Open Journal of Orthopedics
Vol.2 No.4(2012), Article ID:26302,6 pages DOI:10.4236/ojo.2012.24026
Rare Occurrence of Primary Chest Wall Bone Tumours
![]()
1Department of Pathology, Midnapore Medical College & Hospital, Paschim Medinipur, India; 2Department of Pathology, RG Kar Medical College & Hospital, Kolkata, India.
Email: bhaskarmitra12@gmail.com
Received September 18th, 2012; revised October 18th, 2012; accepted November 16th, 2012
Keywords: Primary Chest Wall Tumours; Mesenchymal Chondrosarcoma; Clear Cell Chondrosarcoma; Aggressive Osteoblastoma
ABSTRACT
Primary tumors involving the bony skeleton of the chest wall are uncommon. After a retrospective review of the histopathology archive at our institution from Oct. 08 to Mar. 09 we found three malignant neoplasms affecting the rib, clavicle and the costal cartilages, one mesenchymal chondrosarcoma, one case of clear cell chondrosarcoma and an aggressive osteoblastoma each having distinctive histological diagnosis. Our limited experience in the occurrence of primary chest wall tumors showed that there are occurrences of rare bony lesions. A strong clinical suspicion, clinico-radiological correlation and histopathological confirmation are required for proper evaluation.
1. Introduction
Primary tumors involving the bony skeleton of the chest wall are uncommon. The most common primary malignant tumour of the chest wall is chondrosarcoma [1]. Less common primary bony chest wall tumors include Osteosarcoma and Ewing’s sarcoma/primitive neuroectodermal tumour (PNET). Multiple myeloma is a malignnant tumour of plasmacytes most commonly seen in the bone marrow, frequently involves the chest wall resulting in lytic lesions and bony expansion. After a retrospective review of the histopathology archive at our institution from Oct. 08 to Mar. 09 we found three malignant neoplasms affecting the bony skeleton of the chest wall and the costal cartilages, one mesenchymal chondrosarcoma, one case of clear cell chondrosarcoma and an aggressive osteoblastoma. All of the three cases have distinctive histopathological features allowing a histological diagnosis to be suggestive.
2. Case Reports
Clear cell chondrosarcoma is a rare mesenchymal neoplasm of unclear differentiation. Besides having a chondrogenic nature an osteogenic differentiation was also proposed. We have got a case of Clear cell chondrosarcoma in a 55 year male with chest wall swelling. The patient presented with a well defined lytic lesion of right upper chest wall grossly which was attached with rib and soft tissue measuring 3 × 3 × 0.2 cm. Contrast enhanced CT scan showing mass arising from rib with destruction of attached rib, mass showing heterogenous contrast enhancement with evidence of compression of mediastinal structures. (Figure 1(a)) On gross examination cut surface shows, bluish white, myxoid and gelatinous areas. Foci of hemorrhage and necrosis also noted. Microscopically the tumor was composed of malignant chondrocytes with an abundant clear cytoplasm and sharply defined border with areas of calcification (Figures 1(b)- (d)). Histological features are conclusive for clear cell chondrosarcoma.
Our second case, 25 year male presented with a chest wall mass of left side roentogenographically the clavicular lesion showed osteolytic appearances with stippled calcification (Figure 2(a)). Grossly a mass of 6 × 5 cm with bluish cartilaginous areas and chalky white calcification at places seen, in a surgically excised specimen, received in our department Microscopically the tumor is characterized by a dimorphic pattern in which areas of well differentiated cartilage alternate with undifferentiated stroma composed of small cells (Figures 2(b) and (c)). The proportion of two components varies considerably and boundaries between two are abrupt. The small cells show hyperchromatic nuclei and are usually round to oval with areas of frank spindling sometime found. The small cells are arranged around large, gaping, stag horn shaped vascular spaces like hemangiopericytoma (Figure 2(d)). Pleomorphism and mitotic activity are inconspicuous. Chondroid areas were evident. Features are diagnostic of Mesenchymal chondrosarcoma. Immunohistochemical study showed S100 positive chondroid areas
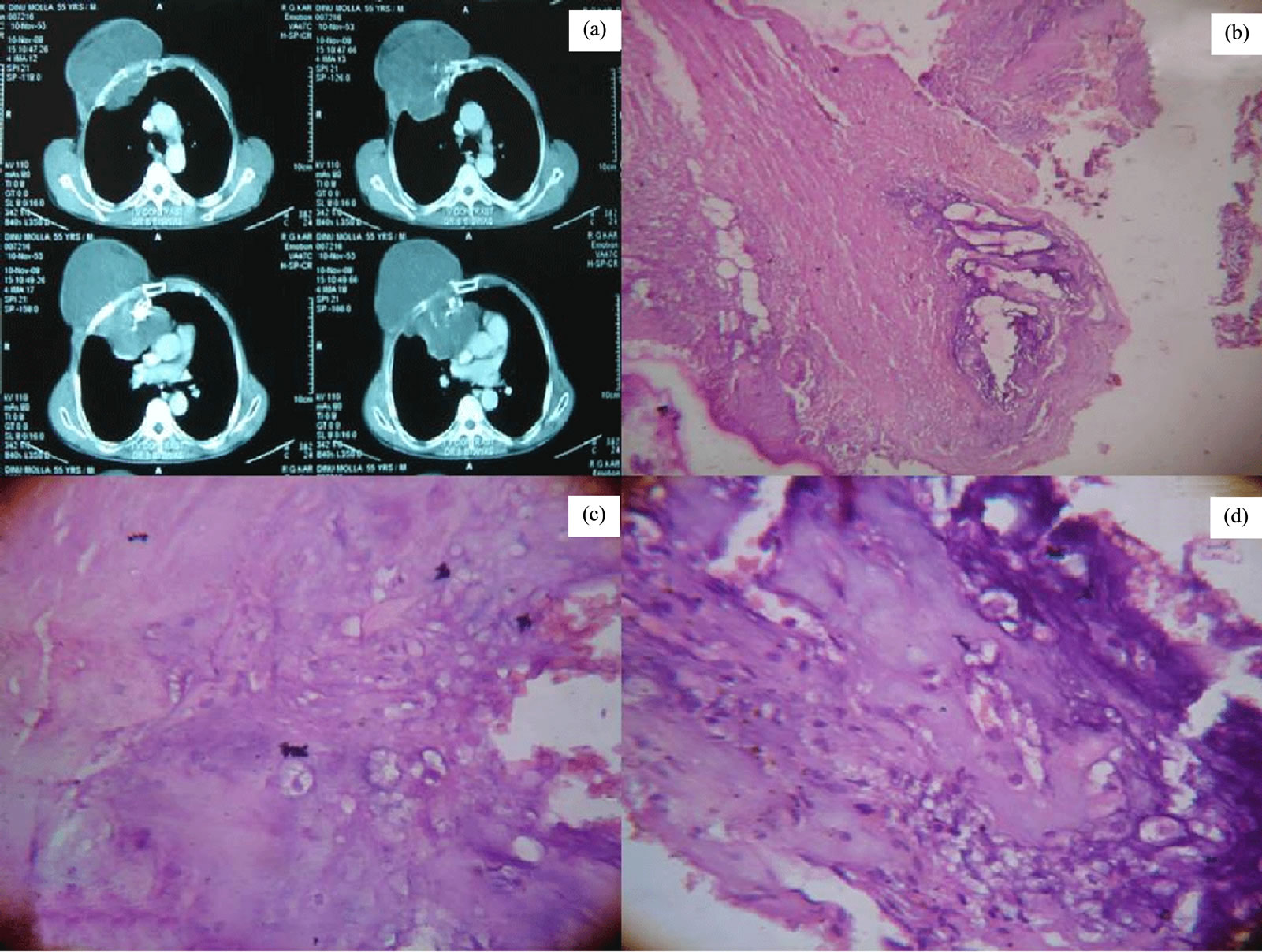
Figure 1. Contrast enhanced CT scan showing mass arising from rib (a). Photomicrograph shows, the tumor with areas of cartilaginous differentiation (Hematoxylin and eosin staining, ×100) (b) and composed of malignant chondrocytes with an abundant clear cytoplasm and sharply defined border (Hematoxylin and eosin staining, ×400) (c) & (d).
and small cell showing CD 99 reactivity, further confirming the histological diagnosis.
The third case was a 20 year female presented with a right chest wall mass of 13 × 6 cm in contrast enhanced CT scan (Figure 3(a)) with a history of recurrence of a similar lesion 8 year back. X ray shows a radiolucent central nidas surrounded by a peripheral sclerotic area. Initially FNAC was done stained smears show clusters of atypical cells with fragments of osteoid formation (Figure 3(b)). Excision of the tumour done, grossly a 12 × 5 cm bony mass with irregular surface seen. Microscopically the tumor was composed a bone-forming tumor with osteoblasts rimming osteoid and bony trabeculae (Figure 3(c)). The tumor consisted of predominantly disorganized osteoid surrounded by numerous large osteoblasts within the vascular stoma (Figure 3(d)). No mitosis or anaplasia was seen. These findings were compatible with aggressive osteoblastoma.
Treatment policy for all three cases was wide resection under general anesthesia, including the affected rib with at least a 4 - 5 cm free margin proximal and distal to the tumor, also resection of portions of the ribs immediately above and below the tumor, the adjacent muscles, and the pleura. In addition, the tissues adherent to the tumor was excised followed by chemo and radiotherapy. Post operative period of all the three cases were uneventful with no evidence of tumour recurrence till date.
3. Discussion
Primary tumors of bony skeleton of chest wall are uncommon, comprising 5% - 10% of all bony tumors. In the literature it is reported that malignant neoplasms are significantly more common than are benign ones [2]. However, Hsu and his friends reported that the ratio of malignant neoplasms to benign neoplasms is similar with the ratio of 1:1 [3]. In this study, 3 patients who were operated for a primary rib lesion were retrospectively evaluated, and their postoperative pathological features were classified. Literature review of such three cases was
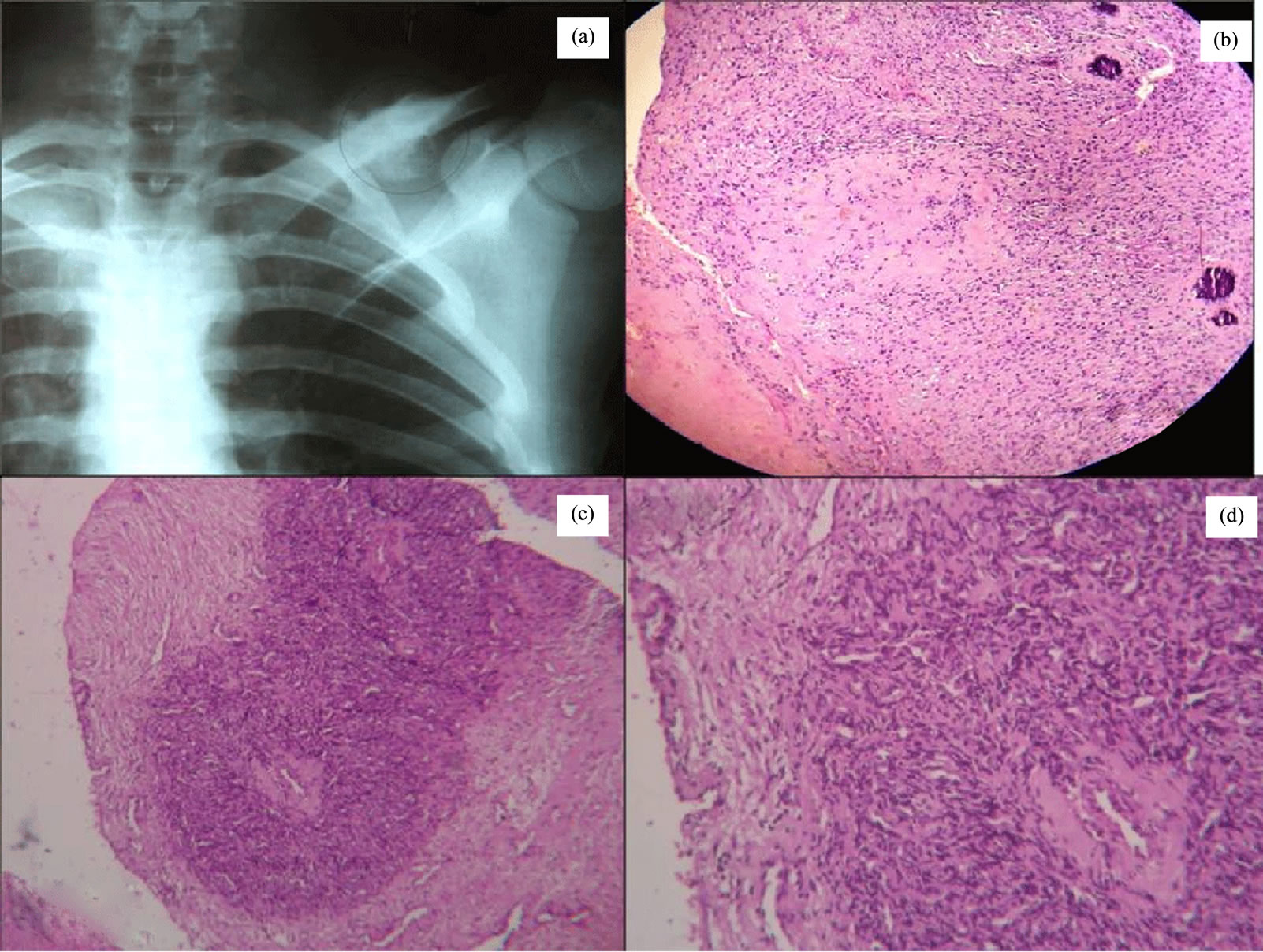
Figure 2. X ray shows, the clavicular osteolytic lesion with stippled calcification (a). Photomicrograph shows, the tumor with dimorphic pattern, areas of well differentiated cartilage alternate with undifferentiated stroma composed of small cells(Hematoxylin and eosin staining, ×100) (b) & (c). The small cells show hyperchromatic nuclei and are usually round to oval arranged around large, gaping, stag horn shaped vascular spaces (Hematoxylin and eosin staining, ×400) (d).
done and different aspect of individual cases was discussed separately.
Clear cell chondrosarcoma is a rare variant that has a relatively good prognosis compared to other variants [4]. Clear cell chondrosarcoma typically affects a younger age group has a better prognosis and is most commonly an epiphyseal lesion but our case showed higher age of incidence. It is a low grade cartilage neoplasm with round cells possessing clear ground glass cytoplasm and sharply defined border often interspersed with small trabeculae of woven bone. The majority of clear cell chondrosarcoma occur in the proximal end of the femur or humerus (in a juxta articular portion) in the third or fourth decades of life in contrast to chest wall swelling in our case. Radiologically these tumors are osteolytic, slightly expansile, sharply marginated and without evidence of matrix mineralization, grossly the tumor shows areas of hyaline cartilage that appear grey and fleshy with areas of cystic changes. Microscopically these tumors are lobulated with benign giant cells usually found at the edge of the lobules. The tumor cells have well defined cytoplasmic borders and a centrally placed round nucleus with occasional prominent nucleolus. Vascularity is a common feature of these tumors and Immunohistochemically exhibits immunoreactivity for S100 protein and collagen type II & X.
Mesenchymal chondrosarcoma is also rare highly malignant tumor first described by Lichtenstein and Bernstein (1959). [5] Most patients are of second or third decades of life with equal sex distribution as seen in our case. The bones most commonly affected are jaw, pelvis, femur, ribs and spine with involvement of some extraosseus structures like orbit, meninges etc. Occurrence in the rib is also quite rare. Radiologically the tumor presents with a permiative type of bone destruction with or without mineralization which was present in our case. Grossly the lesion is pink and fleshy but may show foci of calcification. Huvous et al. [6] have divided mesangial
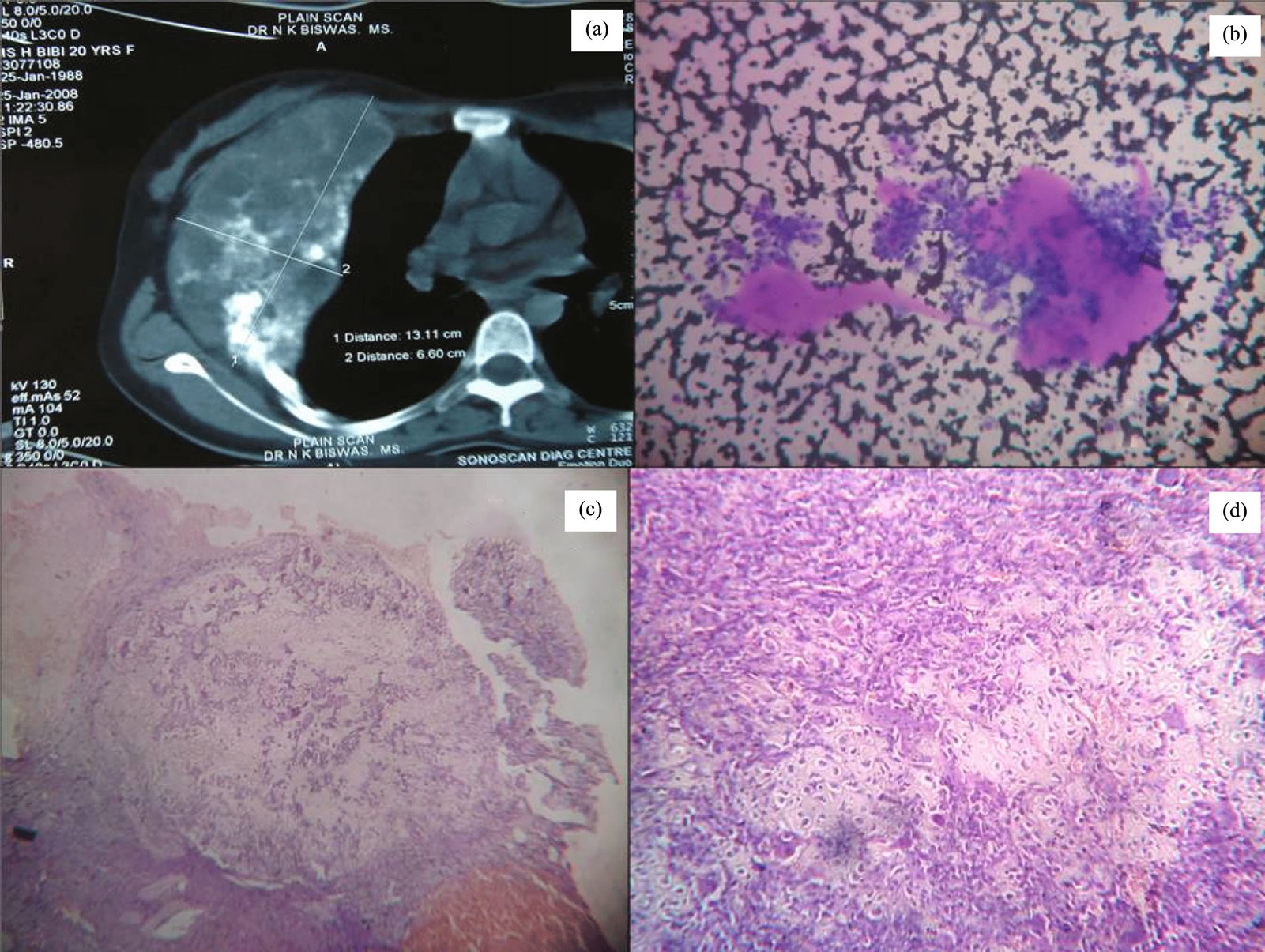
Figure 3. Contrast enhanced CT scan showing the tumour with heterogeneous echogenicity (a). Stained FNAC smears show clusters of atypical cells with fragments of osteoid formation (b). Photomicrographs shows a bone-forming tumor with osteoblasts rimming osteoid and bony trabeculae with disorganized osteoid (Hematoxylin and eosin staining, ×100) (c). Irregular woven bone and osteoid surrounded by activated osteoblasts (Hematoxylin and eosin staining, ×400) (d).
chondrosarcoma into two basic type; hemangiopericytoma like and small dark round cell type. Immunohistochemically, the cartilaginous areas are indistinguishable from other forms of chondrosarcoma. [7] The small cell component is positive for vimentin, CD 99 and Leu 7 [8]. Chondroid areas are positive for S100 protein. Mesenchymal chondrosarcoma is a highly aggressive tumor. In general, they have a metastatic rate higher than other cartilaginous malignancies and have an increased tendency to metastasize to the brain, lung, and liver. Metastasis to the lung is most common. The long-term prognosis is generally poor with 5- and 10-year survival rates of about 50% and 25%, respectively [9]. There are few published cytogenetic studies of mesenchymal chondrosarcoma. Recently, translocations involving chromosomes 13 and 21 ((13; 21) (q10; q10)) have been demonstrated in three tumors (two skeletal, one extraskeletal), possibly representing a chromosomal rearrangement characteristic of this neoplasm [10]. However, a single case of mesenchymal chondrosarcoma has also been reported to show the reciprocal translocation t (11; 22) (q24; q12) typical of Ewing’s sarcoma/peripheral primitive neuroectodermal tumor (PNET) [11], suggesting a possible relationship with this entity.
Osteoblastoma is an uncommon primary bone tumor. The most common sites are the vertebral column and long bone, followed by the feet, skull, clavicle and the ribs. The rib is involved in less than 5% of patients [12-14]. Patients are mostly less than 30 years of age, with approximately 70% of all cases occurring in the second or third decade of life. Males are affected more often than females in a ratio of approximately 2 to 1. Symptoms and signs of osteoblastoma are variable, but the characteristic symptom is a localized, dull pain that does not get worse at night in most cases and cannot be relieved by salicylates [15]. But our patient was a 20 years female. Osteoblastomas can be misdiagnosed as an osteosarcoma if correlation with clinical history, radiology, and histology is not carefully considered or if the several variants of osteoblastoma are not recognized. These variants lie on a morphologic spectrum between conventional osteoblastoma and osteosarcoma. Aggressive osteoblastoma is one such subtype. The histological features of aggressive osteoblastoma may appear malignant, and its biologic behavior may separate it from conventional osteoblastoma. The presence of epithelioid osteoblasts, lace-like or sheet-like osteoid production, and a permeative pattern of tumor growth in osteoblastomas is thought to be associated with an aggressive clinical behavior. The clinically aggressive behavior of osteoblastoma is not related to particular histological features, but rather to the skeletal location. Mitotic activity is not present in osteoblasts in the osteoblastoma. Epithelioid osteoblasts were detected in 14% of the cases without any mitotic activity. Lace-like or sheet-like osteoid was present in 36% of the cases studied [16]. Cytogenetic evaluation of the neoplasm revealed a pseudodiploid clone with a balanced translocation involving chromosomes 4, 7, and 14 [17].
We report an osteoblastoma of the rib occurring in a woman, which was osteolytic, expansile and well circumscribed with variable quantities of calcification or ossification areas. Certain typical CT and histological features of osteoblastoma are well demonstrated, but the ectogenic growth with expansion of the adjacent rib and female occurrence is very rare which has not been previously described.
4. Conclusion
Our limited experience in the management of primary rib tumors showed that there are occurrences of rare bony lesions in the rib. A strong clinical suspicion, clinicoradiological correlation and histopathological confirmation are required for proper evaluation. Consideration of such rare tumours must be there for proper management. Surgery must consist of wide resection with tumor-free margins in order to provide the best chance for cure in both benign and malignant lesions.
5. Acknowledgements
We like to thank all the staffs of department of Pathology, Midnapore Medical College & Hospital and RG Kar Medical College & Hospital.
REFERENCES
- H. Rupprecht, B. M. Spriewald and A. R. Hoffmann, “Successful Removal of Recurrent Giant Chondrosarcoma of the Thoracic Wall in a Patient with Hereditary Multiple Exostoses,” European Journal of Surgical Oncology, Vol. 27, No. 2, 2001, pp. 216-17. doi:10.1053/ejso.2000.1025
- H. I. Pass, “Primary and Metastatic Chest Wall Tumors,” In: J. A. Roth, J. C. Ruckdeschel and T. H. Weisenburger, Eds., Thoracic Oncology, WB Saunders, Philadelphia, 1995, pp. 519-537.
- P. K. Hsu, H. S. Hsu, H. C. Lee, C. C. Hsieh, et. al., “Management of Primary Chest Wall Tumors: 14 Years’ Clinical Experience,” Journal of the Chinese Medical Association, Vol. 69, No. 8, 2006, pp. 377-382. doi:10.1016/S1726-4901(09)70276-X
- J. Bjornsson, K. K. Unni, D. C.Dahlin, J. W. Beabout and F. H. Sim, “Clear Cell Chondrosarcoma of Bone: Observation in 47 Cases,” American journal of surgical pathology, Vol. 8, No. 3, 1984, pp. 223-230. doi:10.1097/00000478-198403000-00009
- L. Lichenstein and D. Berstein, “Unusual Benign and Malignant Chondroid Tumors of Bone. A Survey of Some Mesenchymal Cartilage Tumors and Malignant Chondroblastic Tumors, Including a Few Multicentric Ones, as Well as Many Atypical Benign Chondroblastomas and Chondromyxoid Fibromas,” Cancer, Vol. 12, No. 6, 1959, pp. 1142-1157. doi:10.1002/1097-0142(19830401)51:7<1230::AID-CNCR2820510710>3.0.CO;2-Q
- A. G. Huvous, G. Rosen, M. Dabaska et al., “Mesenchymal Chondrosarcoma. A Clinicopathologic Analysis of 35 Patients with Emphasis on Treatment,” Cancer, Vol. 51, No. 7, 1983, pp. 1230-1237.
- S. W. Weiss and J. R. Goldblum, “Enzinger and Weiss’s Soft Tissue Tumors,” 4th Edition, Mosby, St. Louis, 2001.
- P. E. Swanson, T. J. Lillemoe, J. C. Monivel and M. R. Wick, “Mesenchymal Chondrosarcoma. An Immunohistochemical Study,” The Archives of Pathology & Laboratory Medicine, Vol. 114, No. 9, 1990, pp. 943-948.
- H. D. Dorfman and B. Czerniak, “Malignant Cartilage Tumors,” In: H. D. Dorfman and B. Czerniak, Eds., Bone Tumors, Mosby, St. Louis, 1998, pp. 353-440.
- S. Naumann, P. A. Krallman, K. K. Unni, M. E. Fidler, J. R. Neff and J. A. Bridge, “Translocation der(13;21)(q10;q10) in Skeletal and Extraskeletal Chondrosarcoma,” Modern Pathology, Vol. 15, No. 5, 2002, pp. 572-576. doi:10.1038/modpathol.3880565
- L. Sainati, A. Scapinello, A. Montaldi, S. Blocato, V. Ninfo, M. Carli and G. A. Basso, “Mesenchymal Chondrosarcoma of a Child with the Reciprocal Translocation (11;22)(q24;q12),” Cancer Genetics and Cytogenetics, Vol. 71, No. 2, 1993, pp. 144-147. doi:10.1016/0165-4608(93)90020-M
- D. R. Lucas, K. K. Unni and R. A. McLeod, M. I. O’Connor and F. H. Sim, “Osteoblastoma: Clinicopathologic Study of 306 Cases,” Human Pathology, Vol. 25, No. 2, 1994, pp. 117-134. doi:10.1016/0046-8177(94)90267-4
- M. Berry, H. Mankin, M. Gebhardt, A. Rosenberg and F. Hornicek, “Osteoblastoma: A 30-Year Study of 99 Cases,” Journal of Surgical Oncology, Vol. 98, No. 3, 2008, pp. 179-183. doi:10.1002/jso.21105
- H. P. Tulloh and D. Harry, “Osteoblastoma in a Rib in Childhood,” Clinical Radiology, Vol. 20, No. 3, 1969, pp. 337-338. doi:10.1016/S0009-9260(69)80154-6
- H. M. Kroon and J. Schurmans, “Osteoblastoma: Clinical and Radiologic Findings in 98 New Cases,” Radiology, Vol. 175, No. 3, 1990, pp. 783-790.
- C. Della Rocca and A. G. Huvos, “Osteoblastoma: Varied Histological Presentations with a Benign Clinical Course. An Analysis of 55 Cases,” The American Journal of Surgical Pathology, Vol. 20, No. 7, 1996, pp. 841-850.
- A. C. Baker, L. Rezeanu, M. J. Klein, M. J. Pitt, P. Buecker, J. H. Hersh, J. J. Buchino and G. P. Siegal, “Aggressive Osteoblastoma: A Case Report Involving a Unique Chromosomal Aberration,” International Journal of Surgical Pathology, Vol. 18, No. 3, 2010, pp. 219-224.