Open Journal of Blood Diseases
Vol.4 No.2(2014), Article
ID:47198,8
pages
DOI:10.4236/ojbd.2014.42003
ß-Globin Gene Cluster Haplotypes and Clinical Severity in Sickle Cell Anemia Patients in Southern Brazil
Maria A. L. da Silva1, João R. Friedrisch2, Christina M. Bittar2, Meide Urnau2, Jóice Merzoni1, Vanessa S. Valim1,3, Bruna Amorin1,3, Annelise Pezzi1,3, José Artur B. Chies4, Lúcia M. da Rocha Silla1,2,3*
1Laboratory of Hematopoietic Cell Culture and Molecular Analysis, Center for Experimental Research, Porto Alegre, Brazil
2Hematology and Bone Marrow Transplantation of Hospital de Clinicas de Porto Alegre, Porto Alegre, Brazil
3Post-Graduation in Medicine: Medical Sciences, Federal University of Rio Grande do Sul, Porto Alegre, Brazil
4Laboratory of Immunogenetics, Department of Genetics, UFRGS, Porto Alegre, Brazil
Email: *lsilla@hcpa.ufrgs.br
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 20 April 2014; revised 20 May 2014; accepted 16 June 2014
ABSTRACT
Hematopoietic stem cell transplantation (HSCT) has emerged as a curative strategy for sickle cell anemia (SCA); it is necessary to find markers of SCA clinical severity to spare those SCA patients whose clinical course is mild from the morbidity and mortality associated with HSCT. Haplotypes have been correlated with the severity of clinical manifestations in SCA patients, and fetal hemoglobin (HbF) and socioeconomic status (SeS) have also been described as negative factors. We studied these factors and their impact on clinical manifestations in a population of Southern Brazilian patients attending the Center for Sickle Cell Anemia at Hospital de Clínicas de Porto Alegre/RS, Brazil. Clinical severity was defined as two or more veno-occlusive episodes per year. The βS haplotypes were determined by PCR in 75 SCA patients. Among the 150 βS chromosomes analyzed, 99 (66%) were identified as Bantu (Ban), 41 (27%) as Benin (Ben), and 10 (7%) as other haplotypes. Most patients in our sample (62.7%) belonged to lower SeS groups, precluding meaningful statistical analysis of SeS impact on clinical severity. There was no correlation between haplotypes or HbF level and SCA clinical severity. Gene polymorphisms and environmental issues have to be taken into consideration.
Keywords:Sickle Cell Anemia, ß-Globin, Fetal Hemoglobin and Clinical Severity

1. Introduction
Sickle mutation of the beta-globin locus, which in the homozygous state gives rise to sickle cell anemia (SCA), is caused by the substitution of an adenine (A) for a thymine (T) in the sixth codon of the beta-globin gene, leading to the substitution of glutamic acid for valine and to the production of hemoglobin S (HbS) [1] [2] . HbS polymerizes under low oxygen tension (hypoxia), producing the characteristic sickle-shaped erythrocytes [3] . This event leads to hemolysis that promotes adhesive properties of circulating cells and the vessel wall. Sickle reticulocytes, increased in number in response to hemolysis, express receptors and ligands responsible for their adherence to endothelium and leukocytes. Reticulocyte adherence provides an additional link between hemolytic anemia and vaso-occlusion [4] , which is the hallmark of SCA. These important pathophysiological aspects of SCA trigger painful events and contribute largely to the morbidity and mortality associated with the disease [5] . Some studies have suggested that SCA is a chronic inflammatory disease, with clinical manifestations being influenced by an inflammatory state which contributes to vaso-occlusive events and therefore appears to play an important role in the pathophysiology of the disease [6] -[8] .
Although SCA is a monogenic disease, its clinical picture is highly heterogeneous with some patients having a mild phenotype and others being severely affected [9] . Many of the factors described as contributing to such heterogeneity are genetically determined (bS haplotypes, concomitant α-thalassemia, fetal hemoglobin [HbF] levels) [10] , while others are influenced by environmental or socioeconomic conditions [11] [12] .
The bS haplotypes are defined by the nonrandom association of restriction endonuclease cleavage sites around the beta-globin gene cluster [13] [14] . These polymorphisms have provided important anthropological data concerning the multiple origins of HbS [15] , and evidence suggests that bS mutation has at least five independent origins which correlate with different haplotypes: Senegal, Benin, Central African Republic (CAR) or Bantu, Cameroon, and Arab-Indian [2] [16] [17] . Several studies have suggested that the clinical severity of SCA is correlated with bS haplotypes, with Senegal and Arab-Indian haplotypes associated with a mild clinical course, which has been attributed to higher HbF levels and packed cell volume (PCV) [11] [18] . Bantu has shown the lowest HbF level and the lowest PCV, being associated with severe clinical manifestations [18] [19] , while Benin and Cameroon apparently present intermediate phenotypes [11] [18] . Despite those studies, the influence of bS haplotypes upon SCA clinical and hematologic features is still a controversial issue [19] .
The influence of socioeconomic status (SeS) on the severity of sickle cell disease was evaluated by Okany et al, who found that patients of low SeS had significantly more bone-pain crises over a one-year period [20] . The authors speculated that patients of lower SeS are more likely to live in less comfortable homes with poor sanitation and hence be more exposed to infections that may precipitate pain crises. Furthermore, low SeS has an adverse effect on the nutritional status and hemoglobin concentration of SCA patients [21] . A study conducted in the United Kingdom found that children born into low social classes were more likely to be admitted to the hospital, had longer overall length of hospital stay, and generated higher costs during the first 10 years of life as compared to children born into higher social classes [22] . With the emerging role of hematopoietic stem cell transplantation (HSCT) as a curative strategy for SCA (Michlitsch and Walters, 2008), a reliable marker of clinical severity is of utmost importance.
In the present study, we analyzed the impact of different bS haplotypes, HbF level, and SeS on clinical manifestations in a population of Southern Brazilian SCA patients attending the Center for Sickle Cell Anemia and Hemoglobinopathy at Hospital de Clínicas de Porto Alegre/RS, Brazil.
2. Patients and Methods
SCA Afro-Brazilian patients of all ages regularly attending at the Center for Sickle Cell Anemia and Hemoglobinopathy at Hospital de Clínicas de Porto Alegre/RS, Brazil, were included in this study. They are followed through periodic evaluations and some are using chronic hemotherapy. Also, some patients have recurrent painful crises, priapism, respiratory infections, stroke, osteonecrosis of the humerus and femurs, even chronic complications such as cognitive impairment, gallstones, proteinuria, chronic renal failure, and pulmonary hypertension. In addition to the outpatient treatment often need care due the acute complications in the Emergency Room or Hospital Admission.
The study was approved by the Research Ethics Committee of Hospital de Clínicas de Porto Alegre, under protocol no. 04-438. Written informed consent was obtained from all patients.
The patients’ medical records were reviewed for demographic and clinical data. All recorded clinical data analyzed in this study are from patients who have never received hydroxyurea (HU) or refer to the period before the start of HU treatment, as this drug is known to induce HbF synthesis [23] and to significantly reduce the frequency of vaso-occlusive episodes in most SCA patients. A severe clinical course was defined as two or more (≥2) painful crises per year [24] [25] . The presence of cardiomegaly was determined by echocardiographic parameters obtained from the patients’ medical records, and the presence of vasculopathic events such as cerebrovascular accidents (CVA) was based on the recorded clinical data, since only a few patients had undergone magnetic resonance angiography (MRA). For analysis purposes, such occurrences were all grouped under cardiovascular events.
2.1. Socioeconomic Status (SeS)
SeS was defined based on the responses of patients to a questionnaire designed and validated by the Brazilian Association of Market Research Agencies, including questions on the number of people in the household, TV, washing machine, and similar appliances. The Brazilian government uses this questionnaire to estimate the purchasing power of individuals and families and categorizes the Brazilian population into 5 socioeconomic levels: A to E—with A being the highest and E the lowest level [26] .
2.2. Molecular Studies
Venous blood samples were collected and stored at 4˚C until DNA extraction. Genomic DNA was isolated from blood samples as previously described [27] and stored at −20˚C. The bS haplotypes were determined by analysis of the following polymorphic restriction sites: HindIII-γG, HindIII-γA, HincII-3’ψβ and HpaI-3’β Polymerase chain reaction (PCR) was followed by restriction fragment length polymorphism (RFLP) and agarose gel electrophoresis according to Sutton et al [28] .
2.3. Statistical Analysis
Descriptive statistics were used due to data asymmetry. Quantitative variables were expressed as median and 25th-75th percentile, and qualitative variables as relative and absolute frequencies. Associations among qualitative variables were evaluated by the chi-square test or Fisher’s exact test. Comparisons of age or HbF levels with haplotypes and cardiovascular events were performed using the Kruskal-Wallis test and the Mann-Whitney test, respectively. The level of significance was set at 5% (P < 0.05). The Statistical Package for the Social Sciences (SPSS), version 10.0, was used for data analysis (SPSS Inc., Chicago, Illinois, USA).
3. Results
A total of 75 SCA patients were included in the analysis; of these, 39 (52%) were male, with a median age of 18.5 (2 - 62) years. Fifty (66.7%) patients had 2 or more painful crises per year, and 33 (44%) had some type of cardiovascular event. SeS analysis revealed that 47 (62.7%) patients belonged to C and D socioeconomic levels, and 28 (37.3%) to the B level.
Among the 150 βS chromosomes analyzed, 99 (66%) were identified as Bantu, 41 (27%) as Benin, and 10 (7%) as other haplotypes. As for HbF level, most patients had a concentration above 2 g/dL (Table 1). No significant associations were observed when haplotypes were correlated with pain rate, cardiovascular events, or HbF levels (Table 1). Although not statistically significant, a tendency to an association between haplotypes and HbF levels was observed, in which individuals with haplotypes classified as “others” showed lower HbF levels (P = 0.051) than homozygous (Ban/Ban, Ben/Ben) and heterozygous (Ban/Ben) individuals (Figure 1). As expected, older patients had a higher incidence of cardiovascular events than younger individuals (P = 0.013) (Figure 2). There was no significant correlation between HbF levels and genotype or clinical severity (Table 1 and Table 2).

Table 1. Clinical outcome and hematologic characteristics according to haplotypes.
Ban = Bantu; Ben = Benin; CE = cardiovascular events; HbF = fetal hemoglobin; n = number of individuals. *Number of painful crises per year.
Table 2. Pain rate and fetal hemoglobin (HbF) levels.
n = number of individuals. *Number of painful crises per year. The pain rate was defined as a visit to a medical facility that lasted more than four hours for acute sickling-related pain, which was treated with a parenterally administered narcotic.
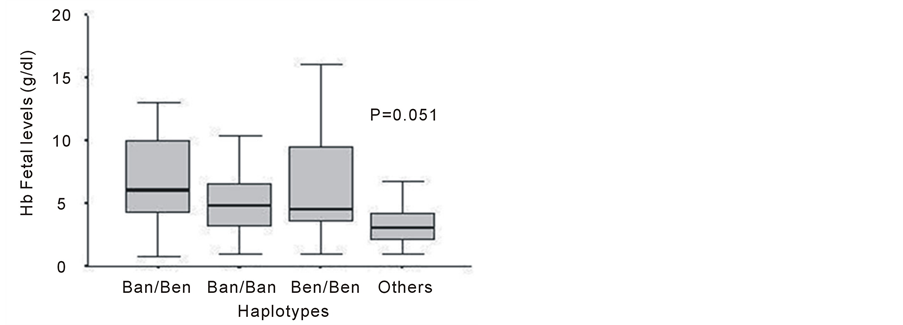
Figure 1. Association between fetal hemoglobin (HbF) levels and haplotypes of patients with sickle cell anemia. Ban: Bantu; Ben: Benin.
4. Discussion
The frequency of βS haplotypes and its correlation with clinical severity, as defined by at least two veno-occlusive episodes per year, HbF concentration, cardiovascular events, and SeS were determined in 75 SCA Southern Brazilian patients who had never received HU.
Our data confirm the prevalence of the Bantu haplotype, followed by Benin, as previously observed by Wagner et al. [29] in the southern region of Brazil, and described in other Brazilian regions [30] . However, our data differ from those reported for North American and Jamaican populations, where Benin haplotypes are more common, reflecting a preference for the traffic of Midwestern Africans to those regions during the British Atlantic slave trade [31] . Such difference is probably due to the African ancestry of our patients. Approximately 2.5 - 4.0 million Africans were brought as slaves to Brazil and distributed in all regions of the country [32] . As a result, most chromosomes with the βS gene have one of the five common haplotypes, and in large series of
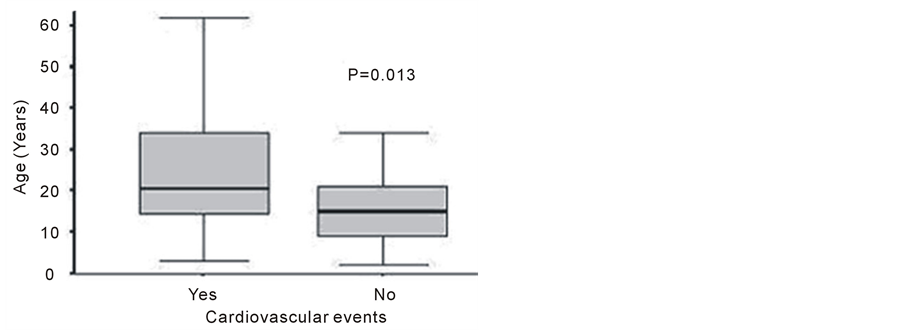
Figure 2. Association between age of patients with sickle cell anemia and cardiovascular events. The median age is indicated in each group.
Brazilian SCA patients, a small proportion of chromosomes (5% - 10%) is associated with less common haplotypes, referred to as atypical haplotypes [33] .
In the present study, 50 (66.7%) patients had a pain rate ≥ 2, which characterizes a severe clinical course (Table 1), and this condition was not associated with the haplotype. There may have been a possible bias because our participants belonged predominantly to the lowest SeS levels, and usually only the severely affected patients regularly attend their appointments at the institution.
In our group of patients, individuals carrying the Bantu/Bantu and Bantu/Benin haplotypes, although not statistically significant, appeared to have a closer relationship with cardiovascular complications than the remaining patients (Table 1). Likewise, as expected, we found a higher frequency of cardiovascular complications among older patients as compared to younger individuals (P = 0.013) (Figure 2).
High HbF levels have been associated with fewer vaso-occlusive events and acute chest syndrome (ACS) episodes and reduced mortality rate [25] . However, no association between HbF levels and number of painful crises (Table 2) or haplotypes was observed among our patients (Table 1). Interestingly, HbF levels were higher among our patients with the Bantu/Bantu genotype (Table 1). Twenty-eight (90.3%) Bantu/Bantu patients showed HbF levels above 2 g/dL, as compared to patients carrying Bantu/Benin, Benin/Benin, or other haplotypes (mean of 4.73, ranging from 3.0 to 7.0). A non-significant, but borderline association indicated that haplotypes referred to here as “others” showed lower HbF levels (P = 0.051) (Figure 1). HbF production is restricted to a small number of erythroid precursors; their progeny in the blood are called F-cells. Both HbF concentration and its distribution amongst erythrocytes are heritable. The distributions of HbF/F-cell among SCA patients are extremely variable regardless of their total HbF level. Even patients with high HbF can have severe disease since HbF is unevenly distributed among F-cells and some cells might have insufficient concentrations to inhibit HbS polymerization (Steinberg, 2013). Elevated HbF levels associated with this disease may also be secondary to the accelerated destruction and rapid regeneration of red blood cells. This state of stress erythropoiesis leads to enhanced production of HbF and F cells [34] . Gonçalves et al. [31] also observed a high HbF concentration in Bantu/Bantu individuals, who usually present average levels below 5% [35] . Also, in a study conducted in Lebanon, a higher HbF level was not considered a marker of low severity in SCA patients, as higher HbF levels were surprisingly associated with more severe clinical cases, suggesting that increased HbF levels and genetic factors other than haplotypes were the major determinants of SCA severity in that population [3] . HbF gene expression is regulated by elements linked to the β-globin gene complex that are associated with the β-globin gene haplotype, and by trans-acting elements associated with the BCL11A gene on chromosome 2p16 and the HBS1L-MYB intergenic region on chromosome 6q23 (Steinberg, 2013). Some authors propose that the distribution of HbF concentrations among F-cells is the most critical element in the pathophysiology of sickle cell anemia (Chang, 1997 and Steinberg, 2013). According to Steinberg et al. 2013, total HbF or F-cell percentages are often not a good predictor of disease severity in an individual with sickle cell anemia because they provide no information on the number of F-cells that have levels of HbF sufficient to protect against polymer-induced damage.
Genetic analysis of HbF regulation is therefore key to understanding the spectrum of SCA severity. Regulation of the expression of gamma-globin and suppression of beta-globin occurs by combined transcriptional regulation, chromosome remodeling, and other epigenetic factors and by the kinetics of red blood cell regeneration and differentiation [36] . Current investigation to identify determinants of HbF regulation and its genetic epidemiology has used thousands of single-nucleotide polymorphisms (SNPs) to screen for regions of interest throughout the genome (genome-wide association studies, GWAS) [36] . Comparative GWAS have revealed three different genes or gene loci as most strongly associated with the variation in HbF levels: BCL11A, HBS1LMYB, and the beta-globin cluster itself [36] [37] .
Finally, it is necessary to acknowledge the importance of a proinflammatory state in SCA patients that appears to play a pathophysiological role as well. Chies et al. [38] observed that an SCA patient became asymptomatic after a kidney transplant surgery and suggested that it could be related to the immunosuppressive therapy received by the patient after the procedure. Although we could not find a significant association between SeS and clinical severity in this study, it is worth pointing out that most of our patients belonged to C and D socioeconomic levels, precluding the evaluation of the actual impact of better sanitation and education on clinical manifestations of the disease, as we have done in an earlier study in which SeS was shown to significantly influence the outcome of bone marrow transplantation in the same hospital (Silla et al. 2009).
5. Conclusions
Our data corroborate the prevalence of Bantu haplotypes among Afro-Brazilians, which is consistent with the origins of the African slaves brought to Brazil during the slave trade. However, no relationship was observed between β haplotypes or HbF levels and the severity of SCA. Moreover, the small number of patients in the upper socioeconomic levels, with good sanitation and higher education level, prevented us from demonstrating a possible protective effect of higher SeS on clinical manifestations. In a time when HSCT is emerging as the only curative treatment for SCA, it is mandatory to find determinants of disease severity, since the procedure is associated with a high morbidity and mortality burden and should be performed early in the course of the disease to avoid end-organ damage before transplantation. Gene polymorphism studies concerning the loci for HbF are underway and will certainly bring new insights into SCA pathophysiology; nevertheless, in this study we showed that HbF levels, in accordance to what was described in Lebanon [3] , might not be an independent marker of clinical severity in all populations of SCA patients.
Studies on genetic polymorphisms are necessary to better understand SCA genetic variations. The genetic modulators identification of disease severity could guide prognosis to determine preventive measures and the best treatment for acute and chronic organ damage. Also, as the proinflammatory state appears to play pathophysiological role in SCA patients and it is related to socioeconomic levels it is important to consider these levels in the bone marrow transplantation scenary.
Acknowledgements
We are thankful to Meide Daniele Urnau, Valéria Doliwa Wislocki, Jóice Merzoni, Rafael Teixeira de Souza for their assistance in data and sample collection, and to Ceres Oliveira for her assistance in the statistical analysis. This study was funded by Research and Event Incentive Fund of Hospital de Clínicas de Porto Alegre (FIPEHCPA), by Hematology Friends Association of Hospital de Clinicas de Porto Alegre (HEMOAMIGOS-HCPA) and by the Laboratory of Im munogenetics, Department of Genetics, Universidade Federal do Rio Grande do Sul (UFRGS).
References
- Steinberg, M.H. and Rodgers, G.P. (2001) Pathophysiology of Sickle Cell Disease: Role of Cellular and Genetic Modifiers. Seminars in Hematology, 38, 299-306. http://dx.doi.org/10.1016/S0037-1963(01)90023-X
- Hanchard, N., Elzein, A., Trafford, C., Rockett, K., Pinder, M., Jallow, M., Harding, R., Kwiatkowski, D. and McKenzie, C. (2007) Classical Sickle Beta-Globin Haplotypes Exhibit a High Degree of Long-Range Haplotype Similarity in African and Afro-Caribbean Populations. BMC Genetics, 8, 52.
- Inati, A., Taher, A., Bou Alawi, W., Koussa, S., Kaspar, H., Shbaklo, H. and Zalloua, P.A. (2003) Beta-Globin Gene Cluster Haplotypes and Hbf Levels Are Not the Only Modulators of Sickle Cell Disease in Lebanon. European Journal of Haematology, 70, 79-83. http://dx.doi.org/10.1034/j.1600-0609.2003.00016.x
- Kato, G.J., Gladwin, M.T. and Steinberg, M.H. (2007) Deconstructing Sickle Cell Disease: Reappraisal of the Role of Hemolysis in the Development of Clinical Subphenotypes. Blood Reviews, 21, 37-47. http://dx.doi.org/10.1016/j.blre.2006.07.001
- Rosse, W.F., Narla, M., Petz, L.D. and Steinberg, M.H. (2000) New Views of Sickle Cell Disease Pathophysiology and Treatment. Hematology/The Education Program of the American Society of Hematology, 2-17. http://dx.doi.org/10.1182/asheducation-2000.1.2
- Chies, J.A. and Nardi, N.B. (2001) Sickle Cell Disease: A Chronic Inflammatory Condition. Medical Hypotheses, 57, 46-50. http://dx.doi.org/10.1054/mehy.2000.1310
- Platt, O.S. (2000) Sickle Cell Anemia as an Inflammatory Disease. Journal of Clinical Investigation, 106, 337-338. http://dx.doi.org/10.1172/JCI10726
- Hebbel, R.P., Osarogiagbon, R. and Kaul, D. (2004) The Endothelial Biology of Sickle Cell Disease: Inflammation and a Chronic Vasculopathy. Microcirculation, 11, 129-151. http://dx.doi.org/10.1080/mic.11.2.129.151
- Ballas, S.K. (1991) Sickle Cell Anemia with Few Painful Crises Is Characterized by Decreased Red Cell Deformability and Increased Number of Dense Cells. American Journal of Hematology, 36, 122-130. http://dx.doi.org/10.1002/ajh.2830360211
- Steinberg, M.H. (2005) Predicting Clinical Severity in Sickle Cell Anaemia. British Journal of Haematology, 129, 465-481. http://dx.doi.org/10.1111/j.1365-2141.2005.05411.x
- Adekile, A.D. (2005) Mild-Phenotype Sickle Cell Disease: Molecular Basis, Clinical Presentation and Management Recommendations. Current Paediatrics, 15, 57-61.
- Zago, M.A. Figueiredo, M.S. and Ogo, S.H. (1992) Bantu βS Cluster Haplotype Predominates among Brazilian Blacks. American Journal of Physical Anthropology, 88, 295-298. http://dx.doi.org/10.1002/ajpa.1330880304
- Steinberg, M.H., Nagel, R.L., Lawrence, C., Swaminathan, V., Lu, Z.H., Plonczynski, M. and Harrell, A. (1996) Beta-Globin Gene Haplotype in Hb SC Disease. American Journal of Hematology, 52, 189-191 http://dx.doi.org/10.1002/(SICI)1096-8652(199607)52:3<189::AID-AJH9>3.0.CO;2-P
- Rusanova, I., Escames, G., Cossio, G., de Borace, R.G., Moreno, B., Chahboune, M., Lopez, L.C., Diez, T. and Acuna-Castroviejo, D. (2010) Oxidative Stress Status, Clinical Outcome, and Beta-Globin Gene Cluster Haplotypes in Pediatric Patients with Sickle Cell Disease. European Journal of Haematology, 85, 529-537. http://dx.doi.org/10.1111/j.1600-0609.2010.01528.x
- Figueiredo, M.S., Kerbauy, J., Goncalves, M.S., Arruda, V.R., Saad, S.T., Sonati, M.F., Stoming, T. and Costa, F.F. (1996) Effect of Alpha-Thalassemia and Beta-Globin Gene Cluster Haplotypes on the Hematological and Clinical Features of Sickle-Cell Anemia in Brazil. American Journal of Hematology, 53, 72-76. http://dx.doi.org/10.1002/(SICI)1096-8652(199610)53:2<72::AID-AJH3>3.0.CO;2-0
- Pagnier, J., Mears, J.G., Dunda-Belkhodja, O., Schaefer-Rego, K.E., Beldjord, C., Nagel, R.L. and Labie, D. (1984) Evidence for the Multicentric Origin of the Sickle Cell Hemoglobin Gene in Africa. Proceedings of the National Academy of Sciences of the United States of America, 81, 1771-1773. http://dx.doi.org/10.1073/pnas.81.6.1771
- Magana, M.T., Ongay, Z., Tagle, J., Bentura, G., Cobian, J.G., Perea, F.J., Casas-Castaneda, M., Sanchez-Lopez, Y.J. and Ibarra, B. (2002) Analysis of βS and βA Genes in a Mexican Population with African Roots. Blood Cells, Molecules, and Diseases, 28, 121-126. http://dx.doi.org/10.1006/bcmd.2002.0498
- Nagel, R.L. and Steinberg, M.H. (2001) Role of Epistatic (Modifier) Genes in the Modulation of the Phenotypic Diversity of Sickle Cell Anemia. Fetal and Pediatric Pathology, 20, 123-136. http://dx.doi.org/10.3109/15513810109168606
- Powars, D., Chan, L.S. and Schroeder, W.A. (1990) The Variable Expression of Sickle Cell Disease Is Genetically Determined. Seminars in Hematology, 27, 360-376.
- Okany, C.C. and Akinyanju, O.O. (1993) The Influence of Socio-Economic Status on the Severity of Sickle Cell Disease. African Journal of Medicine and Medical Sciences, 22, 57-60.
- Animasahun, B.A., Temiye, E.O., Ogunkunle, O.O., Izuora, A.N. and Njokanma, O.F. (2011) The Influence of Socioeconomic Status on the Hemoglobin Level and Anthropometry of Sickle Cell Anemia Patients in Steady State at the Lagos University Teaching Hospital. Nigerian Journal of Clinical Practice, 14, 422-427. http://dx.doi.org/10.4103/1119-3077.91748
- Ellison, A.M. and Bauchner, H. (2007) Socioeconomic Status and Length of Hospital Stay in Children with Vaso-Occlusive Crises of Sickle Cell Disease. Journal of the National Medical Association, 99, 192-196.
- Ware, R.E., Eggleston, B., Redding-Lallinger, R., Wang, W.C., Smith-Whitley, K., Daeschner, C., Gee, B., Styles, L.A., Helms, R.W., Kinney, T.R. and Ohene-Frempong, K. (2002) Predictors of Fetal Hemoglobin Response in Children with Sickle Cell Anemia Receiving Hydroxyurea Therapy. Blood, 99, 10-14. http://dx.doi.org/10.1182/blood.V99.1.10
- Charache, S., Terrin, M.L., Moore, R.D., Dover, G.J., Barton, F.B., Eckert, S.V., McMahon, R.P. and Bonds, D.R. (1995) Effect of Hydroxyurea on the Frequency of Painful Crises in Sickle Cell Anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. New England Journal of Medicine, 332, 1317-1322. http://dx.doi.org/10.1056/NEJM199505183322001
- Kinney, T.R., Helms, R.W., O’Branski, E.E., Ohene-Frempong, K., Wang, W., Daeschner, C., Vichinsky, E., Redding-Lallinger, R., Gee, B., Platt, O.S. and Ware, R.E. (1999) Safety of Hydroxyurea in Children with Sickle Cell Anemia: Results of the HUG-KIDS Study, a Phase I/II Trial. Pediatric Hydroxyurea Group. Blood, 94, 1550-1554.
- (2012) CEB—Critério de Classificacao Economica Brasil. http://www.marketanalysis.com.br/arquivos-download/biblioteca/cceb-1.pdf
- Lahiri, D.K. and Nurnberger Jr., J.I. (1991) A Rapid Non-Enzymatic Method for the Preparation of HMW DNA from Blood for RFLP Studies. Nucleic Acids Research, 19, 5444. http://dx.doi.org/10.1093/nar/19.19.5444
- Sutton, M., Bouhassira, E.E. and Nagel, R.L. (1989) Polymerase Chain Reaction Amplification Applied to the Determination of β-Like Globin Gene Cluster Haplotypes. American Journal of Hematology, 32, 66-69. http://dx.doi.org/10.1002/ajh.2830320113
- Wagner, S.C., Friedrisch, J.R., Job, F. and Hutz, M.H. (1996) Caracterizacao molecular da anemia falciforme em pacientes de Porto Alegre. Revista Brasileira de Genetica, 19, 244.
- Lemos Cardoso, G. and Farias Guerreiro, J. (2006) African Gene Flow to North Brazil as Revealed by HBB*S Gene Haplotype Analysis. American Journal of Human Biology, 18, 93-98. http://dx.doi.org/10.1002/ajhb.20467
- Goncalves, M.S., Bomfim, G.C., Maciel, E., Cerqueira, I., Lyra, I., Zanette, A., Bomfim, G., Adorno, E.V., Albuquerque, A.L., Pontes, A., Dupuit, M.F., Fernandes, G.B. and dos Reis, M.G. (2003) βS-Haplotypes in Sickle Cell Anemia Patients from Salvador, Bahia, Northeastern Brazil. Brazilian Journal of Medical and Biological Research, 36, 1283-1288. http://dx.doi.org/10.1590/S0100-879X2003001000001
- Curtain, P.D. (1972) The Atlantic Slave Trade: A Census. University of Wisconsin Press, Wisconsin.
- Zago, M.A., Silva Jr., W.A., Dalle, B., Gualandro, S., Hutz, M.H., Lapoumeroulie, C., Tavella, M.H., Araujo, A.G., Krieger, J.E., Elion, J. and Krishnamoorthy, R. (2000) Atypical βS Haplotypes Are Generated by Diverse Genetic Mechanisms. American Journal of Hematology, 63, 79-84. http://dx.doi.org/10.1002/(SICI)1096-8652(200002)63:2<79::AID-AJH4>3.0.CO;2-D
- Bank, A. (2006) Regulation of Human Fetal Hemoglobin: New Players, New Complexities. Blood, 107, 435-443. http://dx.doi.org/10.1182/blood-2005-05-2113
- Powars, D.R. (1991) βS-Gene-Cluster Haplotypes in Sickle Cell Anemia. Clinical and Hematologic Features. Hematology/Oncology Clinics of North America, 5, 475-493.
- Green, N.S. and Barral, S. (2011) Genetic Modifiers of HbF and Response to Hydroxyurea in Sickle Cell Disease. Pediatric Blood & Cancer, 56, 177-181. http://dx.doi.org/10.1002/pbc.22754
- Bauer, D.E. and Orkin, S.H. (2011) Update on Fetal Hemoglobin Gene Regulation in Hemoglobinopathies. Current Opinion in Pediatrics, 23, 1-8. http://dx.doi.org/10.1097/MOP.0b013e3283420fd0
- Chies, J.A., Dresch, C., Cruz, M.S., da Silva, G., Barros, E., Bittar, C., Friedrisch, J. and Silla, L.M. (2005) Immunosuppressive Therapy for Kidney Transplant Prevents Vaso-Occlusive Crisis in a Haemoglobin SC Disease Patient. Medical Hypotheses, 64, 174-176. http://dx.doi.org/10.1016/j.mehy.2004.05.004
NOTES

*Corresponding author.