Open Journal of Rheumatology and Autoimmune Diseases
Vol.3 No.2(2013), Article ID:31379,14 pages DOI:10.4236/ojra.2013.32011
Unmet Needs in the Treatment of Rheumatoid Arthritis*
![]()
1Schulich School of Medicine & Dentistry, University of Western Ontario, London, Canada; 2St. Joseph’s Health Care, London, Canada; 3Department of Rheumatology, Lapeyronie Hospital, CHU Montpellier, Montpellier 1 University, Montpellier, France.
Email: janet.pope@sjhc.london.on.ca
Copyright © 2013 Janet Pope, Bernard Combe. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 3rd, 2013; revised May 3rd, 2013; accepted May 11th, 2013
Keywords: Rheumatoid Arthritis; Biologics; Tumor Necrosis Factor Inhibitors; Unmet Need; Interleukin-17A; IL-17 Inhibitors; Treatment Challenges
ABSTRACT
Biologics have greatly improved the management of rheumatoid arthritis (RA), demonstrating efficacy and safety in alleviating symptoms, inhibiting bone erosion, and preventing loss of function. Unmet therapeutic needs in RA remain; however, further advances require an understanding of issues left unaddressed under the current treatment paradigm. Most biologic-naïve and biologic-pretreated patients who initiate a biologic therapy, for example, do not reach American College of Rheumatology 50% (ACR50) response, and few achieve remission. Responses are often not durable, prompting frequent treatment switching. Predictive markers are unavailable to guide therapy selection, and clinical trial data are lacking on whether a tumor necrosis factor inhibitor (TNFi) is the best first-line biologic and on the optimal sequence of use for the different biologics. Risk of serious infection is the major safety concern. Translating preclinical and clinical findings into new therapeutics may help address unmet needs. An increasing body of evidence indicates that the cytokine interleukin (IL)-17A represents an important therapeutic target; ongoing trials with IL-17A inhibitors will determine whether these agents can address some of the unmet needs associated with current biologics.
1. Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease associated with progressive joint damage, disability, and systemic complications [1]. RA has considerable interpatient variability in clinical course and severity, which may be due to genetics or environmental factors. The goal of RA treatment is clinical remission, defined by the absence of signs and symptoms of inflammation. When remission is not possible, the target should be low disease activity, particularly in patients with established disease [2]. To achieve these goals, European League Against Rheumatism (EULAR) guidelines recommend starting therapy with synthetic disease-modifying antirheumatic drugs (DMARDs) as soon as possible after diagnosis [3]. In patients with an inadequate response to synthetic DMARDs, a biologic should be added when poor prognostic features are present (i.e., presence of rheumatoid factor [RF] or anti-cyclic citrullinated peptide [anti-CCP] antibodies, high disease activity despite synthetic DMARD treatment, or early erosions), and in others after failure of more than one DMARD. The Canadian Rheumatology Association (CRA) and the American College of Rheumatology (ACR) have recently published similar treatment recommendations [4,5]. It is common practice to initiate a tumor necrosis factor inhibitor (TNFi) as the first biologic and, if this is ineffective, to switch to another TNFi or to an alternative biologic with a different mechanism of action. The reason for starting biologic therapy with a TNFi may be historical, in that TNFi agents were the first biologic class approved for RA and clinicians now have more than a decade of experience with these agents. Although the advent of biologic therapy has greatly improved RA management, there are still unmet needs. This paper reviews unmet needs with the currently available biologics and then describes an investigational approach targeting interleukin (IL)-17A, which may have the potential to address some of the challenges in the current treatment of RA.
2. Literature Search
Areas of potential unmet needs with respect to efficacy, safety, and treatment persistence were identified based on the authors’ clinical experience, and literature searches were then conducted on Medline to identify relevant articles published in English from January 2000 to June 2012. Search terms included “RA” in combination with biologic by class (“TNF blocker” or “TNF inhibitor”) and by individual drug (“adalimumab”, “certolizumab pegol”, “etanercept”, “golimumab”, “infliximab”, “abatacept”, “rituximab”, “tocilizumab”) and biologic by class or individual drug in combination with “safety”, “infection”, “malignancy”, “autoimmune”, “cardiovascular”, “adherence”, and “persistence”. The term “IL-17” was also searched, with particular attention paid to articles relevant to arthritis. Reference lists in selected articles and recent meeting abstracts were also reviewed. Preference in selecting articles was given to randomized controlled trials, metaanalyses, and large observational registries.
3. Adequacy of Responses to Current Biologics
3.1. Response Rates and Durability of Response
Biologics may be more effective than synthetic DMARDs, but a substantial proportion of patients achieve only partial responses and not remission. In randomized clinical trials, the addition of a TNFi to methotrexate (MTX) produced higher response rates than MTX alone in MTX-inadequate responders [6-9]. The TNFi-MTX combination also significantly improved other efficacy measures: disease activity as assessed by the 28-item Disease Activity Score (DAS28), tender joint counts, swollen joint counts, and C-reactive protein (CRP); function as measured by the Health Assessment Questionnaire-Disability Index (HAQ-DI); and radiographical joint damage as evaluated on serial X-rays. However, most patients treated with a TNFi plus MTX failed to achieve ACR50 or ACR70 responses or remission (Table 1). Radiographical analyses showed the combination slowed or arrested progressive joint damage; however, erosions usually did not heal during the observation period [10-12]. Moreover, good responses were often subsequently lost, with an estimated 20% to 25% of patients per year discontinuing treatment [13-15].
Observational registries suggest that response rates achieved in daily clinical practice may be lower than those reported in randomized clinical trials [16,17]. Several reasons may contribute to this disparity, including differences in patient selection (specific enrollment criteria versus all-comers), use of a washout period in randomized clinical trials (which may increase disease activity at baseline), differences in disease activity (real-world patients often start treatment with lower disease activity and therefore may not achieve high ACR responses), and differences in drug doses and adherence rates [18]. However, for some measures, such as HAQ-DI, the minimally important difference observed in clinical practice is smaller than that observed in randomized clinical trials, indicating that real-world patients indeed experience relevant
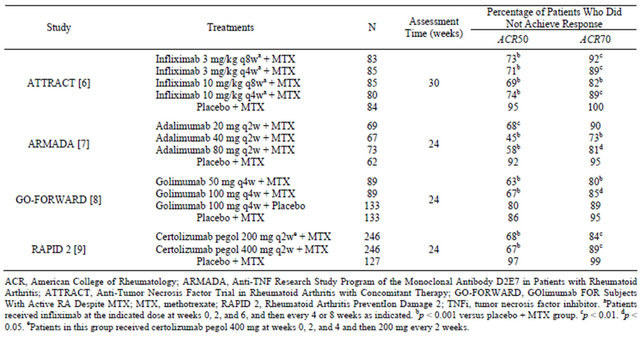
Table 1. Proportion of patients with inadequate response to MTX who did not meet major ACR response criteria in key phase III clinical trials of a TNFi plus MTX.
improvement with available biologic agents [19]. When registry patients meeting clinical trial enrollment criteria are considered separately, response rates approach those achieved in the clinical trials [16,17]. Nevertheless, only a small proportion of patients in clinical practice achieve remission as defined by the new ACR/EULAR Boolean criteria [20].
Several other biologics and small molecules have emerged for RA treatment, including abatacept (a CTLA4-Fc fusion protein that modulates T-cell costimulation), rituximab (an anti-CD20 monoclonal antibody that depletes B cells), tocilizumab (an anti-IL-6 receptor monoclonal antibody), Janus kinase (JAK) inhibitors such as tofacitinib and baricitinib, and the spleen tyrosine kinase (Syk) inhibitor fostamatinib [21]. However, these agents also exhibit a ceiling for response rates, with only a minority of patients reaching ACR50 or ACR70 responses.
3.2. Switching Biologic Therapy
Patients who do not respond to a TNFi or who lose their initial response may still benefit from another TNFi, or they may respond to other biologics with different mechanisms of action. In general, ACR50 and ACR70 response rates are decreased with the next biologic after use of a TNFi. Additionally, the reason for discontinuing the initial TNFi can predict the results of the next treatment. In a meta-analysis of 19 studies, switching to a second TNFi was associated with a slightly higher ACR50 rate if the switch had been prompted by adverse events (43%) rather than by lack of efficacy (31%), although the EULAR responses were similar in both subgroups [22].
Within the TNFi class, differences in molecular structure provide a rationale for switching to a second or even a third TNFi [23]. Choices include the TNF receptor fusion protein etanercept, the chimeric anti-TNFα monoclonal antibody infliximab, the fully human anti-TNFα monoclonal antibodies adalimumab and golimumab, and the pegylated humanized anti-TNFα Fab’ fragment certolizumab pegol. The benefit of switching to a second TNFi is illustrated by the golimumab in patients with active rheumatoid arthritis after treatment with TNFα inhibitors (GO-AFTER) trial, in which 461 patients with active RA who discontinued their previous TNFi were randomized to receive golimumab 50 or 100 mg subcutaneously (SC) or placebo every 4 weeks, with or without background DMARDs. Compared with placebo, golimumab significantly increased ACR50 responses at week 14 (18% versus 6%; p < 0.001) [24]. Fifty-one percent of the patients continued treatment for 3 years. In this subgroup, ACR50 and ACR70 response rates were 40% and 19%, respectively, at the end of the 3-year period, but it is important to recognize that these rates exclude patients who discontinued before 3 years [25].
Rituximab, tocilizumab, and abatacept, other available biologic options with distinct mechanisms of action, are often initiated following TNFi failure. The recombinant IL-1 receptor antagonist anakinra is also approved for use in RA but will not be discussed further here as it is generally less effective than other options and not recommended in EULAR guidelines as a major biologic in RA [3]. Rituximab, tocilizumab, and abatacept significantly increased response rates compared with placebo when added to background MTX in patients who had failed one or more TNFi agents. ACR20, ACR50, and ACR70 response rates at week 24 were 51% versus 18%, 27% versus 5%, and 12% versus 1%, respectively, with rituximab versus placebo (all p < 0.001) in the Randomized Evaluation of Long-Term Efficacy of rituximab in RA (REFLEX) trial [26]; 50% versus 10%, 29% versus 4%, and 12% versus 1%, respectively, with tocilizumab 8 mg/kg versus placebo (all p < 0.001) in the Research on Actemra Determining efficacy after Anti-TNF failures (RADIATE) trial [27]; and 50% versus 20%, 20% versus 4%, and 10% versus 2%, respectively, with abatacept versus placebo (all p ≤ 0.003) in the Abatacept Trial in Treatment of Anti-TNF Inadequate Responders (ATTAIN) trial [28]. Significant improvements in other efficacy measures, including HAQ-DI, DAS28 remission, and health-related quality of life measured by SF-36 were also reported in one or more of these trials. Rituximab also showed a trend for slowing progression of radiographical damage [26].
In a recent meta-analysis, the probability of achieving an ACR50 response did not differ among golimumab, rituximab, tocilizumab, and abatacept in patients with inadequate responses to a prior TNFi (TNF-IR) [29]. As initial biologic therapy in MTX-inadequate responders, TNFi agents were significantly more likely to produce an ACR50 response than abatacept but not rituximab or tocilizumab [29]. However, the results of several recently reported trials call the latter conclusion into question, in that no differences in response rates were seen with abatacept SC compared with adalimumab in MTX-inadequate responders in the Abatacept Versus Adalimumab Comparison in Biologic-Naive rheumatoid arthritis Subjects With Background Methotrexate (AMPLE) trial [30] or between abatacept 10 mg/kg every 4 weeks and infliximab 3 mg/kg every 8 weeks in the Abatacept or infliximab versus placebo, a Trial for Tolerability, Efficacy and Safety in Treating RA (ATTEST) trial [31]. Moreover, monotherapy with tocilizumab (8 mg/kg IV every 4 weeks) was superior to monotherapy with adalimumab in patients who were intolerant of MTX or for whom continued MTX was inappropriate in the ADalimumab ACTemrA (ADACTA) study [32].
In summary, available clinical data indicate the utility of switching therapy in patients who have an inadequate response to their current agent but provide little guidance on selection of the next therapy.
4. Predictive Clinical Characteristics and Biomarkers
Predictive clinical characteristics and biomarkers are needed to help identify which agents should be used in individual patients as initial biologic therapy or following failure of TNFi therapy. Clinical and observational studies indicate that concurrent use of a synthetic DMARD (especially MTX) and nonsmoking status are predictive of better response to TNFi therapy [33-35]. Beyond that, other baseline factors associated with good response depend on the efficacy measure being evaluated. High disease activity at baseline was associated with better ACR50 and ACR70 responses, which is not unexpected given that higher baseline activity offers a greater window for showing a treatment effect [36]. In contrast, low baseline disease activity was associated with DAS28 remission rate, which is also logical given that it is much easier to produce remission when disease is less active. Numerous genetic markers, including TNFα gene polymorphisms, and protein markers, including RF and antiCCP antibodies, have been evaluated, but to date, none has shown robust and consistent predictive value for TNFi response [36] The presence of RF and/or anti-CCP antibodies appears predictive of better responses with rituximab [37]. Other studies suggest that the reason prompting biologic switching and the number of previous biologics may influence treatment response. In the observational Swiss Clinical Quality Management RA cohort, switching to rituximab was more effective than switching to a second or third TNFi when the change in therapy was prompted by inadequate response to the initial TNFi, but switching to rituximab or to another TNFi had comparable efficacy when the switch was prompted by other reasons [38,39]. However, it is important to remember that observational studies are limited by potential patient selection and prescribing biases. Whereas rituximab may be most effective when used after zero to one TNFi, the efficacy of tocilizumab was independent of the number of previous TNFi agents in the RADIATE trial [27]. In a large Canadian observational cohort, abatacept produced similar changes in HAQ-DI as initial biologic therapy and after previous TNFi therapy; however, the durability of response to abatacept was greater when it was used as the first biologic [40].
5. Safety Profiles of Current Agents
5.1. Serious Infection
Serious infection is the most important safety concern with biologic therapy. In a meta-analysis of 160 randomized clinical trials and 46 extension studies, biologics as a group in the standard-dose model were significantly associated with increased risk of serious infection compared with control treatment (odds ratio 1.37, 95% confidence interval [CI] 1.04 - 1.82) [41]. The risk of serious infection with the individual biologics is shown in Figure 1; most showed odds ratios above 1 but had 95% CIs overlapping with unity. However, recent data from several observational registries suggest that TNFi therapy (etanercept, infliximab, and adalimumab) is associated with a small but significantly increased risk of serious infection in daily clinical practice. In an analysis of the Consortium of Rheumatology Researchers in North America (CORRONA) registry involving 7971 RA patients, TNFi use tended to increase the risk of opportunistic infections (incidence rate ratio 1.67, 95% CI 0.95 - 2.94) [42]. Similarly, in the British Society for Rheumatology Biologics Register (BSRBR), involving 15,396 RA patients, TNFi therapy was associated with risk of serious infection compared with nonbiologic DMARDs (hazard ratio [HR] = 1.2, 95% CI 1.1 - 1.5); this association was most pronounced during the first 6 months of treatment (HR = 1.8, 95% CI 1.3 - 2.6) but then declined over time [43]. Finally, in the French RATIO (Recherche Axée sur la Tolérance des Biothérapies) registry, risk of nontuberculosis opportunistic infections was substantially higher with the anti-TNF monoclonal antibodies infliximab and adalimumab than with the TNF receptor fusion protein etanercept [44].
5.2. Tuberculosis Reactivation
TNFi therapy is associated with increased risk of tuberculosis due to reactivation of latent disease [45], with the anti-TNF monoclonal antibodies carrying a higher risk than etanercept [46,47]. As a result, screening for latent tuberculosis before initiation of any TNFi has become standard practice and has resulted in substantial reduction in tuberculosis risk [48]. With this practice in place,

Figure 1. Meta-analysis of serious infection risk with biologics [41]. Reproduced with permission. Copyright © 2010 The Cochrane Collaboration. Published by John Wiley & Sons, Ltd.
tuberculosis risk with newer agents appears low [49,50]. Recommendations are in place for country-specific tuberculosis screening for TNFi agents, tocilizumab, and abatacept, but screening is not necessary for rituximab. TNFi agents are also associated with increased risk of nontuberculosis mycobacterial infections. These infections, which are most commonly caused by Mycobacterium avium, are difficult to diagnose and no screening tests for them are as yet available [51].
5.3. Malignancy
RA patients are at higher risk of certain malignancies, notably, lymphomas and lung cancer [52]. Given that the molecular targets of biologic therapy may be important in tumor surveillance and control, there is concern about increased cancer risk. Evidence supporting this concern is limited. A meta-analysis of clinical trials with infliximab and adalimumab found a higher malignancy risk, particularly with higher doses of these agents [53]. In addition, several studies suggest that TNFi therapy is associated with risk of melanoma and nonmelanoma skin cancers [54,55]. However, large meta-analyses and observational registries suggest that TNFi agents are not associated with increased risk of malignancy overall [41,55-57]. Similarly, pooled analyses of clinical trials suggest that rituximab, tocilizumab, and abatacept are not associated with increased malignancy risk in RA, although additional long-term follow-up data are needed [58-60].
Limited information is available on malignancy risk in patients with a history of cancer, because these patients were typically excluded from randomized clinical trials. The incidence of new cancers was not increased by TNFi agents compared with synthetic DMARDs in a cohort of 293 patients with prior malignancy in the BSRBR, but these findings may have been biased by the fact that patients are more likely to be selected for biologic therapy if a prior malignancy is considered to have a good prognosis [61].
5.4. Immune Reactions
TNFi agents are associated with the production of antinuclear antibodies (ANA) and, less frequently, with antidouble stranded DNA (anti-dsDNA) antibodies [62,63]. Although such antibodies have been associated with the development of systemic lupus erythematosus and other immune conditions, these events are uncommon in patients treated with TNFi agents or abatacept [64,65]. For example, 22 of 156 infliximab-treated patients (14%) developed anti-dsDNA, but only one of them had symptoms suggestive of a drug-induced lupus syndrome [62].
5.5. Cardiovascular Risk
Cardiovascular disease is common in patients with RA. TNFα is involved in all stages of atherosclerosis, from plaque generation through plaque rupture, and may stimulate several cardiovascular risk factors, including dyslipidemia and insulin resistance [66]. Consistent with this profile, multiple observational registries and meta-analyses have shown that TNFi therapy is associated with significant reductions in risk of major cardiovascular events [67-69]. Of note, some evidence suggests that the cardiovascular benefit may be restricted to RA patients who respond to TNFi [70].
The risk of congestive heart failure (CHF) with TNFi therapy remains controversial. TNFα levels are known to be elevated in CHF patients [71], which prompted clinical trials of TNFi agents in this population. Two clinical trials of etanercept in CHF patients were stopped early, with a pooled analysis showing a small, nonsignificant trend toward increased hospitalization and mortality at higher doses [72]. Similarly, infliximab was ineffective in CHF patients, with the higher dose (10 mg/kg) associated with a significant increase in risk of mortality or CHF hospitalization [73]. However, observational studies have not convincingly shown that TNFi agents increase CHF risk in RA patients, particularly in the absence of pre-existing cardiovascular disease [69,74,75]. Nevertheless, caution appears warranted when these agents are used in RA patients with CHF.
Information about the cardiovascular risk profile of the other available biologics is limited. Evidence from longterm extension studies suggests that they do not have a beneficial or a detrimental effect on cardiovascular outcomes [66]. Differences among biologics in associated cardiovascular risk could reflect differences in underlying risk factors. For example, TNFi agents may increase total cholesterol and high-density lipoprotein (HDL) cholesterol [76], and tocilizumab raises total cholesterol and low-density lipoprotein (LDL) lipid parameters with or without increases in HDL [77,78].
6. Persistence and Adherence with Current Therapies
Despite the effectiveness of biologics, patients may discontinue treatment for a variety of reasons, including lack of efficacy, loss of treatment response, adverse events, personal preference, and achievement of remission. Treatment persistence is defined by treatment continuation for a given time period without significant gaps in treatment and/or without switching between biologics. In a recent systematic review, median drug survival across all biologics was typically between 32 and 39 months [79]. For example, median drug survival was 37 months in a cohort of 2364 patients treated initially with infliximab, etanercept, or adalimumab [80]. Median drug survival decreased with each subsequent TNFi to 21 months for the second agent and 13 months for the third agent [80]. In the BSRBR registry, discontinuation of a TNFi due to lack of efficacy was associated with an increased rate of discontinuation of a second TNFi due to lack of efficacy [81]. Similarly, discontinuation due to toxicity from the first agent was associated with an increased rate of discontinuation of the second drug for toxicity. Persistence with abatacept averaged 26.8 months in a Canadian observational cohort and was significantly longer among biologic-naive patients than among those treated previously with TNFi therapy [40].
Whereas persistence measures drug continuation, treatment adherence reflects whether the patient takes a medication according to the prescription. Measures of adherence vary across studies, as do adherence rates. When measured as the proportion of patients with a medication possession ratio ≥ 0.8, adherence rates with TNFi agents ranged from 41% to 81% in four studies, and the mean medication possession ratio for treatment with a biologic plus MTX was 0.64 to 0.72 [79]. In general, higher rates of adherence with biologics in RA are associated with a belief in the medication’s necessity for health and with lower out-of-pocket costs, whereas lower adherence is associated with concern about toxicity [82,83]. Patients who are adherent to the prescribed dosing of a TNFi seem to have a more durable response, consistent with observations relating adherence to treatment benefits in other therapeutic areas.
7. IL-17A Inhibitors: Novel Investigational Agents with Potential to Address Unmet Needs
The TNFi agents were developed at a time when RA was thought to be a Th1-mediated disease leading to the production of monocyte-/macrophage-derived cytokines, such as TNFα, IL-1β, and IL-6. In 2005, a new subset of T-helper cells, known as Th17 cells, was discovered, whose effector functions are mediated predominantly by IL-17A [84,85]. IL-17A plays a key role in host defense against extracellular bacteria and fungi; this role is distinct from the role of the Th1 pathway and its monocyte-derived cytokines in host defense against intracellular pathogens and certain fungi [86].
IL-17A is thought to play an important role in RA according to both preclinical and clinical data and therefore may be a viable therapeutic target. As shown in Figure 2, IL-17A is produced by multiple cell types and causes a range of biological effects culminating in joint inflammation, cartilage degradation, and bone erosion.
These effects are reproduced in experimental models associated with IL-17A overexpression [94], whereas they are blocked in IL-17A-deficient animals [95] and by the use of IL-17A inhibitors [96,97]. In RA patients, IL-17A levels are elevated in serum and synovial fluid and appear to be associated with greater disease activity [98-101]. Moreover, IL-17A has been associated with impaired microvascular function and arterial compliance in RA and therefore may contribute to comorbid cardiovascular risk [101].
IL-17A inhibitors have been developed and evaluated in early clinical trials. Secukinumab (previously known as AIN457), a fully human IgG1α anti-IL-17A monoclonal antibody, was initially assessed in a randomized, placebo-controlled, proof-of-concept trial involving 52 RA patients on stable background MTX [102]. Administered at 10 mg/kg IV at weeks 0 and 3, secukinumab significantly improved the area under the treatment response-time curve for ACR20 (p = 0.011), the adjusted DAS28 score (p = 0.027), and the baseline-adjusted CRP level (p = 0.002), with effects evident by 1 week and maintained throughout the 16-week trial. Secukinumab was subsequently evaluated in a randomized, doubleblind, placebo-controlled phase II trial, in which 237 RA patients with active disease despite stable MTX were allocated to monthly treatment at a dose of 25, 75, 150, or 300 mg SC [103]. Doses of 75 to 300 mg produced higher ACR20 response rates than placebo at the week 16 primary endpoint (47% - 54% versus 36%), although statistical significance was not demonstrated. Patients with ACR20 responses received the same dose of secukinumab through week 52, whereas in patients who did not show a response by week 16, the dose was escalated at week 20. For responders who remained on the 150 mg dose for the entire study, ACR20 response rates improved to 75% at week 24 and to 90% at week 52, with corresponding improvements in ACR70 response rates to 20% and 40%, respectively. Responders who remained on the 150 mg dose also had sustained improvements in DAS28-CRP and HAQ-DI. In contrast, week 16 nonresponders derived little benefit from dose escalation. Adverse events were mostly mild to moderate in severity and led to discontinuation in 6.9% of patients.
Ixekizumab (previously known as LY2439821), a humanized IgG4 anti-IL-17A monoclonal antibody, was evaluated in proof-of-concept and phase II trials. In the former, patients on one or more synthetic DMARDs received ixekizumab 0.2, 0.6, or 2 mg/kg IV at weeks 0, 2, 4, 6, and 8 [104]. Ixekizumab (all doses combined) significantly reduced DAS28 compared with placebo at week 10 (–2.3 versus –1.7; p ≤ 0.05) and showed activity by 1 week that was maintained at the final week 16 assessment. In a subsequent phase II trial, patients on background DMARD therapy received placebo or ixekizumab at doses of 3 to 180 mg SC if naïve to biologic therapy and 80 or 180 mg SC if previously treated with a TNFi [105]. Treatment was given at weeks 0, 1, and 2 and then every other week through week 10. Ixekizumab produced an ACR70 response rate of 14% at the dose levels of 10, 30, and 180 mg in the biologic-naïve cohort

Figure 2. Role of interleukin (IL)-17A in rheumatoid arthritis (RA) pathogenesis. IL-17A is produced by Th17 cells and binds to a receptor complex consisting of IL-17RA and IL-17RC subunits on target cells [86,87]. IL-17A is also produced by cells of the innate immune system, of which mast cells may be the most important in the rheumatoid synovium [86,88-91]. IL-17A acts on synovial fibroblasts to increase chemokine and cytokine production, leading to joint inflammation, on chondrocytes to stimulate release of cartilage-degrading matrix metalloproteinases (MMPs) and block new matrix synthesis, and on osteoblasts and osteoclast precursors to stimulate expression of RANKL and RANK, respectively, leading to bone erosion. The products released promote further effects in the joint (e.g., CCL20 may promote recruitment of Th17 and dendritic cells; tumor necrosis factor (TNF)-α acts on chondrocytes and osteoclast precursors to promote joint damage) [86,92,93]. Secukinumab and ixekizumab are monoclonal antibodies directed against IL-17A, whereas brodalumab is a monoclonal antibody directed against the IL-17RA subunit.
(all p < 0.05 versus placebo) and 10% at a dose of 180 mg in TNFi-pretreated patients (p = 0.11 versus placebo). Although infection rates were slightly higher with ixekizumab than with placebo, no mycobacterial or systemic fungal infections were observed.
Brodalumab (previously AMG-827) differs mechanistically from secukinumab and ixekizumab; it is a fully human monoclonal antibody directed against IL-17RA [106], which is a subunit of the receptor through which IL-17A produces its biological effects [87,107]. IL-17RA also mediates the effects of IL-17F, another Th17 cytokine, with similar but less potent effects than IL-17A, and IL-17RA is also a subunit of the receptor for IL-17E (also known as IL-25), which may be involved in Th2- mediated eosinophil recruitment. A phase II study of brodalumab in biologic-naïve RA patients on background MTX yielded negative results [108]. A total of 252 participants who had previously experienced an inadequate response to MTX were randomized to additional treatment with brodalumab (70, 140, or 210 mg) or placebo subcutaneously at day 1 and weeks 1, 2, 4, 6, 8, and 10. The percentage of patients who achieved ACR50 response at week 12, the primary endpoint, did not significantly differ between any brodalumab-treated group and placebo: 70 mg (16%), 140 mg (16%), 210 mg (10%), placebo (13%). Differences in ACR20 and ACR70 response were nonsignificant for any brodalumab group versus placebo. Incidences of adverse events, including serious adverse events, were similar across treatment groups.
Blocking IL-17A potentially raises the risk of infection and immune-mediated disorders, given the role of IL- 17A in host defense. Several different genetic disorders have been identified that result in a deficiency of IL-17RA or IL-17F or lead to autoantibody production against IL-17A and other Th17 cytokines [109-111]. Affected patients develop chronic mucocutaneous candidiasis characterized by recurrent or persistent skin, nail, and mucosal infections caused by Candida albicans. Therefore, mucocutaneous candidiasis is a theoretical risk with IL-17A inhibitors, but its occurrence should require complete blockade of IL-17A function rather than the partial inhibition likely to be achieved with therapeutic doses.
8. Conclusions
Despite the treatment advances achieved with biologics, most patients do not reach ACR50 or ACR70 responses, and few actually achieve remission. Moreover, responses are often not durable, prompting frequent treatment switching. These issues are evident in both biologic-naïve and TNF-IR patients and underscore the need for new agents that allow a greater proportion of patients to reach treatment goals than currently available biologics (i.e., higher rates of ACR70 response, DAS28 remission, and ACR/ EULAR remission) and that produce longer-lasting responses. The new agents should ideally also improve other features of RA, such as fatigue, function, pain, joint damage, and disability.
On the basis of available data, it is difficult to know which biologic to choose, and in which order, after failure of MTX as well as after failure of a TNFi. Common practice dictates starting biologic therapy with a TNFi, but other drug classes may be perfectly acceptable alternatives, as illustrated by meta-analyses and recent clinical trial findings [29,30]. With multiple biologic options, there is a need for strong predictive biomarkers to determine which drug is most likely to be effective (and which will likely be ineffective), safe, and durable in a given individual. The fact that available biologics are not effective in all patients attests to the heterogeneity of RA and implies that the underlying pathophysiology likely varies across patients and disease stages. Seropositivity appears to predict better responses to rituximab [37], but overall, robust predictive markers for use in clinical practice remain elusive.
From a safety perspective, the risks of serious infection and malignancy are significant concerns with available biologics. Most safety data focus on the TNFi class, and specifically on the first three members that were available—etanercept, infliximab, and adalimumab. In general, the safety data from meta-analyses and observational registries suggest that TNFi agents increase the risk of serious infection and skin cancer but not that of malignancy overall. Importantly, the adoption of appropriate clinical practices, such as tuberculosis screening prior to initiating biologic therapy, has helped to reduce serious infection risk. With the newer biologics, pooled analyses of clinical trial databases provide an initial step in assessing their safety, but additional long-term data are needed to adequately define their overall safety profiles. On the basis of available safety information, there is still a need for new agents with a lower risk of serious infection, particularly for patients with certain comorbidities (e.g., diabetes, bronchiectasis, prednisone use), and a lower risk of potential immune-mediated adverse events. Given the potential cardioprotective effects of TNFi agents, it would be preferable for new agents to retain this feature, particularly because cardiovascular disease is increased in RA. However, it is unknown whether effective RA treatment is sufficient for lowering cardiovascular risk and whether intrinsic factors associated with TNFi contribute to risk reduction.
To address these unmet needs, innovation is needed to translate preclinical and clinical evidence into viable therapeutics. Along these lines, significant progress has been made in identifying IL-17A as a therapeutic target and in developing IL-17 inhibitors for RA. Early clinical trial results are as yet mixed, and further evaluation is ongoing in phase II and III studies. It is still too soon to determine whether the IL-17 inhibitors will address some of the unmet needs associated with current agents.
9. Acknowledgements
This article was supported by a grant from Novartis Pharma AG. BioScience Communications, New York, USA, provided writing and editorial assistance in the development of this manuscript, supported by Novartis Pharma AG.
REFERENCES
- I. B. McInnes and G. Schett, “The Pathogenesis of Rheumatoid Arthritis,” New England Journal of Medicine, Vol. 365, No. 23, 2011, pp. 2205-2219. doi:10.1056/NEJMra1004965
- J. S. Smolen, D. Aletaha, J. W. Bijlsma, F. C. Breedveld, D. Boumpas, G. Burmester, et al., “Treating Rheumatoid Arthritis to Target: Recommendations of an International Task Force,” Annals of the Rheumatic Diseases, Vol. 69, No. 4, 2010, pp. 631-637. doi:10.1136/ard.2009.123919
- J. S. Smolen, R. Landewé, F. C. Breedveld, M. Dougados, P. Emery, C. Gaujoux-Viala, et al., “EULAR Recommendations for the Management of Rheumatoid Arthritis with Synthetic and Biological Disease-Modifying Antirheumatic Drugs,” Annals of the Rheumatic Diseases, Vol. 69, No. 6, 2010, pp. 964-975. doi:10.1136/ard.2009.126532
- V. P. Bykerk, P. Akhavan, G. S. Hazlewood, O. Schieir, A. Dooley, B. Haraoui, et al., “Canadian Rheumatology Association Recommendations for Pharmacological Management of Rheumatoid Arthritis with Traditional and Biologic Disease-Modifying Antirheumatic Drugs,” Journal of Rheumatology, Vol. 39, No. 8, 2012, pp. 1559- 1582. doi:10.3899/jrheum.110207
- J. A. Singh, D. E. Furst, A. Bharat, J. R. Curtis, A. F. Kavanaugh, J. M. Kremer, et al., “2012 Update of the 2008 American College of Rheumatology Recommendations for the Use of Disease-Modifying Antirheumatic Drugs and Biologic Agents in the Treatment of Rheumatoid Arthritis,” Arthritis Care & Research, Vol. 64, No. 5, 2012, pp. 625-639. doi:10.1002/acr.21641
- R. Maini, E. W. St. Clair, F. Breedeveld, D. Furst, J. Kalden, M. Weisman, et al., “Infliximab (Chimeric AntiTumour Necrosis Factor Alpha Monoclonal Antibody) versus Placebo in Rheumatoid Arthritis Patients Receiving Concomitant Methotrexate: A Randomized Phase III Trial,” Lancet, Vol. 354, No. 9194, 1999, pp. 1932-1939. doi:10.1016/S0140-6736(99)05246-0
- M. E. Weinblatt, E. C. Keystone, D. E. Furst, L. W. Moreland, M. H. Weisman, C. A. Birbara, et al., “Adalimumab, a Fully Human Anti-Tumor Necrosis Factor α Monoclonal Antibody, for the Treatment of Rheumatoid Arthritis in Patients Taking Concomitant Methotrexate: The ARMADA Trial,” Arthritis and Rheumatism, Vol. 48, No. 1, 2003, pp. 35-45. doi:10.1002/art.10697
- E. C. Keystone, M. C. Genovese, L. Klareskog, E. C. Hsia, S. T. Hall, P. C. Miranda, et al., “Golimumab, a Human Antibody to Tumour Necrosis Factor α Given by Monthly Subcutaneous Injections, in Active Rheumatoid Arthritis Despite Methotrexate Therapy: The GOFORWARD Study,” Annals of the Rheumatic Diseases, Vol. 68, No. 6, 2009, pp. 789-796. doi:10.1136/ard.2008.099010
- J. Smolen, R. B. Landewé, P. Mease, J. Brzezicki, D. Mason, K. Luijtens, et al., “Efficacy and Safety of Certolizumab Pegol plus Methotrexate in Active Rheumatoid Arthritis: The RAPID 2 Study. A Randomised Controlled Trial,” Annals of the Rheumatic Diseases, Vol. 68, No. 6, 2009, pp. 797-804. doi:10.1136/ard.2008.101659
- R. N. Maini, F. C. Breedveld, J. R. Kalden, J. S. Smolen, D. Furst, M. H. Weisman, et al., “Sustained Improvement over Two Years in Physical Function, Structural Damage, and Signs and Symptoms among Patients with Rheumatoid Arthritis Treated with Infliximab and Methotrexate,” Arthritis and Rheumatism, Vol. 50, No. 4, 2004, pp. 1051-1065. doi:10.1002/art.20159
- E. C. Keystone, A. F. Kavanaugh, J. T. Sharp, H. Tannenbaum, Y. Hua, L. S. Teoh, et al., “Radiographic, Clinical and Functional Outcomes of Treatment with Adalimumab (a Human Anti-Tumor Necrosis Factor Monoclonal Antibody) in Patients with Active Rheumatoid Arthritis Receiving Concomitant Methotrexate Therapy: A Randomized, Placebo-Controlled, 52-Week Trial,” Arthritis and Rheumatism, Vol. 50, No. 5, 2004, pp. 1400-1411. doi:10.1002/art.20217
- E. Keystone, D. van der Heijde, D. Mason Jr., R. Landewé, R. V. Vollenhoven, B. Combe, et al., “Certolizumab Pegol plus Methotrexate Is Significantly More Effective than Placebo plus Methotrexate in Active Rheumatoid Arthritis: Findings of a Fifty-Two-Week, Phase III, Multicenter, Randomized, Double-Blind, PlaceboControlled, Parallel-Group Study,” Arthritis and Rheumatism, Vol. 58, No. 11, 2008, pp. 3319-3329. doi:10.1002/art.23964
- A. Finckh, J. F. Simard, C. Gabay, P. A. Guerne and SCQM Physicians, “Evidence for Differential Acquired Drug Resistance to Anti-Tumour Necrosis Factor Agents in Rheumatoid Arthritis,” Annals of the Rheumatic Diseases, Vol. 65, No. 6, 2006, pp. 746-752. doi:10.1136/ard.2005.045062
- L. Barra, J. E. Pope and M. Payne, “Real-World AntiTumor Necrosis Factor Treatment in Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis: CostEffectiveness Based on Number Needed to Treat to Improve Health Assessment Questionnaire,” Journal of Rheumatology, Vol. 36, No. 7, 2009, pp. 1421-1428. doi:10.3899/jrheum.081122
- J. D. Greenberg, G. Reed, D. Decktor, L. Harrold, D. Furst, A. Gibofsky, et al., “A Comparative Effectiveness Study of Adalimumab, Etanercept and Infliximab in Biologically Naive and Switched Rheumatoid Arthritis Patients: Results from the US CORRONA Registry,” Annals of the Rheumatic Diseases, Vol. 71, No. 7, 2012, pp. 1134-1142. doi:10.1136/annrheumdis-2011-150573
- A. Zink, A. Strangfeld, M. Schneider, P. Herzer, F. Hierse, M. Stoyanova-Scholz, et al., “Effectiveness of Tumor Necrosis Factor Inhibitors in Rheumatoid Arthritis in an Observational Cohort Study: Comparison of Patients according to Their Eligibility for Major Randomized Clinical Trials,” Arthritis and Rheumatism, Vol. 54, No. 11, 2006, pp. 3399-3407. doi:10.1002/art.22193
- W. Kievit, J. Fransen, A. J. Oerlemans, H. H. Kuper, M. A. van der Laar, D. J. de Rooij, et al., “The Efficacy of Anti-TNF in Rheumatoid Arthritis, a Comparison between Randomised Controlled Trials and Clinical Practice,” Annals of the Rheumatic Diseases, Vol. 66, No. 11, 2007, pp. 1473-1478. doi:10.1136/ard.2007.072447
- R. Caporali, F. B. Pallavicini, M. Filippini, R. Gorla, A. Marchesoni, E. G. Favalli, et al., “Treatment of Rheumatoid Arthritis with Anti-TNF-Alpha Agents: A Reappraisal,” Autoimmunity Reviews, Vol. 8, No. 3, 2009, pp. 274-280. doi:10.1016/j.autrev.2008.11.003
- J. E. Pope, D. Khanna, D. Norrie and J. M. Ouimet, “The Minimally Important Difference for the Health Assessment Questionnaire in Rheumatoid Arthritis Clinical Practice Is Smaller than in Randomized Controlled Trials,” Journal of Rheumatology, Vol. 36, No. 2, 2009, pp. 254-259. doi:10.3899/jrheum.080479
- S. H. Shahouri, K. Michaud, T. R. Mikuls, L. Caplan, T. S. Shaver, J. D. Anderson, et al., “Remission of Rheumatoid Arthritis in Clinical Practice: Application of the American College of Rheumatology/European League Against Rheumatism 2011 Remission Criteria,” Arthritis and Rheumatism, Vol. 63, No. 11, 2011, pp. 3204-3215. doi:10.1002/art.30524
- M. H. Buch and P. Emery, “New Therapies in the Management of Rheumatoid Arthritis,” Current Opinion in Rheumatology, Vol. 23, No. 3, 2011, pp. 245-251. doi:10.1097/BOR.0b013e3283454124
- A. Rémy, J. Avouac, L. Gossec and B. Combe, “Clinical Relevance of Switching to a Second Tumour Necrosis Factor-Alpha Inhibitor after Discontinuation of a First tumour Necrosis Factor-Alpha Inhibitor in Rheumatoid Arthritis: A Systematic Literature Review and MetaAnalysis,” Clinical and Experimental Rheumatology, Vol. 29, No. 1, 2011, pp. 96-103.
- M. H. Buch, “Sequential Use of Biologic Therapy in Rheumatoid Arthritis,” Current Opinion in Rheumatology, Vol. 22, No. 3, 2010, pp. 321-329. doi:10.1097/BOR.0b013e328337bd01
- J. S. Smolen, J. Kay, M. K. Doyle, R. Landewé, E. L. Matteson, J. Wollenhaupt, et al., “Golimumab in Patients with Active Rheumatoid Arthritis after Treatment with Tumour Necrosis Factor Alpha Inhibitors (GO-AFTER study): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase III Trial,” Lancet, Vol. 374, No. 9685, 2009, pp. 210-221. doi:10.1016/S0140-6736(09)60506-7
- J. S. Smolen, J. Kay, R. Landewé, N. Gaylis, J. Wollenhaupt, F. T. Murphy, et al., “Golimumab in Patients with Active Rheumatoid Arthritis Who Have Previous Experience with Tumour Necrosis Factor Inhibitors: Results of a Long-Term Extension of the Randomised, Double-Blind, Placebo-Controlled GO-AFTER Study through Week 160,” Annals of the Rheumatic Diseases, Vol. 71, No. 10, 2012, pp. 1671-1679. doi:10.1136/annrheumdis-2011-200956
- S. B. Cohen, P. Emery, M. W. Greenwald, M. Dougados, R. A. Furie, M. C. Genovese, et al., “Rituximab for Rheumatoid Arthritis Refractory to Anti-Tumor Necrosis Factor Therapy: Results of a Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase III Trial Evaluating Primary Efficacy and Safety at Twenty-Four Weeks,” Arthritis and Rheumatism, Vol. 54, No. 9, 2006, pp. 2793-2806. doi:10.1002/art.22025
- P. Emery, E. Keystone, H. P. Tony, A. Cantagrel, R. van Vollenhoven, A. Sanchez, et al., “IL-6 Receptor Inhibition with Tocilizumab Improves Treatment Outcomes in Patients with Rheumatoid Arthritis Refractory to AntiTumour Necrosis Factor Biologicals: Results from a 24- Week Multicentre Randomised Placebo-Controlled Trial,” Annals of the Rheumatic Diseases, Vol. 67, No. 11, 2008, pp. 1516-1523. doi:10.1136/ard.2008.092932
- M. C. Genovese, J. C. Becker, M. Schiff, M. Luggen, Y. Sherrer, J. Kremer, et al., “Abatacept for Rheumatoid arthritis Refractory to Tumor Necrosis Factor α Inhibition,” New England Journal of Medicine, Vol. 353, No. 11, 2005, pp. 1114-1123. doi:10.1056/NEJMoa050524
- C. Salliot, A. Finckh, W. Katchamart, Y. Lu, Y. Sun, C. Bombardier, et al., “Indirect Comparisons of the Efficacy of Biological Antirheumatic Agents in Rheumatoid Arthritis in Patients with an Inadequate Response to Conventional Disease-Modifying Antirheumatic Drugs or to an Anti-Tumour Necrosis Factor Agent: A Meta-Analysis,” Annals of the Rheumatic Diseases, Vol. 70, No. 2, 2011, pp. 266-271. doi:10.1136/ard.2010.132134
- M. Schiff, R. Fleischmann, M. Weinblatt, R. Valente, D. van der Heijde, G. Citera, et al., “Abatacept sc versus Adalimumab on Background Methotrexate in RA: One Year Results from the AMPLE Study,” Annals of the Rheumatic Diseases, Vol. 71, Suppl. 3, 2012, p. 60.
- M. Schiff, M. Keiserman, C. Codding, S. Songcharoen, A. Berman, S. Nayiager, et al., “Efficacy and Safety of Abatacept or Infliximab vs Placebo in ATTEST: A Phase III, Multi-Centre, Randomised, Double-Blind, PlaceboControlled Study in Patients with Rheumatoid Arthritis and an Inadequate Response to Methotrexate,” Annals of the Rheumatic Diseases, Vol. 67, No. 8, 2008, pp. 1096- 1103. doi:10.1136/ard.2007.080002
- C. Gabay, P. Emery, R. van Vollenhoven, A. Dikranian, R. Alten, M. Klearman, et al., “Tocilizumab (TCZ) Monotherapy Is Superior to Adalimumab (ADA) Monotherapy in Reducing Disease Activity in Patients with Rheumatoid Arthritis (RA): 24-Week Data from the Phase 4 ADACTA Trial,” Annals of the Rheumatic Diseases, Vol. 71, Suppl. 3, 2012, p. 152.
- K. L. Hyrich, K. D. Watson, A. J. Silman, D. P. Symmons and British Society for Rheumatology Biologics Register, “Predictors of Response to Anti-TNF-α Therapy among Patients with Rheumatoid Arthritis: Results from the British Society for Rheumatology Biologics Register,” Rheumatology, Vol. 45, No. 12, 2006, pp. 1558-1565. doi:10.1093/rheumatology/kel149
- L. Mancarellla, F. Bobbio-Pallavicini, F. Ceccarelli, P. C. Falappone, A. Ferrante, D. Malesci, et al., “Good Clinical Response, Remission, and Predictors of Remission in Rheumatoid Arthritis Patients Treated with Tumor Necrosis Factor-Alpha Blockers: The GISEA Study,” Journal of Rheumatology, Vol. 34, No. 8, 2007, pp. 1670- 1673.
- L. E. Kristensen, M. C. Kapetanovic, A. Gülfe, M. Söderlin, T. Saxne and P. Geborek, “Predictors of Response to Anti-TNF Therapy according to ACR and EULAR Criteria in Patients with Established RA: Results from the South Swedish Arthritis Treatment Group Register,” Rheumatology, Vol. 47, No. 4, 2008, pp. 495-499. doi:10.1093/rheumatology/ken002
- P. Emery and T. Dörner, “Optimising Treatment of Rheumatoid Arthritis: A Review of Potential Biological Markers of Response,” Annals of the Rheumatic Diseases, Vol. 70, No. 12, 2011, pp. 2063-2070. doi:10.1136/ard.2010.148015
- K. Chatzidionysiou, E. Lie, E. Nasonov, G. Lukina, M. L. Hetland, U. Tarp, et al., “Highest Clinical Effectiveness of Rituximab in Autoantibody-Positive Patients with Rheumatoid Arthritis and in Those for Whom No more than One Previous TNF Antagonist Has Failed: Pooled Data from 10 European Registries,” Annals of the Rheumatic Diseases, Vol. 70, No. 9, 2011, pp. 1575-1580. doi:10.1136/ard.2010.148759
- A. Finckh, A. Ciurea, L. Brulhart, D. Kyburz, B. Möller, S. Dehler, et al., “B Cell Depletion May Be more Effective than Switching to an Alternative Anti-Tumor Necrosis Factor Agent in Rheumatoid Arthritis Patients with Inadequate Response to Anti-Tumor Necrosis Factor Agents,” Arthritis and Rheumatism, Vol. 56, No. 5, 2007, pp. 1417-1423. doi:10.1002/art.22520
- A. Finckh, A. Ciurea, L. Brulhart, B. Möller, U. A. Walker, D. Courvoiser, et al., “Which Subgroup of Patients with Rheumatoid Arthritis Benefits from Switching to Rituximab versus Alternative Anti-Tumour Necrosis Factor (TNF) Agents after Previous Failure of an Anti-TNF Agent?” Annals of the Rheumatic Diseases, Vol. 69, No. 2, 2010, pp. 387-393. doi:10.1136/ard.2008.105064
- J. Pope, E. Rampakakis, J. Sampalis and O. Desjardins, “The Effectiveness of Abatacept in a Large RA Real World Practice: Changes in the HAQ over Time and Durability of Response [Abstract and Poster],” Arthritis and Rheumatism, Vol. 63, No. S10, 2011, pp. S476-S477.
- J. A. Singh, G. A. Wells, R. Christensen, E. Tajong-Ghogomu, L. Maxwell, J. K. Macdonald, et al., “Adverse Effects of Biologics: A Network Meta-Analysis and Cochrane Overview,” Cochrane Database of Systematic Reviews, Vol. 16, No. 2, 2011, Article ID: CD008794.
- J. D. Greenberg, G. Reed, J. M. Kremer, E. Tindall, A. Kavanaugh, C. Zheng, et al., “Association of Methotrexate and Tumour Necrosis Factor Antagonists with Risk of Infectious Outcomes including Opportunistic Infections in the CORRONA Registry,” Annals of the Rheumatic Diseases, Vol. 69, No. 2, 2010, pp. 380-386. doi:10.1136/ard.2008.089276
- J. B. Galloway, K. L. Hyrich, L. K. Mercer, W. G. Dixon, B. Fu, A. P. Ustianowski, et al., “Anti-TNF Therapy Is Associated with an Increased Risk of Serious Infections in Patients with Rheumatoid Arthritis especially in the First 6 Months of Treatment: Updated Results from the British Society for Rheumatology Biologics Register with Special Emphasis on Risks in the Elderly,” Rheumatology (Oxford), Vol. 50, No. 1, 2011, pp. 124-131. doi:10.1093/rheumatology/keq242
- D. Salmon-Ceron, F. Tubach, O. Lortholary, O. Chosidow, S. Bretagne, N. Nicolas, et al., “Drug-Specific Risk of Non-Tuberculosis Opportunistic Infections in Patients Receiving Anti-TNF Therapy Reported to the 3-Year Prospective French RATIO Registry,” Annals of the Rheumatic Diseases, Vol. 70, No. 4, 2011, pp. 616-623. doi:10.1136/ard.2010.137422
- J. J. Gómez-Reino, L. Carmona, V. R. Valverde, E. M. Mola and M. D. Montero(BIOBADASER Group), “Treatment of Rheumatoid Arthritis with Tumor Necrosis Factor Inhibitors may Predispose to Significant Increase in Tuberculosis Risk: A Multicenter Active-Surveillance Report,” Arthritis and Rheumatism, Vol. 48, No. 8, 2003, pp. 2122-2127. doi:10.1002/art.11137
- F. Tubach, D. Salmon, P. Ravaud, Y. Allanore, P. Goupille, M. Bréban, et al., “Risk of Tuberculosis Is Higher with Anti-Tumor Necrosis Factor Monoclonal Antibody Therapy than with Soluble Tumor Necrosis Factor Receptor Therapy: The Three-Year Prospective French Research Axed on Tolerance of Biotherapies Registry,” Arthritis and Rheumatism, Vol. 60, No. 7, 2009, pp. 1884-1894. doi:10.1002/art.24632
- W. G, Dixon, K. L. Hyrich, K. D. Watson, M. Lunt, J. Galloway, A. Ustianowski, et al., “Drug-Specific Risk of Tuberculosis in Patients with Rheumatoid Arthritis Treated with Anti-TNF Therapy: Results from the British Society for Rheumatology Biologics Register (BSRBR),” Annals of the Rheumatic Diseases, Vol. 69, No. 3, 2010, pp. 522-528. doi:10.1136/ard.2009.118935
- L. Carmona, J. J. Gómez-Reino, V. Rodriguez-Valverde, D. Montero, E. Pascual-Gómez, E. M. Mola, et al., “Effectiveness of Recommendations to Prevent Reactivation of Latent Tuberculosis Infection in Patients Treated with Tumor Necrosis Factor Antagonists,” Arthritis and Rheumatism, Vol. 52, No. 6, 2005, pp. 1766-1772. doi:10.1002/art.21043
- C, Salliot, M. Dougados and L. Gossec, “Risk of Serious Infections during Rituximab, Abatacept and Anakinra Treatments for Rheumatoid Arthritis: Meta-Analyses of Randomised Placebo-Controlled Trials,” Annals of the Rheumatic Diseases, Vol. 68, No. 1, 2009, pp. 25-32. doi:10.1136/ard.2007.083188
- L. Campbell, C. Chen, S. S. Bhagat, R. A. Parker and A. J. Östör, “Risk of Adverse Events including Serious Infections in Rheumatoid Arthritis Patients Treated with Tocilizumab: A Systematic Literature Review and MetaAnalysis of Randomized Controlled Trials,” Rheumatology (Oxford), Vol. 50, No. 3, 2011, pp. 552-562. doi:10.1093/rheumatology/keq343
- K. L. Winthrop, E. Chang, S. Yamashita, M. F. Iademarco and P. A. LoBue, “Nontuberculous Mycobacteria Infections and Anti-Tumor Necrosis Factor-α Therapy,” Emerging Infectious Diseases, Vol. 15, No. 15, 2009, pp. 1556-1561. doi:10.3201/eid1510.090310
- A. L. Smitten, T. A. Simon, M. C. Hochberg and S. Suissa, “A Meta-Analysis of the Incidence of Malignancy in Adult Patients with Rheumatoid Arthritis,” Arthritis Research & Therapy, Vol. 10, No. 2, 2008, p. R45. doi:10.1186/ar2404
- T. Bongartz, A. J. Sutton, M. J. Sweeting, I. Buchan, E. L. Matteson and V. Montori,” Anti-TNF Antibody Therapy in Rheumatoid Arthritis and the Risk of Serious Infections and Malignancies: Systematic Review and Meta-Analysis of Rare Harmful Effects in Randomized Controlled Trials,” Journal of the American Medical Association, Vol. 295, No. 19, 2006, pp. 2275-2285. doi:10.1001/jama.295.19.2275
- F. Wolfe and K. Michaud, “Biologic Treatment of Rheumatoid Arthritis and the Risk of Malignancy: Analyses from a Large US Observational Study,” Arthritis and Rheumatism, Vol. 56, No. 9, 2007, pp. 2886-2895. doi:10.1002/art.22864
- X. Mariette, M. Matucci-Cerinic, K. Pavelka, P. Taylor, R. van Vollenhoven, R. Heatley, et al., “Malignancies Associated with Tumor Necrosis Factor Inhibitors in Registries and Prospective Observational Studies: A Systematic Review and Meta-Analysis,” Annals of the Rheumatic Diseases, Vol. 70, No. 11, 2011, pp. 1895-1904. doi:10.1136/ard.2010.149419
- J. Askling, E. Baecklund, F. Granath, P. Geborek, M. Fored, C. Backlin, et al., “Anti-Tumour Necrosis Factor Therapy in Rheumatoid Arthritis and Risk of Malignant Lymphomas: Relative Risks and Time Trends in the Swedish Biologics Register,” Annals of the Rheumatic Diseases, Vol. 68, No. 5, 2009, pp. 648-653. doi:10.1136/ard.2007.085852
- A. E. Thompson, S. W. Rieder and J. E. Pope, “Tumor Necrosis Factor Therapy and the Risk of Serious Infection and Malignancy in Patients with Early Rheumatoid Arthritis: A Meta-Analysis of Randomized Controlled Trials,” Arthritis and Rheumatism, Vol. 63, No. 6, 2011, pp. 1479-1485. doi:10.1002/art.30310
- R. F. van Vollenhoven, P. Emery, C. O. Bingham III, E. C. Keystone, R. Fleischmann, D. E. Furst, et al., “LongTerm Safety of Patients Receiving Rituximab in Rheumatoid Arthritis Clinical Trials,” Journal of Rheumatology, Vol. 37, No. 3, 2010, pp. 558-567. doi:10.3899/jrheum.090856
- M. H. Schiff, J. M. Kremer, A. Jahreis, E. Vernon, J. D. Isaacs and R. F. van Vollenhoven, “Integrated Safety in Tocilizumab Clinical Trials,” Arthritis Research & Therapy, Vol. 13, No. 5, 2011, p. R141. doi:10.1186/ar3455
- [61] T. A. Simon, A. L. Smitten, J. Franklin, J. Askling, D. Lacaille, F. Wolfe, et al., “Malignancies in the Rheumatoid Arthritis Abatacept Clinical Development Programme: An Epidemiological Assessment,” Annals of the Rheumatic Diseases, Vol. 68, No. 12, 2009, pp. 1819- 1826. doi:10.1136/ard.2008.097527
- [62] W. G. Dixon, K. D. Watson, M. Lunt, L. K. Mercer, K. L. Hyrich, D. P Symmons, et al., “Influence of Anti-Tumor Necrosis Factor Therapy on Cancer Incidence in Patients with Rheumatoid Arthritis Who Have Had a Prior Malignancy: Results from the British Society for Rheumatology Biologics Register,” Arthritis Care & Research, Vol. 62, No. 6, 2010, pp. 755-763. doi:10.1002/acr.20129
- [63] P. J. Charles, R. J. Smeenk, J. De Jong, M. Feldmann and R. N. Maini, “Assessment of Antibodies to DoubleStranded DNA Induced in Rheumatoid Arthritis Patients Following Treatment with Infliximab, a Monoclonal Antibody to Tumor Necrosis Factor α: Findings In OpenLabel and Randomized Placebo-Controlled Trials,” Arthritis and Rheumatism, Vol. 43, No. 11, 2000, pp. 2383- 2390. doi:10.1002/1529-0131(200011)43:11<2383::AID-ANR2>3.0.CO;2-D
- [64] F. Atzeni, M. Turiel, F. Capsoni, A. Doria, P. Meroni and P. Sarzi-Puttini, “Autoimmunity and Anti-TNF-α Agents,” Annals of the New York Academy of Science, Vol. 1051, 2005, pp. 559-569. doi:10.1196/annals.1361.100
- [65] H. Bacquet-Deschryver, F. Jouen, M. Quillard, J. F. Mé- nard, V. Goëb, T. Lequerré, et al., “Impact of Three Anti-TNFα Biologics on Existing and Emergent Autoimmunity in Rheumatoid Arthritis and Spondylarthropathy Patients,” Journal of Clinical Immunology, Vol. 28, No. 5, 2008, pp. 445-455. doi:10.1007/s10875-008-9214-3
- [66] J. Sibilia and R. Westhovens, “Safety of T-cell Co-Stimulation Modulation with Abatacept in Patients with Rheumatoid Arthritis,” Clinical and Experimental Rheumatology, Vol. 25, No. S5, 2007, pp. S46-S56.
- [67] J. D. Greenberg, V. Furer and M. E. Farkouh, “Cardiovascular Safety of Biologic Therapies for the Treatment of RA,” Nature Reviews. Rheumatology, Vol. 8, No. 1, 2012, pp. 13-21.
- [68] C, Barnabe, B. J. Martin and W. A. Ghali, “Systematic Review and Meta-Analysis: Anti-Tumor Necrosis Factor α Therapy and Cardiovascular Events in Rheumatoid Arthritis,” Arthritis Care & Research, Vol. 63, No. 4, 2011, pp. 522-529. doi:10.1002/acr.20371
- [69] J. D. Greenberg, J. M. Kremer, J. R. Curtis, M. C. Hochberg, G. Reed, P. Tsao, et al., “Tumour Necrosis Factor Antagonist Use and Associated Risk Reduction of Cardiovascular Events among Patients with Rheumatoid Arthritis,” Annals of the Rheumatic Diseases, Vol. 70, No. 4, 2011, pp. 576-582. doi:10.1136/ard.2010.129916
- [70] S. L. Westlake, A. N. Colebatch, J. Baird, N. Curzen, P. Kiely, M. Quinn, et al., “Tumour Necrosis Factor Antagonists and the Risk of Cardiovascular Disease in Patients with Rheumatoid Arthritis: A Systemic Literature Review,” Rheumatology (Oxford), Vol. 50, No. 3, 2011, pp. 518-531. doi:10.1093/rheumatology/keq316
- [71] W. G. Dixon, K. D. Watson, M. Lunt, K. L. Hyrich, A. J. Silman and D. P. Symmons, “Reduction in the Incidence of Myocardial Infarction in Patients with Rheumatoid Arthritis who Respond to Anti-Tumor Necrosis Factor α Therapy: Results from the British Society for Rheumatology Biologics Register,” Arthritis and Rheumatism, Vol. 56, No. 9, 2007, pp. 2905-2912. doi:10.1002/art.22809
- [72] B. Levine, J. Kalman, L. Mayer, H. M. Fillit and M. Packer, “Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure,” New England Journal of Medicine, Vol. 323, No. 4, 1990, pp. 236-241. doi:10.1056/NEJM199007263230405
- [73] D. L. Mann, J. J. McMurray, M. Packer, K. Swedberg, J. S. Borer, W. S. Colucci, et al., “Targeted Anticytokine Therapy in Patients with Chronic Heart Failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL),” Circulation, Vol. 109, No. 13, 2004, pp. 1594-1602. doi:10.1161/01.CIR.0000124490.27666.B2
- [74] E. S. Chung, M. Packer, K. H. Lo, A. A. Fasanmade and J. T. Willerson, “Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, Double-Blind, Placebo-Controlled, Pilot Trial of Infliximab, a Chimeric Monoclonal Antibody to Tumor Necrosis Factor-α, in Patients with Moderate-to-Severe Heart Failure: Results of the Anti-TNF Therapy Against Congestive Heart Failure (ATTACH) Trial,” Circulation, Vol. 107, No. 25, 2003, pp. 3133-3140. doi:10.1161/01.CIR.0000077913.60364.D2
- [75] F. Wolfe and K. Michaud, “Heart Failure in Rheumatoid Arthritis: Rates, Predictors, and the Effect of Anti-Tumor Necrosis Factor Therapy,” American Journal of Medicine, Vol. 116, No. 5, 2004, pp. 305-311. doi:10.1016/j.amjmed.2003.09.039
- [76] J. Listing, A. Strangfeld, J. Kekow, M. Schneider, A. Kapelle, S. Wassenberg, et al., “Does Tumor Necrosis Factor α Inhibition Promote or Prevent Heart Failure in Patients with Rheumatoid Arthritis?” Arthritis and Rheumatism, Vol. 58, No. 3, 2008, pp. 667-677. doi:10.1002/art.23281
- [77] C. I. Daïen, Y. Duny, T. Barnetche, J. P. Daurès, B. Combe and J. Morel, “Effect of TNF Inhibitors on Lipid Profile in Rheumatoid Arthritis: A Systematic Review with Meta-Analysis,” Annals of the Rheumatic Diseases, Vol. 71, No. 6, 2012, pp. 862-868. doi:10.1136/annrheumdis-2011-201148
- [78] R. N. Maini, P. C Taylor, J. Szechinski, K. Pavelka, J. Bröll, G. Balint, et al., “Double-Blind Randomized Controlled Clinical Trial of the Interleukin-6 Receptor Antagonist, Tocilizumab, in European Patients with Rheumatoid Arthritis who Had an Incomplete Response to Methotrexate,” Arthritis and Rheumatism, Vol. 54, No. 9, 2006, pp. 2817-2829. doi:10.1002/art.22033
- [79] M. C. Genovese, J. D. McKay, E. L. Nasonov, E. F. Mysler, N. A. da Silva, E. Alecock, et al., “Interleukin-6 Receptor Inhibition with Tocilizumab Reduces Disease Activity in Rheumatoid Arthritis with Inadequate Response to Disease-Modifying Antirheumatic Drugs: the Tocilizumab in Combination with Traditional Disease-Modifying Antirheumatic Drug Therapy Study,” Arthritis and Rheumatism, Vol. 58, No. 10, 2008, pp. 2968-2980. doi:10.1002/art.23940
- [80] M. A. Blum, D. Koo and J. A. Doshi, “Measurement and Rates of Persistence with and Adherence to Biologics for Rheumatoid Arthritis,” Clinical Therapeutics, Vol. 33, No. 7, 2011, pp. 901-913. doi:10.1016/j.clinthera.2011.06.001
- [81] S. M, Du Pan, S. Dehler, A. Ciurea, H. R. Ziswiler, C. Gabay, A. Finckh, et al., “Comparison of Drug Retention Rates and Causes of Drug Discontinuation between Anti-Tumor Necrosis Factor Agents in Rheumatoid Arthritis,” Arthritis and Rheumatism, Vol. 61, No. 5, 2009, pp. 560-568. doi:10.1002/art.24463
- [82] K. L, Hyrich, M. Lunt, K. D. Watson, D. P. Symmons and A. J. Silman, “British Society for Rheumatology Biologics Register. Outcomes after Switching from One Anti-Tumor Necrosis Factor α Agent to a Second Anti-Tumor Necrosis Factor α Agent in Patients with Rheumatoid Arthritis: Results from a Large UK National Cohort Study,” Arthritis and Rheumatism, Vol. 56, No. 1, 2007, pp. 13-20. doi:10.1002/art.22331
- [83] S. Curkendall, V. Patel, M. Gleeson, R. S. Campbell, M. Zagari and R. Dubois, “Compliance with Biologic Therapies for Rheumatoid Arthritis: Do Patient Out-of-Pocket Payments Matter?” Arthritis and Rheumatism, Vol. 59, No. 10, 2008, pp. 1519-1526. doi:10.1002/art.24114
- [84] J. L. Barton, “Patient Preferences and Satisfaction in the Treatment of Rheumatoid Arthritis with Biologic Therapy,” Patient Preference and Adherence, Vol. 3, 2009, pp. 335-344. doi:10.2147/PPA.S5835
- [85] L. E. Harrington, R. D. Hatton, P. R. Mangan, H. Turner, T. L. Murphy, K. M. Murphy, et al., “Interleukin 17-Producing CD4+ Effector T Cells Develop a Lineage Distinct from the T Helper Type 1 and 2 Lineages,” Nature Immunology, Vol. 6, No. 11, 2005, pp. 1123-1132.
- [86] C. L. Langrish, Y. Chen, W. M. Blumenschein, J. Mattson, B. Basham, J. D. Sedgwick, et al., “IL-23 Drives a Pathogenic T Cell Population that Induces Auto-Immune Inflammation,” Journal of Experimental Medicine, Vol. 201, No. 2, 2005, pp. 233-240. doi:10.1084/jem.20041257
- [87] P. Miossec, T. Korn and V. K. Kuchroo, “Interleukin-17 and Type 17 Helper T Cells,” New England Journal of Medicine, Vol. 361, No. 9, 2009, pp. 888-898. doi:10.1056/NEJMra0707449
- [88] S. L. Gaffen, “Structure and Signalling in the IL-17 Receptor Family,” Nature Reviews. Immunology, Vol. 9, No. 8, 2009, pp. 556-567. doi:10.1038/nri2586
- [89] R. M. Onishi and S. L. Gaffen, “Interleukin-17 and Its Target Genes: Mechanisms of Interleukin-17 Function in Disease,” Immunology, Vol. 129, No. 3, 2010, pp. 311- 321. doi:10.1111/j.1365-2567.2009.03240.x
- [90] A. J. Hueber, D. L. Asquith, A. M. Miller, J. Reilly, S. Kerr, J. Leipe, et al., “Mast Cells Express IL-17A in Rheumatoid Arthritis Synovium,” Journal of Immunology, Vol. 184, No. 7, 2010, pp. 3336-3340. doi:10.4049/jimmunol.0903566
- [91] A. M. Lin, C. J. Rubin, R. Khandpur, J. Y. Wang, M. Riblett, S. Yalavarthi, et al., “Mast Cells and Neutrophils Release IL-17 through Extracellular Trap Formation in Psoriasis,” Journal of Immunology, Vol. 187, No. 1, 2011, pp. 490-500. doi:10.4049/jimmunol.1100123
- [92] C. T. Weaver, R. D. Hatton, P. R. Mangan and L. E. Harrington, “IL-17 Family Cytokines and the Expanding Diversity of Effector T Cell Lineages,” Annual Review of Immunology, Vol. 25, 2007, pp. 821-852. doi:10.1146/annurev.immunol.25.022106.141557
- [93] E. Lubberts, “Th17 Cytokines and Arthritis,” Seminars in Immunopathology, Vol. 32, No. 1, 2010, pp. 43-53. doi:10.1007/s00281-009-0189-9
- [94] S. Kotake, T. Yago, M. Kawamoto and Y. Nanke, “Role of Osteoclasts and Interleukin-17 in the Pathogenesis of Rheumatoid Arthritis: Crucial ‘Human Osteoclastology’,” Journal of Bone and Mineral Metabolism, Vol. 30, No. 2, 2012, pp. 125-135.
- [95] E. Lubberts, L. A. Joosten, B. Oppers, L. van den Bersselaar, C. J. Coenen-de Roo, J. K. Kolls, et al., “IL-1-Independent Role of IL-17 in Synovial Inflammation and Joint Destruction during Collagen-Induced Arthritis,” Journal of Immunology, Vol. 167, No. 2, 2001, pp. 1004-1013.
- [96] S. Nakae, A. Nambu, K. Sudo and Y. Iwakura, “Suppression of Immune Induction of Collagen-Induced Arthritis in IL-17-Deficient Mice,” Journal of Immunology, Vol. 171, No. 11, 2003, pp. 6173-6177.
- [97] E. Lubberts, M. I. Koenders, B. Oppers-Walgreen, L. van den Bersselaar, C. J. Coenen-de Roo, L. A. B. Joosten, et al., “Treatment with a Neutralizing Anti-Murine Interleukin-17 Antibody after the Onset of Collagen-Induced Arthritis Reduces Joint Inflammation, Cartilage Destruction, and Bone Erosion,” Arthritis and Rheumatism, Vol. 50, No. 2, 2004, pp. 650-659. doi:10.1002/art.20001
- [98] M. I. Koenders, E. Lubberts, B. Oppers-Walgreen, L. van den Bersselaar, M. M. Helsen, F. E. Di Padova, et al., “Blocking of Interleukin-17 during Reactivation of Experimental Arthritis Prevents Joint Inflammation and Bone Erosion by Decreasing RANKL and Interleukin-1,” American Journal of Pathology, Vol. 167, No. 1, 2005, pp. 141-149. doi:10.1016/S0002-9440(10)62961-6
- [99] M. Ziolkowska, A. Koc, G. Luszczykiewicz, K. Ksiezopolska-Pietrzak, E. Klimczak, H. Chwalinska-Sadowska, et al., “High Levels of IL-17 in Rheumatoid Arthritis Patients: IL-15 Triggers in vitro IL-17 Production via Cyclosporine A-Sensitive Mechanism,” Journal of Immunology, Vol. 164, No. 5, 2000, pp. 2832-2838.
- [100] S. A. Metawi, D. Abbas, M. M. Kamal and M. K. Ibrahim, “Serum and Synovial Fluid Levels of Interleukin-17 in Correlation with Disease Activity in Patients with RA,” Clinical Rheumatology, Vol. 30, No. 9, 2011, pp. 1201- 1207. doi:10.1007/s10067-011-1737-y
- [101] J. Suurmond, A. L. Dorjée, M. R. Boon, E. F. Knol, T. W. Huizinga, R. E. Toes, et al., “Mast Cells Are the Main Interleukin 17-Positive Cells in Anticitrullinated Protein Antibody-Positive and -Negative Rheumatoid Arthritis and Osteoarthritis Synovium,” Arthritis Research & Therapy, Vol. 13, No. 5, 2011, p. R150. doi:10.1186/ar3466
- [102] W. Marder, S. Khalatbari, J. D. Myles, R. Hench, S. Yalavarthi, S. Lustig, et al., “Interleukin 17 as a Novel Predictor of Vascular Function in Rheumatoid Arthritis,” Annals of the Rheumatic Diseases, Vol. 70, No. 9, 2011, pp. 1550-1555. doi:10.1136/ard.2010.148031
- [103] W. Hueber, D. D. Patel, T. Dryja, A. M. Wright, I. Koroleva, G. Bruin, et al., “Effects of AIN457, a Fully Human Antibody to Interleukin-17A, on Psoriasis, Rheumatoid Arthritis, and Uveitis,” Science Translational Medicine, Vol. 2, No. 52, 2010, p. 52ra72. doi:10.1126/scitranslmed.3001107
- [104] M. C. Genovese, P. Durez, H. B. Richards, J. Supronik, E. Dokoupilova, J. A. Aelion, et al., “One Year Efficacy and Safety Results of a Phase II Trial of Secukinumab in Patients with Rheumatoid Arthritis [abstract],” Arthritis and Rheumatism, Vol. 63, No. S10, 2011, pp. S149-S150.
- [105] M. C. Genovese, F. Van den Bosch, S. A. Roberson, S. Bojin, I. M. Biagini, P. Ryan, et al., “LY2439821, a Humanized Anti-Interleukin-17 Monoclonal Antibody, in the Treatment of Patients with Rheumatoid Arthritis. A Phase I Randomized, Double-Blind, Placebo-Controlled, Proof-of-Concept Study,” Arthritis and Rheumatism, Vol. 62, No. 4, 2010, pp. 929-939. doi:10.1002/art.27334
- [106] M. C. Genovese, M. W. Greenwald, C. S. Cho, A. Berman, L. Jin, G. Cameron, et al., “A Phase 2 Study of Multiple Subcutaneous Doses of LY2439821, an Anti-IL-17 Monoclonal Antibody, in Patients with Rheumatoid Arthritis in Two Populations: Naïve to Biologic Therapy or Inadequate Responders to Tumor Necrosis Factor Alpha Inhibitors [abstract],” Arthritis and Rheumatism, Vol. 63, No. S10, 2011, p. S1017.
- [107] K. A. Papp, C. Leonardi, A. Menter, J. P. Ortonne, J. G. Krueger, G. Kricorian, et al., “Brodalumab, an Anti-Interleukin-17—Receptor Antibody for Psoriasis,” New England Journal of Medicine, Vol. 366, No. 13, 2012, pp. 1181-1189. doi:10.1056/NEJMoa1109017
- [108] Y. Iwakura, H. Ishigame, S. Saijo and S. Nakae, “Functional Specialization of Interleukin-17 Family Members,” Immunity, Vol. 34, No. 2, 2011, pp. 149-162. doi:10.1016/j.immuni.2011.02.012
- [109] K. Pavelka, Y. Chon, R. Newmark, N. Erondu and S. L. Lin, “A Randomized, Double-Blind, Placebo-Controlled, Multiple-Dose Study to Evaluate the Safety, Tolerability, and Efficacy of Brodalumab (AMG 827) in Subjects with Rheumatoid Arthritis and an Inadequate Response to Methotrexate,” Arthritis and Rheumatism, Vol. 64, No. S10, 2012, p. S362.
- [110] A. Puel, R. Döffinger, A. Natividad, M. Chrabieh, G. Barcenas-Morales, C. Picard, et al., “Autoantibodies against IL-17A, IL-17F, and IL-22 in Patients with Chronic Mucocutaneous Candidiasis and Autoimmune Polyendocrine Syndrome Type I,” Journal of Experimental Medicine, Vol. 207, No. 2, 2010, pp. 291-297. doi:10.1084/jem.20091983
- [111] A. Puel, S. Cypowyj, J. Bustamante, J. F. Wright, L. Liu, H. K. Lim, et al., “Chronic Mucocutaneous Candidiasis in Humans with Inborn Errors of Interleukin-17 Immunity,” Science, Vol. 332, No. 6025, 2011, pp. 65-68. doi:10.1126/science.1200439
- [112] K. Kisand, A. S. Bøe Wolff, K. T. Podkrajsek, L. Tserel, M. Link, K. V. Kisand, et al., “Chronic Mucocutaneous Candidiasis in APECED or Thymoma Patients Correlates with Autoimmunity to Th17-Associated Cytokines,” Journal of Experimental Medicine, Vol. 207, No. 2, 2010, pp. 299-308. doi:10.1084/jem.20091669
NOTES
*Declaration of Interest: Dr. Pope has no conflicts of interest to declare. Dr. Combe has received compensation for speaking, consultancy, and/ or advisory board participation from BMS, Celgene, Merck, Novartis, Pfizer, Roche, and UCB and received grant support from Pfizer, Roche, and UCB.