Open Journal of Rheumatology and Autoimmune Diseases
Vol.05 No.03(2015), Article ID:58283,5 pages
10.4236/ojra.2015.53011
Current Approach to Treatment of Juvenile Idiopathic Arthritis: Case Report of Hiperimmunglobulin E Syndrome Developed Juvenile Idiopathic Arthritis
Tuba Tülay Koca1*, Aydın Arslan2
1Malatya State Hospital, Physical Medicine and Rehabilitation Clinic, Malatya, Turkey
2Malatya State Hospital, Orthopedics and Traumatology Clinic, Malatya, Turkey
Email: *tuba_baglan@yahoo.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 9 May 2015; accepted 21 July 2015; published 24 July 2015
ABSTRACT
Introduction: Juvenile idiopathic arthritis (JIA) represents a heterogeneous group of childhood chronic arthritic conditions. The pathogenesis of JIA remains incompletely understood. This disease can lead to a significant morbidity including joint deformity, growth impairment and a persistence of active arthritis into adulthood. The past two decades have witnessed significant advances in treatment and improved outcomes for affected children. With the current use of biologics, more target-specific, better tolerated, safer and more effective treatments have become possible. However, continuing, comprehensive follow-up is needed to characterize the long-term effects of such treatments. Hyperimmunoglobulin E syndrome (hyper-IgE, or Job’s syndrome) is a rare immune deficiency characterized by high IgE levels, atopic chronic eczema, tendency towards recurrent pyogenic infection, neutrophil chemotaxis disorder and varying T-cell function impairment. Case Report: The case of a 17-year-old male patient with hyper-IgE who develops the oligoarticular subtype of JIA over a period of four years is discussed. The course of JIA is unfavorable, causing severe deformity of numerous joints (left elbow, right 3rd metacarpophalangeal, left knee, right ankle) and a fungal infection scar on the left eye. Blood tests show an ESR of 89 mm/h, rheumatoid factor (RF) 8.3 IU/mL (0 - 20) and positive antinuclear antibody (ANA). To improve gait, corrective surgery is performed on the right ankle, followed by rehabilitation and physical therapy. Conclusion: Developments in the near future will be crucial for understanding JIA pathophysiology and improving treatment.
Keywords:
Juvenile Idiopathic Arthritis, Hyperimmunoglobulin E Syndrome, Treatment

1. Introduction
The term juvenile idiopathic arthritis (JIA) indicates all types of arthritis of unknown origin with onset under sixteen years of age and a duration of at least six weeks [1] .
The disease is classified into seven distinct groups in order to develop more specific treatments for different disease forms, as to a better understanding of its pathophysiology. The pathogenesis of JIA remains incompletely understood. Environmental factors and their interactions with multiple genes have been held responsible for the development of JIA [2] .
JIA may cause marked morbidity including joint deformity, growth impairment and active arthritis persisting into adulthood [3] [4] . The last two decades have witnessed an improvement in the treatment of children with chronic arthritis and its outcomes [5] . Notable progress has been recorded in the understanding of the pathogenesis of JIA, the definition of disease control and its treatment by biologics [2] .
Hyperimmunoglobulin E syndrome (hyper-IgE, or Job’s syndrome) is a rare immune deficiency characterized by high IgE levels, atopic chronic eczema, tendency towards recurrent pyogenic infection, neutrophil chemotaxis disorder and varying T-cell function impairment [6] .
Here, we discuss a case of a 17 year-old boy with hyper IgE syndromefor a longduration, whom is diagnosed with JIA disease four years ago. We aim the contribution of such a demonstrative case to the knowledge of the literature.
2. Case Report
A 17-year-old male patient presented with joint movement restriction in the elbow, wrist, phalanges, hip, knee and ankle. The patient had been diagnosed with immune deficiency (hyper IgE syndrome) in his childhood following studies for persistent skin infections and was placed under monthly IVIg treatment. Diagnosed at first with inflammatory rheumatism, the patient received steroids and methotrexate (MTX). As the complaints persisted, adalimumab (ADA) was started. Following the second dose of this drug, swelling and redness of the left eye developed. A fungal infection (fungus ball) was suspected and amphotericin B (Ambisome®), 50 mg/day IV was administered for two months, followed by intraocular amphotericin B once or twice daily. After interrupting ADA, 40 mg sc every two weeks about 18 months ago, the current treatment with weekly etanercept (ETN), 50 mg was started.
Physical examination findings included: left corneal opacity related to prior fungal infection ( Picture 1 ); subcutaneous soft tissue swelling of the right wrist; boutonnière deformity of the right 2nd finger; soft tissue hypertrophy, contracture in flexion and swelling of the para-olecranon subcutaneous soft tissue; soft tissue
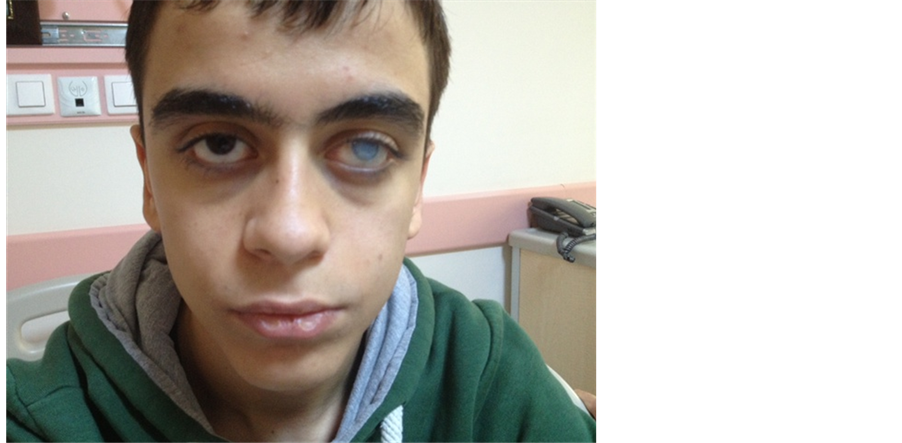
Picture 1. Coarse facial features, a wide root of the nose and a corneal opacity due to infection of the left eye can be seen.
hypertrophy and equinovarus of the right ankle.
Conventional x-ray showed the following: 2nd proximal interphalangeal (PIP) joint subluxation and boutonnière deformity, calcification in the soft tissue around the joint. (Figure 1), Left elbow; calcification in the para- olecranon subcutaneous soft tissue and contracture in flexion (Figure 2).
Laboratory values were as follows: WBC 5.58 × 10³/μL (4.6 - 10.2), hemoglobin 11.1 g/dL (12.2 - 18.1), platelets 220 × 10³/μL, ESR 89 mm/h (0 - 20), CRP 20.4 mg/dL (0 - 0.8), rheumatoid factor (RF) 8.3 IU/mL (0 - 20), ANA positive, HLA B27 negative, uric acid 5.3 mg/dL, serum calcium 9.4 mg/dL, blood sugar 78 mg/dL, alkaline phosphatase 110 IU/L, total vitamin D 14 ng/mL (10 - 44).
The patient’s medical treatment consisted of ETN, 50 mg sc weekly, to which we added MTX, 10 mg weekly. The patient also received methylprednisolone, 120 mg as a slow infusion to treat a right suprapatellar bursitis

Figure 1. Conventional x-ray imaging of both hands and wrists: Right 2nd digit PIP subluxation, pinching of right intercarpal intervals, increased sclerosis, calcifications in the periarticular soft tissue.

Figure 2. Advanced destruction of the left elbow joint and calcifications in the soft tissue.
with 8/10 pain (on a 10-cm VAS) and high acute phase reactants. After the active clinical picture receded, a tibiotalocalcaneal arthrodesis were performed on the patient’s ankle joint in 5 degrees valgus, neutral flexion and 5 degrees external rotation. Intra-operative observation showed the calcified deposits around the ankle joint to be white, with a creamy consistency ( Picture 2 ). At the post-operative examination, the ankle joint was observed to be fixed in neutral position at 90˚ ( Picture 3 ).
Orthese application, electrotherapy, muscle tensing and strengthening exercises, proprioceptive training, gait training and a rehabilitation program including the use of supportive equipment for workaday life were sche-
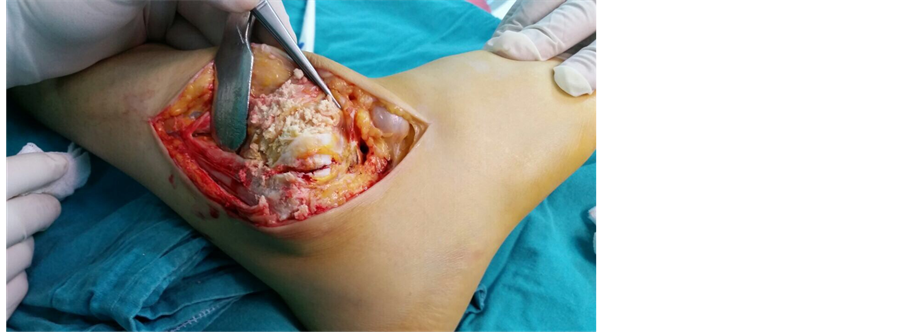
Picture 2. Intra-operative examination of ankle joint shows the soft tissue calcifications previously observed on x-ray.
![]()
Picture 3. Post-arthrodesis view of the patient’s ankle joint.
duled for the postoperative period.
3. Discussion
Hyper-IgE syndrome, believed to be generally a hereditary autosomal dominant condition but also sometimes seen sporadically, is a rare immune deficiency [6] . It was first described in 1972 by Buckley in two cases of male children with coarse facial features, peripheral eosinophilia and high IgE levels [7] [8] .
Coarse facial features, wide nasal bridge, prominent nose and imbalance between cheek and chin dimensions are among the distinctive features of the condition [8] . Our patient displayed the described physical characteristics ( Picture 1 ). He presented no evidence of an active focal infection or skin lesion that may suggest a pyogenic infection.
JIA is a heterogeneous group of chronic childhood arthritic conditions [2] . The pathogenesis of JIA has yet to be elucidated; environmental factors and their interaction with numerous genes are the most commonly accepted etiopathology [1] [2] [5] . Chronic inflammatory arthritis developing on a background of immune deficiency was hypothesized in the reported case.
In recent years, the realization that JIA covers more than one disease has been gaining in strength. The aim of classifying JIA is to understand the pathophysiology of its different forms and to develop better, more specific treatment methods [5] .
JIA is subdivided into seven subtypes by the International League of Associations for Rheumatology (ILAR) classification: systemic arthritis, oligoarthritis, rheumatoid factor RF-negative polyarthritis, RF-positive polyarthritis, psoriatic arthritis, enthesitis-related arthritis, and undifferentiated arthritis. Uveitis is very rare in patients with systemic onset JIA and in juvenile-onset rheumatoid arthritis. Chronic uveitis is a complication observed mainly in patients with ANA-positive oligoarticular involvement [9] [10] .
Considering the oligoarticular involvement pattern, the seronegativity and the ANA positivity of our patient, we classified him as an oligoarticular JIA. However, he showed metacarpophalangeal and PIP involvement, as well as an ocular fungal infection, which differ from the classic oligoarticular form. The patient’s knee joint involvement and sacro-iliitis may also place him in the enthesitis-relative arthritis subgroup. His testing negative for HLA B27 and his upper extremity involvement do not, however, strictly fit this subgroup. The patient had no active skin problem, including psoriasis. The clinical course of his arthritis had been very severe due to the contributing effect of the immune deficiency, resulting in serious joint deformities.
Therapeutic choices for JIA include drug treatment, rehabilitation and corrective joint surgery [11] . The discovery of biologics now allows successful long-term treatment of JIA [2] [3] [5] [9] -[12] . Studies have shown that the efficacy of the biologics used in the treatment of JIA has steadily been increasing in the last ten years. The reason for this observation is the inadequacy of direct, head-to-head comparisons of these agents, as most studies compare efficiency indirectly [13] .
4. Conclusion
While satisfactory solutions have been achieved since the discovery of biologics and long-term follow-up, increased experience and studies comparing the new agents head-to-head are still needed.
Cite this paper
Tuba TülayKoca,AydınArslan, (2015) Current Approach to Treatment of Juvenile Idiopathic Arthritis: Case Report of Hiperimmunglobulin E Syndrome Developed Juvenile Idiopathic Arthritis. Open Journal of Rheumatology and Autoimmune Diseases,05,62-67. doi: 10.4236/ojra.2015.53011
References
- 1. Rigante, D., Bosco, A. and Esposito, S. (2014) The Etiology of Juvenile Idiopathic Arthritis. Clinical Reviews in Allergy & Immunology.
http://dx.doi.org/10.1007/s12016-014-8460-9 - 2. Huang, J.L. (2012) New Advances in Juvenile Idiopathic Arthritis. Chang Gung Medical Journal, 35, 1-14.
http://dx.doi.org/10.4103/2319-4170.106171 - 3. Kahn, P. (2009) Juvenile Idiopathic Arthritis—Current and Future Therapies. Bulletin of the NYU Hospital for Joint Diseases, 67, 291-302.
- 4. Kahn, P. (2012) Juvenile Idiopathic Arthritis: An Update for the Clinician. Bulletin of the NYU Hospital for Joint Diseases, 70, 152-166.
- 5. Eisenstein, E.M. and Berkun, Y. (2014) Diagnosis and Classification of Juvenile Idiopathic Arthritis. Journal of Autoimmunity, 48-49, 31-33.
http://dx.doi.org/10.1016/j.jaut.2014.01.009 - 6. Grimbacher, B., Schäffer, A.A., Holland, S.M., Davis, J., Gallin, J.I., et al. (1999) Genetic Linkage of Hyper-IgE Syndrome to Chromosome 4. American Journal of Human Genetics, 65, 735-744. http://dx.doi.org/10.1086/302547
- 7. Buckley, R.H., Wray, B.B. and Belmaker, E.Z. (1972) Extreme Hyperimmunoglobulinemia E and Undue Susceptibility to Infection. Pediatrics, 49, 59-70.
- 8. Grimbacher, B., Holland, S.M., Gallin, J.I., Greenberg, F., Hill, S.C., et al. (1999) Hyper-IgE Syndrome with Recurrent Infections—An Autosomal Dominant Multisystem Disorder. New England Journal of Medicine, 340, 692-702.
http://dx.doi.org/10.1056/NEJM199903043400904 - 9. Tugal-Tutkun, I., Quartier, P. and Bodaghi, B. (2014) Disease of the Year: Juvenile Idiopathic Arthritis-Associated Uveitis—Classification and Diagnostic Approach. Ocular Immunology & Inflammation, 22, 56-63.
http://dx.doi.org/10.3109/09273948.2013.871565 - 10. Oktayoglu, P. and Tekeoglu, I. (2011) Juvenilidiopatikartritte tedavi ve biyolojik ajanlar [Juvenileidiopathicarthri- tistreatmentandbiologicagents]. Sakarya medical journal, 3, 76-85.
- 11. Horneff, G. (2013) Update on Biologicals for Treatment of Juvenile Idiopathic Arthritis. Expert Opinion on Biological Therapy, 13, 361-376.
http://dx.doi.org/10.1517/14712598.2013.735657 - 12. Herlin, T. (2008) Biological Therapy Treatment of Juvenile Idiopathic Arthritis. Ugeskrift for Læger, 170, 2105-2108.
- 13. Otten, M.H., Anink, J., Spronk, S. and van Suijlekom-Smit, L.W. (2013) Efficacy of Biological Agents in Juvenile Idiopathic Arthritis: A Systematic Review Using Indirect Comparions. Annals of the Rheumatic Diseases, 72, 1806- 1812.
http://dx.doi.org/10.1136/annrheumdis-2012-201991
NOTES

*Corresponding author.