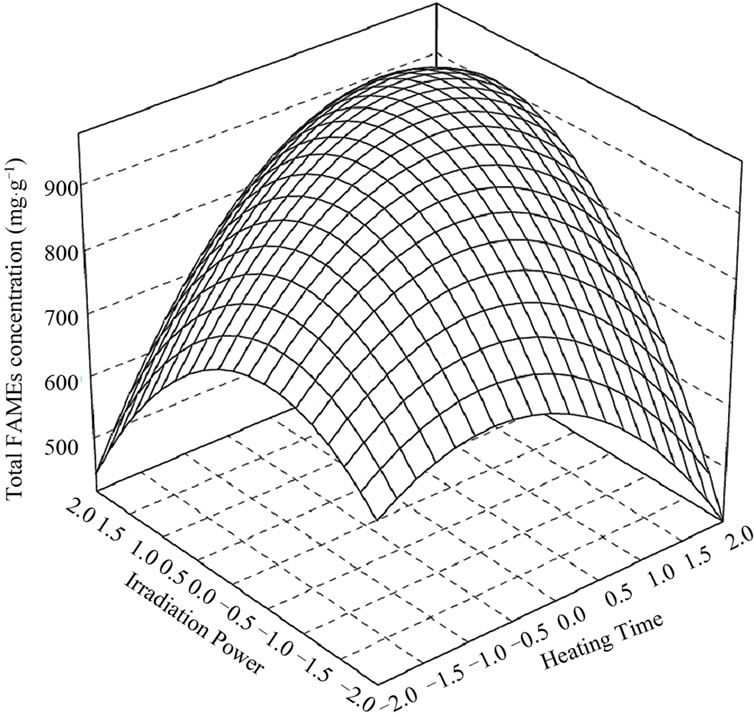
Figure 1. Response surface and contour plots for effect of heating time and irradiation power on FAMEs concentration.
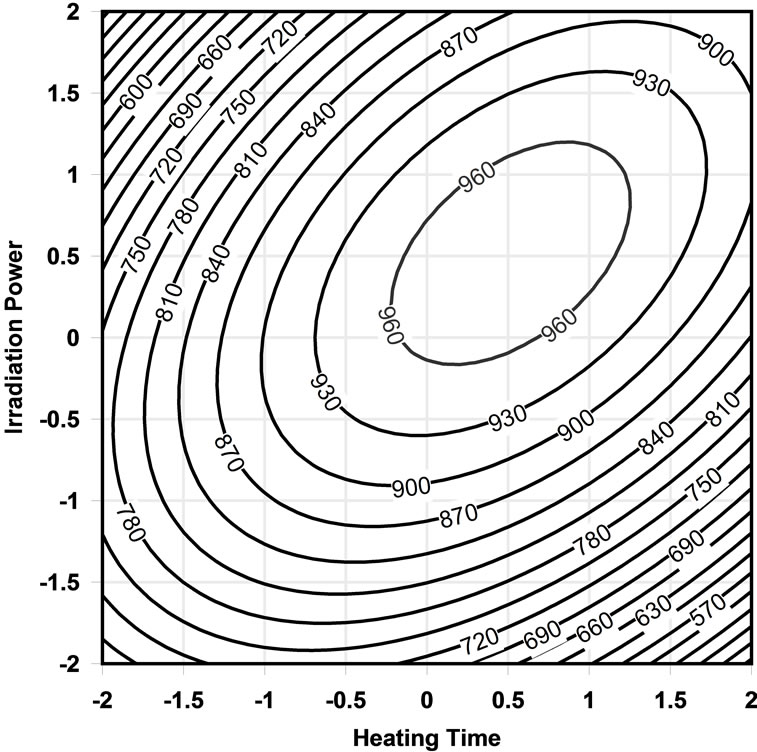
Figure 2. Contour plots for effect of heating time and irradiation power on FAMEs concentration.
From the canonical analysis of Equation (3), when C is equal to zero, we obtain the equation:
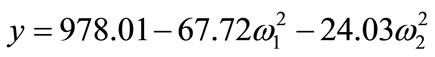 (5)
(5)
which indicates that the maximum concentration of FAME is 978.01 mg∙mL–1 when the base volume is 1.5 mL. Thus, considering the practicality and agreement with the method proposed by Joseph and Ackman [16], the selected conditions were time of 60 s, 60% of maximum power (220 W), and base volume of 1.5 mL.
When Armstrong et al. [15] proposed a method for preparing FAME from plasma phospholipids, using boron trifluoride in methanol and heating by microwave irradiation, they determined that total fatty acid concentration tended to be lower with microwave irradiation when compared to convectional heat. For the heating time of 45 s, the total contents of monounsaturated and polyunsaturated fatty acids were significantly lower in relation to those obtained by convectional heating, indicating that microwave heating requires further assessment. In our experiments, we did not observe any influence on monounsaturated and polyunsaturated composition when microwave irradiation was used.
In another study with microwave irradiation, Jeyashoke, Krisnangkura, and Chen [28] found that the FAME of canola, olive, soybean, rice, and sunflower oils can be prepared in about 15 s with 1.0% of sodium methoxide and a methanol/toluene mixture of 1:3 (v/v). When the ratio of the methanol/toluene mixture was 1:10 (v/v), the time required for the transesterification was 40 s.
When the selected condition was used for transesterification with heating by microwave irradiation for soybean oil fortified with 10% linoleic acid, we obtained the total FAME concentration of 964 ± 12.3 mg∙g–1. The conventional heating proposed by Joseph and Ackman [16] gave a concentration of 970 ± 9.2 mg∙g–1. These results were not significantly different when compared by Student-t test (p > 0.05).
The individual concentrations of FAME in the soybean oil sample obtained by microwave irradiation and by water bath heating ranged from 0.74 to 471 mg∙g–1 and showed a linear relationship and a correlation coefficient of 0.9997 (p < 0.001) (Figure 3). This high correlation coefficient indicates that the results obtained by these methods correlate strongly with the results obtained by the original method.
The addition of linoleic acid to the soybean oil sample (Table 2), followed by the preparation of FAME by conventional heating and by microwave heating, resulted in recovery levels of 92.40 and 91.73%, respectively, without a significant difference (p > 0.05). These results also indicate that the quantification of the linoleic acid methyl ester was sufficiently accurate, since the range of 80.0% - 110.0% is considered acceptable in method validation studies [29].
Table 3 shows the recovery values for derivatization of the mixture of standards: tripalmitin (TG 16:0), tristearin

Figure 3. Correlation between individual concentration of FAMEs from soybean oil prepared by conventional heating and microwave irradiation.
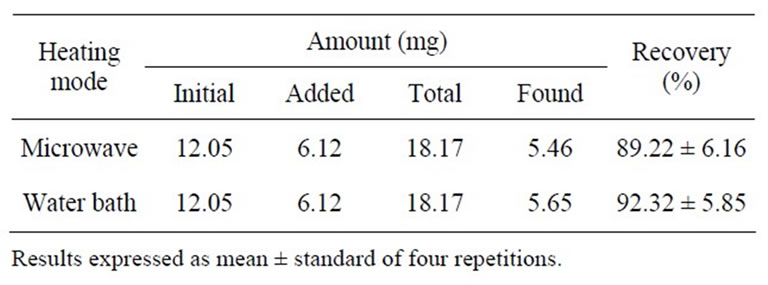
Table 2. Recovery percentage of linoleic acid added to soybean oil with FAME preparation by heating in water bath and microwave.
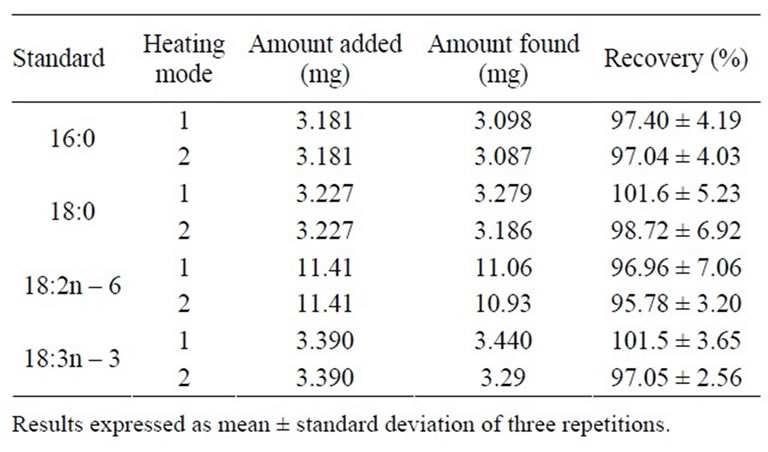
Table 3. Recovery percentage of different standards by conventional heating (1) and microwave heating (2).
(TG 18:0), trilinolein (TG 18:3n – 3), and linoleic acid (18:2n – 6) heated in a water bath and by microwave irradiation. The recovery percentage ranged from 95.96% to 101.6% for water bath heating and from 95.78% to 98.72% for microwave irradiation heating. There was no significant difference (p > 0.05) between the recovery percentages of triacylglycerols and linoleic acid by both heating methods. These results indicate that the proposed FAME preparation method by microwave irradiation heating produces results that are equivalent to those of the conventional method. Moreover, the method accuracy is appropriate for the preparation of FAME from triacylglycerols and free fatty acids.
4. Conclusion
Thus, due to the ease of the proposed method and the excellent results obtained, as compared to the conventional methodology, the derivatization of triacylglycerols and free fatty acids, with the use of a solution of 0.5 mol∙L–1 sodium hydroxide in methanol, which followed by reaction with a 12% solution of boron trifluoride in methanol and heating by microwave irradiation in a closed system, was proven to be a very convenient and fast methodology, significantly reducing the analysis time.
5. Acknowledgements
The authors are grateful to CAPES, CNPq, and Fundação Araucária for the financial support.
REFERENCES
- S. Sudheendran, C. C. Chang and R. J. Deckelbaum, “n – 3 vs. Saturated Fatty Acids: Effects on the Arterial Wall,” Prostaglandins, Leukotrienes and Essential Fatty Acids, Vol. 82, No. 4-6, 2010, pp. 205-209. doi:10.1016/j.plefa.2010.02.020
- M. Ledoux, L. Laloux and R. L. Wolff, “Analytical Methods for Determination of Trans-C18 Fatty Acids Isomers in Milk Fat,” Analysis, Vol. 28, No. 5, 2000, pp. 402-412. doi:10.1051/analusis:2000125
- M. A. De la Fuente, P. Luna and M. Juarez, “Chromatographic Techniques to Determine Conjugated Linoleic Acid Isomers,” Trends in Analytical Chemistry, Vol. 25, No. 9, 2006, pp. 917-926. doi:10.1016/j.trac.2006.04.012
- C. Mazzocchia, G. Módica, A. Kaddouri and R. Nannicini, “Fatty Acid Methyl Esters Synthesis from Triglycerides over Heterogeneous Catalysts in the Presence of Microwaves,” Comptes Rendus Chimie, Vol. 7, No. 6-7, 2004, pp. 601-605. doi:10.1016/j.crci.2003.12.004
- N. C. Shantha and G. E. Napolitano, “Gas-Chromatography of Fatty-Acids,” Journal of Chromatography A, Vol. 624, No. 1-2, 1992, pp. 37-51. doi:10.1016/0021-9673(92)85673-H
- L. D. Metcalfe and A. A. Schmitz, “Rapid Preparation of Fatty Acid Esters for Gas Chromatographic Analysis,” Analytical Chemistry, Vol. 33, No. 3, 1961, pp. 363-364. doi:10.1021/ac60171a016
- W. R. Morrison and L. M. Smith, “Preparation of Fatty Acid Methyl Esters and Dimethylacetals from Lipids with Boron Fluoride-Methanol,” Journal of Lipid Research, Vol. 5, No. 1-3, 1964, pp. 600-608.
- “Official Methods of Analysis,” 15th Edition, AOAC, Arlington, 1990.
- American Oil Chemists’ Society, “Official and Tentative Methods of the American Oil Chemists’ Society, 1969,” The Analyst, Vol. 72, No. 853, 1989, p. 157.
- International Union of Pure and Applied Chemistry (IUPAC), “Standard Methods for the Analysis of Oils Fats and Derivatives, Applied Chemistry Division, Comission on Oils, Fats and Derivatives,” 6th Edition, Pergamon Press, Oxford, 1979.
- W. K. Fulk and M. S. Shorb, “Production of an Artifact during Methanolysis of Lipids by Boron TrifluorideMethanol,” Journal of Lipid Research, Vol. 11, No. 3, 1970, pp. 276-277.
- P. Banerjee, G. Dawson and A. Dasgupta, “Enrichment of Saturated Fatty-Acid Containing Phospholipids in Sheep Brain-Serotonin Receptor Preparations—Use of Microwave Irradiation for rapid Transesterification of Phospholipids,” Biochimica et Biophysica Acta, Vol. 1110, No. 1, 1992, pp. 65-74. doi:10.1016/0005-2736(92)90295-W
- D. Bas and I. H. Boyaci, “Modeling and Optimization I: Usability of Response Surface Methodology,” Journal of Food Engineering, Vol. 78, No. 3, 2007, pp. 836-845. doi:10.1016/0005-2736(92)90295-W
- D. C. Montgomery, “Design and Analysis of Experiments,” 5th Edition, John Wiley and Sons, New York, 1991.
- J. M. Armstrong, A. H. Metherel and K. D. Stark, “Direct Microwave Transesterification of Fingertip Prick Blood Samples for Fatty Acid determinations,” Lipids, Vol. 43, No. 2, 2008, pp. 187-196. doi:10.1007/s11745-007-3141-6
- J. D. Joseph and R. G. Ackman, “Capillary Column GasChromatographic Method for Analysis of Encapsulated Fish Oils and Fish Oil Ethyl-Esters—Collaborative Study,” Journal of AOAC International, Vol. 75, No. 3, 1992, pp. 488-506.
- J. V. Visentainer and M. R. B. Franco, “Ácidos Graxos em Óleos e Gorduras: Identificação e Quantificação,” Varela, São Paulo, 2006.
- R. H. Thompson, “Simplifying Fatty Acid Analyses in Multicomponent Foods with a Standard Set of Isothermal GLC Conditions Coupled with ECL Determinations,” Journal of Chromatographic Science, Vol. 34, No. 11, 1996, pp. 495-504.
- K. Strànsky, T. Jursik and A. Vitek, “Standard Equivalent Chain Length Values of Monoenic and Polyenic (Methylene Interrupted) Fatty Acids,” Journal of High Resolution Chromatography, Vol. 20, No. 3, 1997, pp. 143-158. doi:10.1002/jhrc.1240200305
- J. V. Visentainer, M. D. Noffs, P. D. O. Carvalho, V. V. de Almeida, C. C. de Oliveira and N. E. de Souza, “Lipid Content and fatty Acid Composition of 15 Marine Fish Species from the Southeast Coast of Brazil,” Journal of the American Oil Chemists’ Society, Vol. 84, No. 6, 2007, pp. 543-547.
- D, Cantellops, A. P. Reid, R. R. Eitenmiller and A. R. Long, “Determination of Lipids in Infant Formula Powder by Direct Extraction Methylation of Lipids and Fatty Acid Methyl Esters (FAME) Analysis by Gas Chromatography,” Journal AOAC International, Vol. 82, No. 5, 1999, pp. 1128-1139.
- C. D. Bannon, J. D. Craske and A. E. Hilliker, “Analysis of Fatty-Acid Methyl-Esters with high-accuracy and Reliability. V. Validation of Theoretical Relative Response Factors of Unsaturated Esters in the Flame Ionization Detector,” Journal of the American Oil Chemists’ Society, Vol. 63, No. 1, 1986, PP. 105-110.
- C. A. Martin, C. C. de Oliveira, J. V. Visentainer, M. Matsushita and N. E. de Souza, “Optimization of the Selectivity of a Cyanopropyl Stationary Phase for the Gas Chromatographic Analysis of Trans Fatty Acids,” Journal of Chromatography A, Vol. 1194, No. 1, 2008, pp. 111- 117. doi:10.1016/j.chroma.2008.04.033
- A. C. Aguiar, S. M. Cottica, M. Boroski, C. C. De Oliveira, E. G. Bonafe, P. B. França, N. E. Souza and J. V. Visentainer, “Quantification of Essential Fatty Acids in the Heads of Nile Tilapia (Oreochromis Niloticus) Fed with Linseed Oil,” Journal of the Brazilian Computer Society, Vol. 22, No. 4, 2011, pp. 643-647. doi:10.1590/S0103-50532011000400005
- Analytical Methods Committee, “Recommendations for the Definition, Estimation and Use of the Detection Limit,” Analyst, Vol. 112, No. 2, 1987, pp. 199-204. doi:10.1039/an9871200199
- Statsoft Inc., “Statistica for Windows,” Statsoft Inc., Tulsa, 2005.
- Consortium Scilab (Digiteo), “Scilab for Windows,” Le Chesnay, Cedex, France, 2010.
- N. Jeyashoke, K, Krisnangkura and S. T. Chen, “Microwave Induced Rapid Transmethylation of Fatty Acids for Analysis of Food Oil,” Journal of Chromatography A, Vol. 818, No. 1, 1998, pp. 133-137. doi:10.1016/S0021-9673(98)00545-7
- R. Caulcutt and R. Boddy, “Statistics for Analytical Chemists,” Chapman and Hall, London, 1983.
NOTES
*Corresponding author.