Open Journal of Endocrine and Metabolic Diseases
Vol.05 No.04(2015), Article ID:55917,9 pages
10.4236/ojemd.2015.54006
Hyperglycemia Induced Changes in Vascular AKT3 May Inhibit Pressure-Induced Apoptosis in the Rat Inferior Venae Cavae
Kevin M. Rice1, Ravi Kumar Arvapalli1, Eric R. Blough1,2
1Center for Diagnostic Nanosystems, Marshall University, Huntington, WV, USA
2Department of Pharmacy Science and Research, School of Pharmacy, Marshall University, Huntington, WV, USA
Email: rice9@marshall.edu
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 27 March 2015; accepted 19 April 2015; published 23 April 2015
ABSTRACT
Background: Vein graft failure after bypass surgery is greatly increase in patients with diabetes mellitus. The cellular mechanisms underlying the cause of this failure are largely unexplored. Protein kinase B/AKT is a mechanically sensitive regulator of cellular growth and apoptosis. Herein we examine whether diabetes affects the regulation of AKT in response to increased venous loading. Methods: Inferior venae cavae (IVC) from the non-diabetic lean (LNZ) and the diabetic obese syndrome X Zucker(OSXZ) rats were isolated and incubated ex vivo under basal or pressurized conditions (120 mmHg). Protein expression, basal activation and the ability of increased pressure to activate AKT3 and apoptosis-related signaling were evaluated by immunoblot analysis. Results: Compared to that seen in the non-diabetic lean animals, increased venous pressure in the OSXZ rats was not characterized by increases in APAF-1 concentration, XIAP proteolysis, AIF cleavage, or Bad phosphorylation. This evidence of decreased apoptotic signaling was associated with increased basal p-AKT3 levels (+136% ± 13% P < 0.05 higher in the OSXZ vs. LNZ IVC). Conclusion: These data suggest that diabetes-associated increases in p-AKT3 may alter the ability of the IVC to undergo pressure induced apoptosis-related signaling. Further investigation is required to determine whether these changes are associated with the increased vein graft attrition seen in the diabetic population.
Keywords:
Diabetes, Zucker Rat, Inferior Venae Cavae, AKT3

1. Introduction
Diabetes is a primary risk factor for the development of coronary artery disease and venous graft failure due to stenosis [1] - [6] . Although it is currently unclear why diabetes may adversely affect vascular function, recent data has suggested that diabetes may affect how endothelial and smooth muscle cells (SMC) “sense” and “respond” to external stimuli [7] - [9] . Consistent with this notion, other data from our laboratory has demonstrated that the insulin resistant (IR) inferior venae cavae (IVC) of the obese syndrome X Zucker (OSXZ) rat differs from its lean (LNZ) counterpart in its ability to activate mitogen activated protein kinase (MAPK) signaling in response to an increase in vascular pressure [8] . Whether diabetes may affect the regulation of other mechanically-sensitive signaling pathways is currently unclear.
The protein kinase B (PKD/AKT) protein is thought to play an important role in the regulation of cell growth, proliferation, differentiation, motility, survival, metabolism and protein synthesis [10] - [13] . The phosphorylation (activation) of AKT can be induced by mechanical stimuli (stretch) or via growth factor stimulation [14] . Three AKT isoforms have been identified to date, AKT1, AKT2 and AKT3, each of which is encoded by a separate gene [15] . Of the three, AKT1 is the predominant isoform and is found in many different tissue types where it is thought to play a role in cell growth and survival [16] . Conversely, AKT2 is highly expressed in insulin-respon- sive tissues like muscle and adipocytes where it functions to regulate the uptake of glucose following insulin stimulation [17] . The distribution of AKT3 appears to be quite restricted as its expression is mainly confined to the testes and brain [17] . The role that the AKT1 and AKT3 isoforms may play in diabetes is not fully understood, however, recent data has reported that increased AKT1 levels can function to protect the cardiovascular system against pathological stimuli [18] , while AKT3 has been posited to play a role in brain development, the inhibition of foam cell formation [19] and mitochondrial biogenesis in endothelial cells [20] [21] .
Given the increased venous graft failure rate due to occlusion seen with diabetes and the potential role that the mechanically-sensitive AKT protein may play in regulating cell death, this study was designed to investigate if diabetes altered the regulation of increased load-induced (elevations in vascular pressure) AKT and apoptotic signaling in the rat inferior venae cavae (IVC). Similar to our previous work, we hypothesized that diabetes was associated with alterations in how the IVC responded to a loading stimulus.
2. Methods
2.1. Animals
All procedures were conducted in strict accordance with the Public Health Service policy on animal welfare. All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals as approved by the Council of the American Physiological Society and the Animal Use Review Board of Marshall University. Young (10 week, n = 12) male normal lean Zucker (non-diabetic, LNZ) (276 ± 19 g) and young (10 week, n = 12) male obese syndrome-X Zucker (diabetic, OSXZ) (347 ± 31 g) rats were obtained from the Charles River Laboratories and barrier housed one per cage in an AAALAC approved vivarium. Housing conditions consisted of a 12H: 12H dark-light cycle and temperature was maintained at 22˚C ± 2˚C. Animals were provided food and water ad libitum. Rats were allowed to recover from shipment for at least two weeks before experimentation during which time the animals were carefully observed and weighed weekly.
2.2. Materials
Antibodies against apoptotic protease activating factor 1 (APAF-1) [cat #4452], apoptosis inducing factor (AIF) [cat #4642], Cytochrome C [cat #4272], Caspase 12 [cat #2202], Bad [cat #9239], p-Bad (Ser 136) [cat #9295], B-cell lymphoma-extra large(BCL-XL) [cat #2762], X-linked inhibitor of apoptosis protein(XIAP) [cat #2042], pan AKT [cat #9272], p-AKT (Ser 308) [cat #9275], p-AKT (Thr 473) [cat #9271], Bad [cat #9292],, p70 S6 Kinase Control Cell Extracts [cat #9203], mouse IgG and rabbit IgG antibodies were purchased from Cell Signaling Technology (Beverly, MA). Antibody against B-cell lymphoma 3-encoded protein (Bcl-3) (H-146) [sc-13038] and AKT 3 (M-14) [sc-11521] were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Precast 10% and 15% SDS-PAGE gels were procured from Lonza (Rockland, ME). Enhanced chemiluminescence (ECL) western blotting detection reagent was from Amersham Biosciences (Piscataway, NJ). Restore western blot stripping buffer was obtained from Thermo scientific (Rockford, IL) and 3T3 cell lysates were from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals were purchased from Sigma (St. Louis, MO).
2.3. Inferior Venae Cavae Preparation
Rats were anesthetized with a ketamine-xylazine (4:1) cocktail (50 mg/kg ip) and supplemented as necessary for reflexive response. Collection and isolation of the inferior venae cavae were performed as described previously [8] . Inferior venae cavae were cannulated onto polystyrene tubing (outside diameter 3.0 mm; inner diameter 2.6 mm) with the aid of a dissection microscope. Mounted vessels were equilibrated, perfused, and subjected to 120 mm Hg of pressure for 30 minutes as described previously [8] .
2.4. Immunoblot Analysis
Vessels were immediately snap-frozen in liquid nitrogen at the end of each experiment. Protein isolates were prepared from collected venae cavae by pulverizing the samples under liquid nitrogen using a mortar and pestle and washed three times with ice cold phosphate buffered saline (PBS). T-
2.5. Immunoprecipitation of AKT3
Tissue lysates (2 ug/ul) were precleared using protein A Sepharose beads for 1 h at 4˚C on a rotatory shaker. AKT3 antibody conjugated to magnetic beads was added to the lysate and incubated overnight at 4˚C with rotation. Beads were magnetically separated and heated in SDS sample buffer and ran on a SDS-PAGE (10%) for immunoblotting.
2.6. Data Analysis
Results are presented as mean ±
3. Results
3.1. Verification of Loading Stimulus
Light microscope analysis demonstrated no apparent differences in gross morphology between venae cavae obtained from lean and obese Zucker rats (data not shown). Isolated vessels responded to stretch in a passive manner (data not shown). Continuous recording of the vena cava loading pressure was monitored throughout the loading procedure. The vessel was immediately discarded if fluctuations in stretch induced loading occurred.
3.2. Diabetes Alters the Pressure-Induced Activation of AKT3.
The ratio of p-AKT (Thr308) to total AKT was not different between the LNZ and OSXZ animals (Figure 1(a) and Figure 1(d)). The ratio of p-AKT (Ser 473) to total AKT was lower in the OSXZ IVC compared to that observed in the LNZ animals (28% ± 8%, P ≤ 0.05) (Figure 1(b) and Figure 1(d)). The ratio of p-AKT (Ser 473) to total AKT was decreased with pressurization in the lean (31% ± 6%, P ≤ 0.05) but not OSXZ IVC (Figure 1(b) and Figure 1(d)).
The ratio of p-AKT3 (Ser 473) to total AKT3 was higher in the OSXZ IVC (136% ± 13%, P ≤ 0.05) (Figure 1(c) and Figure 1(d)) compared to the LNZ IVC. The ratio of p-AKT3 (Ser 473) to total AKT3 was unchanged in the LNZ IVC with loading (Figure 1(c) and Figure 1(d)) and decreased in the OSXZ IVC (120% ± 16%, P ≤ 0.05) (Figure 1(c) and Figure 1(d)).
3.3. Diabetes Attenuates Pressure Induced Cytochrome C Release into the Cytosol
Compared to observations in the IVC obtained from the lean animals, diabetes did not alter the cytosolic content of cytochrome-C. Pressurization of the IVC resulted in an increase in cytosolic cytochrome-C in both the lean (99% ± 9.9%, P ≤ 0.05) and obese (51% ± 14.7%, P ≤ 0.05); however, the obese response was significantly less than that seen in the lean animals (Figure 2).

 (a) (b)
(a) (b)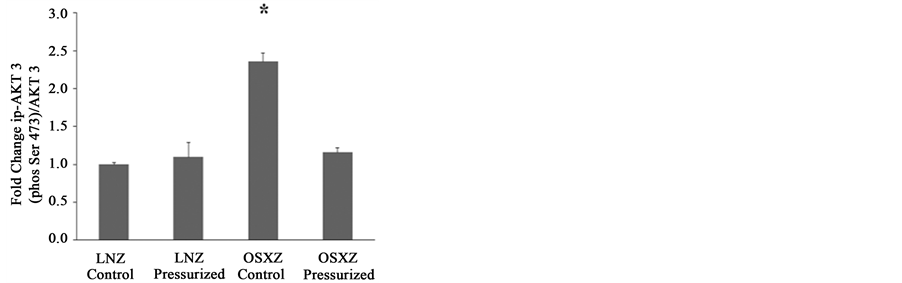
 (c) (d)
(c) (d)
Figure 1. AKT expression in normal and diabetic rat inferior venae cavae. Immunoblot analyses indicating protein content of (a) pan-p-AKT (Thr308); (b) pan-p-AKT (Ser 473); (c) p-AKT3 (Ser 473) and (d) their respective bands in venae cavae from non-diabetic lean Zucker (LNZ) and diabetic obese syndrome X Zucker (OSXZ) rats. *Significantly different from the non-diabetic control venae cavae (P ≤ 0.05), n = 6/group.
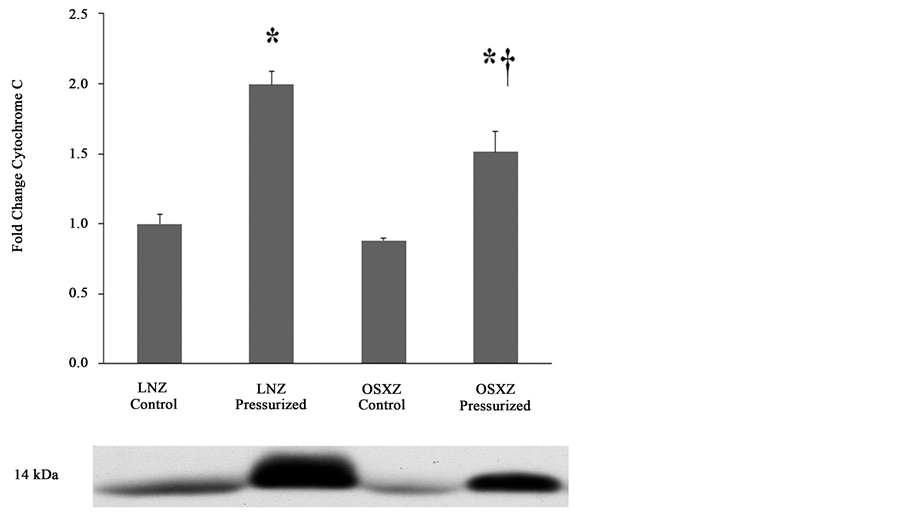
Figure 2. Diabetes attenuates loading-induced cytochrome-C release in the IVC. The basal (control) and pressure-induced cytochrome-C in venae cavae from non-diabetic lean Zucker (LNZ) and diabetic obese syndrome X Zucker (OSXZ) rats. *Significantly different from unloaded venae cavae within the same group (P < 0.05). †Significantly different from corresponding LNZ venae cavae (P ≤ 0.05), n = 6/group.
3.4. Diabetes Alters Regulation of Apoptotic Molecules in Pressurized Vena Cava
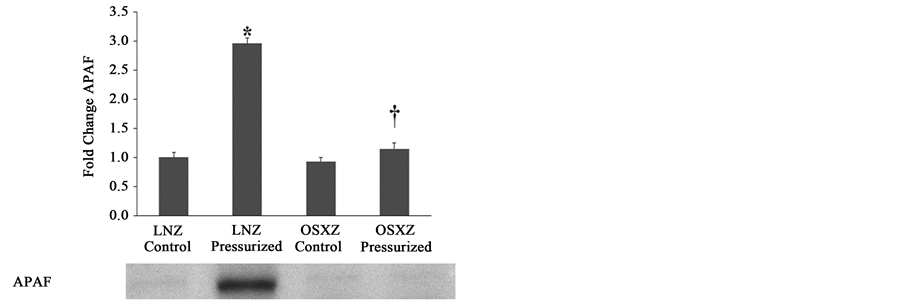
To investigate the influence of pressurization on the non-diabetes and diabetes on the expression and regulation of apoptotic associated proteins in the inferior venae cavae, we used immunoblotting to determine the total amount of APAF-1, AIF, and XIAP present in pressurized vena cava and non-pressurized (control) vena cava obtained from non-diabetic (LNZ) animals and diabetic (OSXZ) animals.
Compared to observations in the lean IVC preparations, diabetes had no effect on the cytosolic expression of APAF-1 (Figure 3(a)), however pressurization of the IVC in the control elicited an increase in cytosolic APAF-1 (196% ± 9.4%, P ≤ 0.05), whereas the pressurization in the diabetic IVC was not able to stimulate an increase in the cytosolic content of APAF-1 (Figure 3(a)).
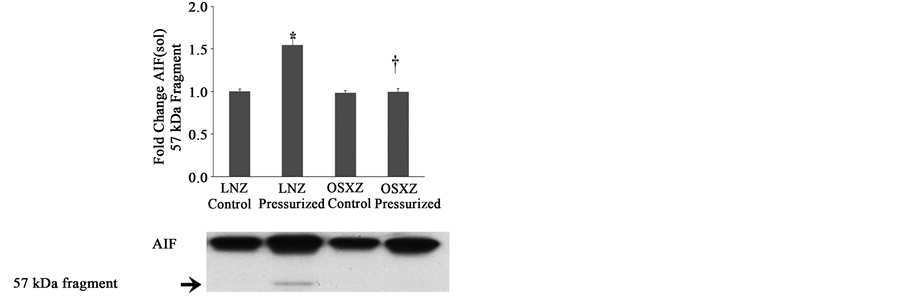
Compared to IVC preparations from lean animals, diabetes had no effect on the cytosolic content of AIF (67 kDa) or on AIF (sol) (57 kDa). Pressurization of the IVC obtained from lean animals resulted in an increase in the cytosolic content of AIF (32% ± 2.5%, P ≤ 0.05) and AIF (sol) (54% ± 8.8%, P ≤ 0.05) (Figure 3(b) and Figure 3(c)). Pressurization in the diabetic IVC resulted in an increase in AIF (24% ± 6.0%, P ≤ 0.05) cytosolic content with no detectable cytosolic change in AIF (sol) (Figure 3(b) and Figure 3(c)), however this induction of AIF was significantly less than seen in the lean animals.
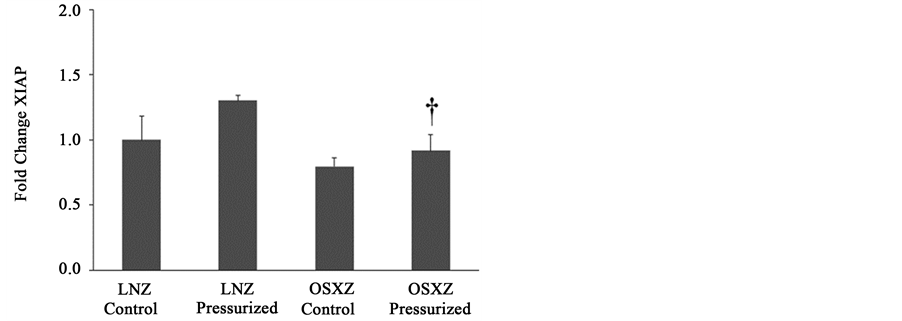
When compared to lean, diabetes did not alter the expression of XIAP or any fragments of XIAP (Figures 4(a)-(c)). Pressurization of the IVC from the lean and obese animals resulted in no change in XIAP expression. XIAP fragmentation was detected in pressurized IVC. Pressurization resulted in XIAP fragments of 45 kDa (338% ± 7.2%, p ≤ 0.05) when compared to non-pressurize IVC and 30 kDa (40% ± 9.5%, P ≤ 0.05) when compared to non-pressurized IVC in the lean, however in the obese only the 30 kDa fragment was detectable (30% ± 15.9%, P ≤ 0.05) (Figures 4(a)-(c)).
 (a)
(a)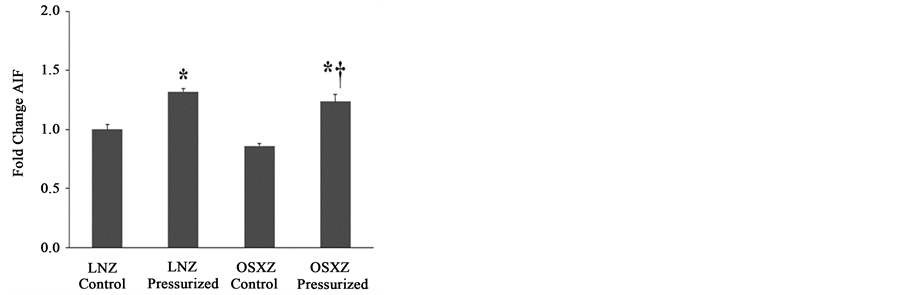
 (b) (c)
(b) (c)
Figure 3. Diabetes related alterations in loading-induced apoptotic associated proteins. The basal (control) and pressure- induced (a) APAF-1; (b) AIF; and (c) AIF 57 kDa fragment, in venae cavae from non-diabetic lean Zucker (LNZ) and diabetic obese syndrome X Zucker (OSXZ) rats. *Significantly different from unloaded venae cavae within the same group (P < 0.05). †Significantly different from corresponding non-diabetic venaecavae (P < 0.05). n = 6/group.
 (a)
(a)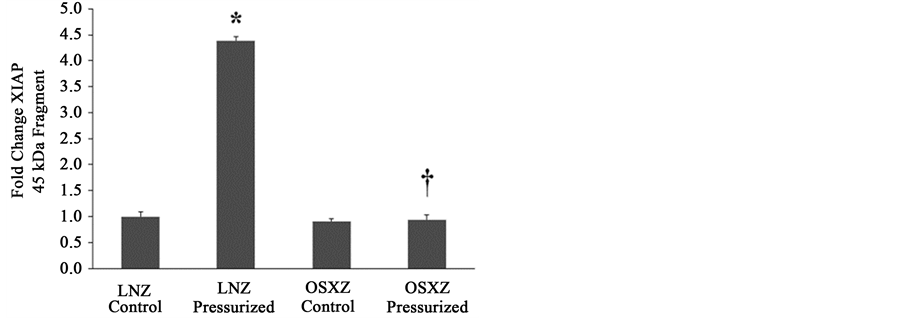 (b)
(b)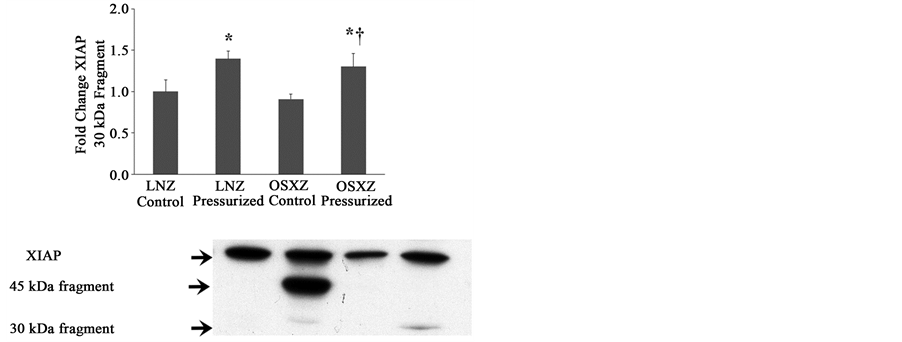 (c)
(c)
Figure 4. Diabetes related alterations in loading-induced XIAP cleavage. The basal (control) and pressure-induced (a) XIAP full length; (b) 45 kDa XIAP fragment; and (c) 30 kDa XIAP fragmentin venae cavae from non-diabetic lean Zucker (LNZ) and diabetic obese syndrome X Zucker (OSXZ) rats. *Significantly different from unloaded venae cavae within the same group (P < 0.05). †Significantly different from corresponding non-diabetic venae cavae (P < 0.05). n = 6/group.
4. Discussion
Diabetes is associated with increased vein graft neointimal hyperplasia and higher graft failure rates [22] . The findings of this study, and our previous [8] , may provide insight as to why differences may exist. Consistent with our past study we found that elevated vascular loading in the LNZ IVC, but not the OSXZ IVC, were associated with evidence of increased apoptotic signaling. These findings may have clinical application as they suggest that OSXZ IVC may be “hard wired” to respond differently to mechanical stimuli than that seen in the normal, non-obese animals. Whether or not we might demonstrate similar findings with the use of human venous preparations awaits further experimentation.
One of the primary findings of this study was that insulin resistance appeared to be associated with the up regulation of AKT3 protein levels in the rat IVC. AKT3 is the third member of the AKT serine-threonine protein kinase family. It has been reported that AKT3 deficient mice have normal glucose metabolism and normal body weight but that they exhibit a reduction in brain mass [23] . The different roles that AKT3 may play in maintaining cellular function have yet to be fully characterized. AKT3 has been found to mediate resistance to apoptosis in melanoma cells [24] and undergo amplification in some types of human breast cancer [25] . Constitutively active AKT3 has also been shown to decrease doxorubicin-induced cleavage of caspase-3 [26] . Why AKT3 expression may be increased in the OSXZ IVC is currently unclear but may be related to elevations in blood insulin levels as hyperinsulinemia has been found to be associated with increased tumor formation [27] - [29] . Supporting this contention, the obese Zucker rat is not only hyperinsulinemic but it also shows an increased susceptibility to DMBA-induced mammary tumor development [30] . Previous research has indicated that AKT3 may play a room in mitochondrial biogenesis in endothelial cells [31] . It has also been reported that the ability of AKT3 to regulate mitochondrial dynamics may be due to AKT3 regulation of chromosome maintenance regioin-1 (CRM-1) dependent nuclear export [20] . One report has indicated that AKT3 maybe localized in both the nucleus and nuclear membrane where it is regulated [32] . Additionally in humans the AKT3 gene has been shown to have a strong association with angiogenesis and high altitude sickness [33] . Whether the increased AKT3 expression we observed in the OSXZIVC is a compensatory response to elevations circulating in sulin levels, as a preventive mechanism to prevent tumor development, or as an adaptive measure to ensure adequate mitochondrial function under conditions of diminished carbohydrate availability is currently unclear and will required additional study.
To examine if the alterations in AKT3 were associated with differences in apoptotic signaling we first examined how increased loading affected cytosolic cytochrome-C levels. With increased IVC pressurization we found evidence of diminished levels of cytosolic cytochrome-C, apoptotic protease activating factor 1 (APAF-1), decreased cleavage of apoptosis inducing factor (AIF), and reduced levels of cleaved X-linked inhibitor of apoptosis protein (XIAP) in the OSXZ IVC compared to that observed in the IVC samples obtained from the LNZ animals (Figure 4). Cytochrome-C is a water soluble heme protein that is released from the mitochondria membrane into the cytosol where it assists APAF-1 and caspase-9 in forming the apoptosome [34] . The AIF is a mitochondrial localized flavor protein that is released from the mitochondria where it then triggers chromatin condensation and DNA degradation. It is thought that release of AIF is regulated, at least in part, following its cleavage by the calcium-dependent protease, calpain [35] [36] . Similar to AIF, XIAP is also regulated by cleavage. When intact, XIAP functions to inhibit apoptotic cell death by binding to and inhibiting caspases 3, 7, and 9 [37] .
5. Conclusion
These data, taken together, are consistent with our previous data showing an absence of load-induced caspase-3 fragmentation in the pressurized diabetic IVC [8] , suggesting that insulin resistance may render the IVC resistant to load-induced apoptosis. Whether these apoptotic-related differences cellular signaling between LNZ and OSXZ IVC are due solely to changes in the regulation of AKT3 alone or via alterations in other signaling pathways is currently unclear and awaits further study.
Acknowledgements
This work was supported in part from DOE grant (DE-PS02-09ER-01 to E.R.B).
Conflicting Interest
The authors declare that they have no competing interests. KMR, RKA carried out the molecular immunoassay studies, participated in the data analysis and drafted the manuscript. KMR, ERB carried out the animal experimentations, participated in the design of the study, performed the statistical analysis, conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
References
- Hambly, R.I., Sherman, L., Mehta, J. and Aintablian, A. (1976) Reappraisal of the Role of the Diabetic State in Coronary Artery Disease. Chest, 70, 251-257. http://dx.doi.org/10.1378/chest.70.2.251
- Waller, B.F., Palumbo, P.J., Lie, J.T. and Roberts, W.C. (1980) Status of the Coronary Arteries at Necropsy in Diabetes Mellitus with Onset after Age 30 Years. Analysis of 229 Diabetic Patients with and without Clinical Evidence of Coronary Heart Disease and Comparison to 183 Control Subjects. The American Journal of Medicine, 69,498-506. http://dx.doi.org/10.1016/S0149-2918(05)80002-5
- Woodfield, S.L., Lundergan, C.F., Reiner, J.S., Greenhouse, S.W., Thompson, M.A., Rohrbeck, S.C., Deychak, Y., Simoons, M.L., Califf, R.M., Topol, E.J., et al. (1996) Angiographic Findings and Outcome in Diabetic Patients Treated with Thrombolytic Therapy for Acute Myocardial Infarction: The GUSTO-I Experience. Journal of the American College of Cardiology, 28, 1661-1669. http://dx.doi.org/10.1016/S0735-1097(96)00397-X
- (2000) Correction: The Effect of Previous Coronary-Artery Bypass Surgery on the Prognosis of Patients with Diabetes Who Have Acute Myocardial Infarction. The New England Journal of Medicine, 343, 980. http://dx.doi.org/10.1056/NEJM200009283431320
- Detre, K.M., Lombardero, M.S., Brooks, M.M., Hardison, R.M., Holubkov, R., Sopko, G., Frye, R.L. and Chaitman, B.R. (2000) The Effect of Previous Coronary-Artery Bypass Surgery on the Prognosis of Patients with Diabetes Who Have Acute Myocardial Infarction. Bypass Angioplasty Revascularization Investigation Investigators. The New England Journal of Medicine, 342, 989-997. http://dx.doi.org/10.1056/NEJM200004063421401
- The BARI Investigators (1997) Influence of Diabetes on 5-Year Mortality and Morbidity in a Randomized Trial Comparing CABG and PTCA in Patients with Multivessel Disease: The Bypass Angioplasty Revascularization Investigation (BARI). Circulation, 96, 1761-1769.
- Mompeo, B., Popov, D., Sima, A., Constantinescu, E. and Simionescu, M. (1998) Diabetes-Induced Structural Changes of Venous and Arterial Endothelium and Smooth Muscle Cells. Journal of Submicroscopic Cytology and Pathology, 30, 475-484.
- Rice, K.M., Desai, D.H., Kakarla, S.K., Katta, A., Preston, D.L., Wehner, P. and Blough, E.R. (2006) Diabetes Alters Vascular Mechanotransduction: Pressure-Induced Regulation of Mitogen Activated Protein Kinases in the Rat Inferior Vena Cava. Cardiovascular Diabetology, 5, 18. http://dx.doi.org/10.1186/1475-2840-5-18
- Pei-Ling Chiu, A., Wang, F.L., Lal, N., Wang, Y., Zhang, D.H., Hussein, B., Wan, A. Vlodavsky, I. and Rodrigues, B. (2014) Endothelial Cells Respond to Hyperglycemia by Increasing the LPL Transporter GPIHBP1. American Journal of Physiology―Endocrinology and Metabolism, 306, E1274-E1283. http://dx.doi.org/10.1152/ajpendo.00007.2014
- Martelli, A.M., Tabellini, G., Bressanin, D., Ognibene, A., Goto, K., Cocco, L. and Evangelisti, C. (2012) The Emerging Multiple Roles of Nuclear AKT. Biochimica et Biophysica Acta (BBA)―Molecular Cell Research, 1823, 2168- 2178.http://dx.doi.org/10.1016/j.bbamcr.2012.08.017
- Buitenhuis, M. (2011) The Role of PI3K/Protein Kinase B (PKB/c-AKT) in Migration and Homing of Hematopoietic Stem and Progenitor Cells. Current Opinion in Hematology, 18, 226-230. http://dx.doi.org/10.1097/MOH.0b013e32834760e5
- Zhou, H.Y. and Huang, S.L. (2011) Role of mTOR Signaling in Tumor Cell Motility, Invasion and Metastasis. Current Protein & Peptide Science, 12, 30-42. http://dx.doi.org/10.2174/138920311795659407
- Pillai, V.B., Sundaresan, N.R. and Gupta, M.P. (2014) Regulation of AKT Signaling by Sirtuins: Its Implication in Cardiac Hypertrophy and Aging. Circulation Research, 114, 368-378. http://dx.doi.org/10.1161/CIRCRESAHA.113.300536
- Krepinsky, J.C., Li, Y., Chang, Y., Liu, L., Peng, F., Wu, D., Tang, D., Scholey, J. and Ingram, A.J. (2005) AKT Mediates Mechanical Strain-Induced Collagen Production by Mesangial Cells. Journal of the American Society of Nephrology, 16, 1661-1672. http://dx.doi.org/10.1681/ASN.2004100897
- Walker, K.S., Deak, M., Paterson, A., Hudson, K., Cohen, P. and Alessi, D.R. (1998) Activation of Protein Kinase B Beta and Gamma Isoforms by Insulin in Vivo and by 3-Phosphoinositide-Dependent Protein Kinase-1 in Vitro: Comparison with Protein Kinase B Alpha. Biochemical Journal, 331, 299-308.
- Tuttle, R.L., Gill, N.S., Pugh, W., Lee, J.P., Koeberlein, B., Furth, E.E., Polonsky, K.S., Naji, A. and Birnbaum, M.J. (2001) Regulation of Pancreatic Beta-Cell Growth and Survival by the Serine/Threonine Protein Kinase AKT1/PKBα. Nature Medicine, 7, 1133-1137. http://dx.doi.org/10.1038/nm1001-1133
- Chen, W.S., Peng, X.D., Wang, Y., Xu, P.Z., Chen, M.L., Luo, Y., Jeon, S.M., Coleman, K., Haschek, W.M., Bass, J., et al. (2009) Leptin Deficiency and Beta-Cell Dysfunction Underlie Type 2 Diabetes in Compound AKT Knockout Mice. Molecular and Cellular Biology, 29, 3151-3162. http://dx.doi.org/10.1128/MCB.01792-08
- O’Neill, B.T. and Abel, E.D. (2005) AKT1 in the Cardiovascular System: Friend or Foe? Journal of Clinical Investigation, 115, 2059-2064. http://dx.doi.org/10.1172/JCI25900
- Ding, L., Biswas, S., Morton, R.E., Smith, J.D., Hay, N., Byzova, T.V., Febbraio, M. and Podrez, E.A. (2012) AKT3 Deficiency in Macrophages Promotes Foam Cell Formation and Atherosclerosis in Mice. Cell Metabolism, 15, 861- 872. http://dx.doi.org/10.1016/j.cmet.2012.04.020
- Corum, D.G., Tsichlis, P.N. and Muise-Helmericks, R.C. (2014) AKT3 Controls Mitochondrial Biogenesis and Autophagy via Regulation of the Major Nuclear Export Protein CRM-1. The FASEB Journal, 28, 395-407. http://dx.doi.org/10.1096/fj.13-235382
- Wright, G.L., Maroulakou, I.G., Eldridge, J., Liby, T.L., Sridharan, V., Tsichlis, P.N. and Muise-Helmericks, R.C. (2008) VEGF Stimulation of Mitochondrial Biogenesis: Requirement of AKT3 Kinase. The FASEB Journal, 22, 3264- 3275. http://dx.doi.org/10.1096/fj.08-106468
- Nishio, K., Fukui, T., Tsunoda, F., Kawamura, K., Itoh, S., Konno, N., Ozawa, K. and Katagiri, T. (2005) Insulin Resistance as a Predictor for Restenosis after Coronary Stenting. International Journal of Cardiology, 103, 128-134. http://dx.doi.org/10.1016/j.ijcard.2004.08.039
- Easton, R.M., Cho, H., Roovers, K., Shineman, D.W., Mizrahi, M., Forman, M.S., Lee, V.M., Szabolcs, M., de Jong, R., Oltersdorf, T., et al. (2005) Role for AKT3/Protein Kinase Bγ in Attainment of Normal Brain Size. Molecular and Cellular Biology, 25, 1869-1878. http://dx.doi.org/10.1128/MCB.25.5.1869-1878.2005
- Shao, Y. and Aplin, A.E. (2010) AKT3-Mediated Resistance to Apoptosis in B-RAF-Targeted Melanoma Cells. Cancer Research, 70, 6670-6681. http://dx.doi.org/10.1158/0008-5472.CAN-09-4471
- Kirkegaard, T., Witton, C.J., Edwards, J., Nielsen, K.V., Jensen, L.B., Campbell, F.M., Cooke, T.G. and Bartlett, J.M. (2010) Molecular Alterations in AKT1, AKT2 and AKT3 Detected in Breast and Prostatic Cancer by FISH. Histopathology, 56, 203-211. http://dx.doi.org/10.1111/j.1365-2559.2009.03467.x
- Gagnon, V., Van Themsche, C., Turner, S., Leblanc, V. and Asselin, E. (2008) AKT and XIAP Regulate the Sensitivity of Human Uterine Cancer Cells to Cisplatin, Doxorubicin and Taxol. Apoptosis, 13, 259-271. http://dx.doi.org/10.1007/s10495-007-0165-6
- Novosyadlyy, R., Lann, D.E., Vijayakumar, A., Rowzee, A., Lazzarino, D.A., Fierz, Y., Carboni, J.M., Gottardis, M.M., Pennisi, P.A., Molinolo, A.A., et al. (2010) Insulin-Mediated Acceleration of Breast Cancer Development and Progression in a Nonobese Model of Type 2 Diabetes. Cancer Research, 70, 741-751. http://dx.doi.org/10.1158/0008-5472.CAN-09-2141
- Ferguson, R.D., Novosyadlyy, R., Fierz, Y., Alikhani, N., Sun, H., Yakar, S. and Leroith, D. (2012) Hyperinsulinemia Enhances c-Myc-Mediated Mammary Tumor Development and Advances Metastatic Progression to the Lung in a Mouse Model of Type 2 Diabetes. Breast Cancer Research, 14, R8. http://dx.doi.org/10.1186/bcr3089
- Ferguson, R.D., Gallagher, E.J., Cohen, D., Tobin-Hess, A., Alikhani, N., Novosyadlyy, R., Haddad, N., Yakar, S. and LeRoith, D. (2013) Hyperinsulinemia Promotes Metastasis to the Lung in a Mouse Model of Her2-Mediated Breast Cancer. Endocrine-Related Cancer, 20, 391-401. http://dx.doi.org/10.1530/ERC-12-0333
- Hakkak, R., Korourian, S. and Melnyk, S. (2013) Obesity, Oxidative Stress and Breast Cancer Risk. Journal of Cancer Science and Therapy, 5, e129.
- Fieber, C.B., Eldridge, J., Taha, T.A., Obeid, L.M. and Muise-Helmericks, R.C. (2006) Modulation of Total AKT Kinase by Increased Expression of a Single Isoform: Requirement of the Sphingosine-1-Phosphate Receptor, Edg3/S1P3, for the VEGF-Dependent Expression of AKT3 in Primary Endothelial Cells. Experimental Cell Research, 312, 1164- 1173. http://dx.doi.org/10.1016/j.yexcr.2006.01.022
- Santi, S.A. and Lee, H. (2010) The AKT Isoforms Are Present at Distinct Subcellular Locations. AJP: Cell Physiology, 298, C580-C591. http://dx.doi.org/10.1152/ajpcell.00375.2009
- Buroker, N.E., Ning, X.H., Zhou, Z.N., Li, K., Cen, W.J., Wu, X.F., Zhu, W.Z., Scott, C.R. and Chen, S.H. (2012) AKT3, ANGPTL4, eNOS3, and VEGFA Associations with High Altitude Sickness in Han and Tibetan Chinese at the Qinghai-Tibetan Plateau. International Journal of Hematology, 96, 200-213. http://dx.doi.org/10.1007/s12185-012-1117-7
- Liu, X., Kim, C.N., Yang, J., Jemmerson, R. and Wang, X. (1996) Induction of Apoptotic Program in Cell-Free Extracts: Requirement for dATP and Cytochrome c. Cell, 86, 147-157. http://dx.doi.org/10.1016/S0092-8674(00)80085-9
- Norberg, E., Karlsson, M., Korenovska, O., Szydlowski, S., Silberberg, G., Uhlen, P., Orrenius, S. and Zhivotovsky, B. (2010) Critical Role for Hyperpolarization-Activated Cyclic Nucleotide-Gated Channel 2 in the AIF-Mediated Apoptosis. The EMBO Journal, 29, 3869-3878. http://dx.doi.org/10.1038/emboj.2010.253
- Polster, B.M., Basanez, G., Etxebarria, A., Hardwick, J.M. and Nicholls, D.G. (2005) Calpain I Induces Cleavage and Release of Apoptosis-Inducing Factor from Isolated Mitochondria. Journal of Biological Chemistry, 280, 6447-6454. http://dx.doi.org/10.1074/jbc.M413269200
- Deveraux, Q.L. and Reed, J.C. (1999) IAP Family Proteins―Suppressors of Apoptosis. Genes & Development, 13, 239-252. http://dx.doi.org/10.1101/gad.13.3.239