Open Journal of Endocrine and Metabolic Diseases
Vol.3 No.5A(2013), Article ID:35963,23 pages DOI:10.4236/ojemd.2013.35A002
Molecular Insights into Appetite Control and Neuroendocrine Disease as Risk Factors for Chronic Diseases in Western Countries
1Centre of Excellence in Alzheimer’s Disease Research and Care, School of Medical Sciences, Edith Cowan University, Joondalup, Australia
2School of Psychiatry and Clinical Neurosciences, The University of Western Australia, Nedlands, Australia
3McCusker Alzheimer’s Research Foundation, Hollywood Medical Centre, Nedlands, Australia
Email: *i.martins@ecu.edu.au
Copyright © 2013 I. J. Martins et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received June 13, 2013; revised July 13, 2013; accepted August 10, 2013
Keywords: Stress; Metabolic Syndrome; Neuroendocrine Disease; Appetite; Fatty Liver
ABSTRACT
Environmental factors such as stress, anxiety and depression are important to consider with the global increase in chronic diseases such as cardiovascular diseases, cancer, stroke, obesity, diabetes and neurodegenerative diseases. Brain metabolic diseases associated with conditions such as obesity and diabetes require early intervention with diet, lifestyle and drug therapy to prevent diseases to various organs such as the liver with non alcoholic fatty liver disease (NAFLD) and other organs such as the heart, lungs thyroid, pancreas, brain, kidneys and reproductive systems. Behavioural stress and the molecular mechanisms that are involved in neuroendocrine diseases such as insulin resistance in obesity require attention since associated inflammatory processes early in the disease process have been associated with neurodegenerative diseases. Molecular neuroendocrine disturbances that cause appetite dysregulation and hyperphagia are closely linked to hyperinsulinemia, dyslipidaemia and reduced lifespan. The origins of metabolic diseases that afflict various organs possibly arise from hypothalamic disturbances with loss of control of peripheral endocrine hormones and neuropeptides released from the brain. Diet and drug therapies that are directed to the autonomic nervous system, neuroendocrine and limbic systems may help regulate and integrate leptin and insulin signals involving various neuropeptides associated with chronic diseases such as obesity and diabetes. The understanding of brain circuits and stabilization of neuroanatomical structures in the brain is currently under investigation. Research that is involved in the understanding of diet and drugs in the stabilization of brain structures such as frontostriatal limbic circuits, hypothalamus brainstem circuits and parasympathetic nervous system is required. Information related to neuropeptides and neurotransmitters that are released from the brain and their regulation by therapeutic drugs requires further assessment. The promise of appropriate diets, lifestyle and drugs that target the CNS and peripheral tissues such as the adipose tissue, liver and pancreas may improve the prognosis of chronic diseases such as obesity and diabetes that are also closely associated with neurodegeneration.
1. Introduction
In Western and third world countries, the global obesity epidemic has been reported to affect at least 10% of the global population [1,2]. In the United States, the projected healthcare costs for obesity related conditions have been reported to be 344 billion dollars to the year 2018, which account for 21% of all health costs. Obesity and diabetes are endocrine disorders associated hyperinsulinemia and other hormonal imbalance; resulting in inflammatory complications of various organs including the brain, thyroid, parathyroid, adrenal gland and pancreas [3,4]. Cardiovascular disease is the major chronic disease and the reported increase in cases can be linked to the global obesity epidemic [5-8]. Stress, fatigue, anxiety and depression disorders are closely linked to other chronic diseases and the molecular mechanisms that are involved in neuroendocrine disturbances can induce or result from insulin resistance and lead to obesity and diabetes [9-16]. Anxiety disorders include appetite dysregulation, habit disoders, obsessive compulsive disorders, phobias, mood disorders and social phobias. Anxiety disorders affect mental health and induce changes in the brain associated with hormone dysregulation and biological clock alterations affecting tissues such as the liver and adipose tissue [17-19]. In 2013, the world health organization (WHO 2013) indicated that the number of global deaths due to chronic disease was 63% and of these 48% were due to cardiovascular disease, 21% to cancer and 12% to chronic respiratory conditions. In Western countries, the increase in non alcoholic fatty liver disease (NAFLD) and its comorbidity with other conditions such as rheumatoid arthritis have stimulated research into the molecular mechanisms in neuroendocrine disorders, which may contribute to various chronic diseases (Figure 1).
Consumption of excess dietary fat and calories contributes to the development of obesity with an increase in both size and number of fat cells. This results in an increase in lipid mediated oxidative stress, which is associated with chronic disease [20,21]. Interest in adipose tissue and free fatty acid metabolism has increased as elevated fatty acids are known to affect the release of adipose tissue derived cytokines such as tumour necrosis factor (TNF) and interleukin 6 (IL-6), which may have deleterious effects on glucose and lipid metabolism; contributing to the inflammatory state of the disease [22-24]. Inflammation in endocrine disorders such as occurring in obesity and diabetes may cause increased oxidative stress and therefore increases thge risk for cardiovascular disease [25-31]. Development of dietary, lifestyle and pharmacological strategies early in childhood may prevent chronic diseases involving insulin and leptin resistance and may delay the acceleration in the rate of chronic diseases that has become a concern in many countries including the United States [32-34]. The increase in childhood obesity is not only seen in industrialized countries but also in poor and developing countries [35,36]. Neuroendocrine disturbances involving insulin resistance include thyroid dysfunction in obesity/diabetes [37-41] and this is closely related to lipid abnormalities seen in the metabolic syndrome characterised by raised plasma triglyceride (TG) levels and low density lipoprotein (LDL) cholesterol and decreased high density lipoprotein (HDL) cholesterol levels. The complex interactions involved in neuroendocrine disease induce appetite dysregulation that involves abnormal elevation in inflammatory mediators with biochemical disorders of neuropeptides and hormones released from various tissues and the gastrointestinal tract in these chronic diseases (Figure 2).
Lipidomics is the large-scale study of cellular lipid pathways and can identify numerous individual molecular lipids, which may assist in unravelling the molecular mechanisms of lipid-induced neuroendocrine regulation of metabolic diseases [42-52]. Appetite regulation and increased food intake may be influenced by individual lipid species via abnormal gene regulation and may possibly involve early neurodegeneration in chronic diseases such as obesity and diabetes. Insulin and leptin resistance are key features of these conditions and are associated with the abnormal organ pathophysiology and disturbed lipid metabolism.
2. Dyslipidemia, Insulin Resistance Syndrome and Obesity
The metabolic syndrome characterized by dyslipidemia and hypertension is often seen in obese individuals with excess abdominal fat accumulation [53,54]. Lipoprotein sub-fractionation of plasma from obese individuals indicate the presence of large very low density lipoproteins (VLDL), small dense LDL and decrease in the HDL2 subfraction of HDL. Other lipid abnormalities seen in obesity are increased circulating free fatty acid levels with excessive lipid accumulation in cells of various tissues, especially the liver with a tendency to develop non-
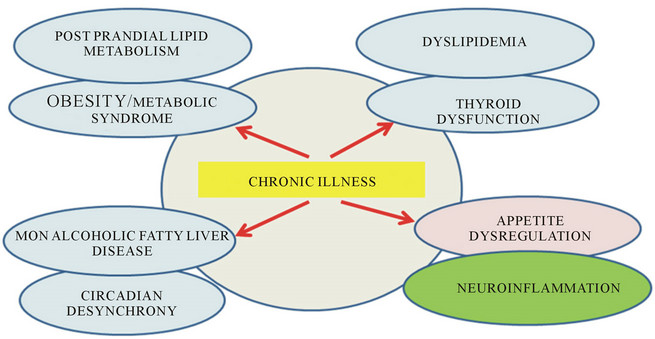
Figure 1. Understanding of chronic illness to global obesity and the metabolic syndrome in western countries.
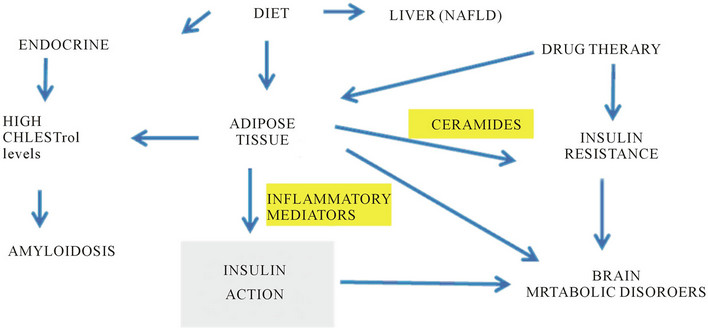
Figure 2. Drug therapy and diet control endocrine events with insulin resistance and brain lipid peroxidation in chronic diseases such as obesity and disabetes linked to neurodegeneration.
alcoholic fatty liver disease (NAFLD). High fat and high glycaemic load diets provide an oversupply of lipids to peripheral tissues and may contribute to the development of insulin resistance. Lipid accumulation, particularly in the abdomen and around the organs in obesity may indicate high fatty acid intake, but can also be a feature of the co-existence of mitochondrial disease associated with diminished mitochondrial lipid oxidation and abnormal lipid metabolism.
Previous studies have characterised lipid molecular species in plasma by liquid chromatography coupled to mass spectrometry to improve the understanding of identification of lipids in the biology of health and disease. Molecular lipid species that represent sphingolipid, glycerolipid, glycerophospholipid, fatty acyl, sterol, prenol classes have been studied [53-55]. Increases in the lipid fraction of lysophosphatidylcholines were found in proinflammatory and proatherogenic conditions with decreases in lipids with antioxidant properties such as the ether phospholipids [53-55]. Insulin resistance was associated with triacylglycerol species containing saturated or monounsaturated fatty acids whereas triacylglycerol species containing linoleic acid (18:2 n-6) were not associated with insulin resistance. Ceramide species were altered in obese and diabetic patients [47-51]. The plasma measurement allows characterization of lipid species but may not reflect tissue specific changes. Fatty acid analysis in NAFLD samples show numerous changes in lipid classes with several alterations in polyunsaturated fatty acids with increased metabolism of essential fatty acids [56].
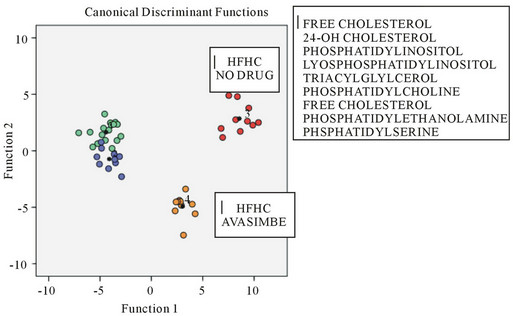
To assess the effects of high fat high cholesterol (HFHC) diets on brain tissue lipid peroxidation, mice were used as a model with apo E knockin mice compared with control mice (Figure 3(a)) and C57BL/6J mice (Figure 3(b)) [57,58]. In Figure 3(a) the heat map of brain lipid classes is shown with increases in cholesteryl esters and lysophosphatidylcholine species in the brain in young apo E4. In aged apo E4 mice increases in cholesteryl esters, lysophosphatidylcholine, sphingolipid, lysophosphatidylinositol (lyso-PI) species were found. In C57BL/6J control mice the effects of HFHC diets were assessed with drug intervention using anti-oxidative acyl coA cholesterol acyltransferase inhibitor Avasimibe (Figure 3(b)). As shown in Figure 3(b) the alterations in brain tissue lipid peroxidation showed a marked decrease by the drug Avasimibe compared with brain lipids from HFHC treated mice where the brain lipid classes were markedly altered and the phosphatidylinositol (PI) was oxidised to the lyso-PI series. The role of HFHC diets in apo E4 KI mice and control mice indicate the generation of brain lipid mediators that are involved in the promotion of the amyloidogenic pathways of amyloid beta [59, 60] with brain neuroinflammation and potential for neuronal death. In both control mice and apo E knockin mice, the HFHC diet induced NAFLD in particular apo E4 KI mice and control mice fatty liver was found with the role of insulin resistance and neuroendocrine disturbances closely associated with NAFLD and brain lipid peroxidetion in these mice (Figures 3(a) and (b)).
Plasma free fatty acids (FFA) might mediate the insulin resistance and impaired glucose tolerance associated with central obesity [42,43,52]. Dietary FFA may promote insulin resistance by downregulation of the insulin signaling pathway in various tissues such as the muscle and liver with the promotion of TG storage in the liver (NAFLD) and interference with insulin action and glucose disposal. Chronically elevated plasma glucose and FFA results in lipotoxicity that contributes to insulin resistance and associated chronic diseases which in turn are connected to appetite dysregulation leading to the development of obesity, diabetes and neurodegenerative diseases. Alterations in brain lipid species such as phospholipids and sphingolipids in mice induced by the HFHC diets indicated a variety of cellular pathways for sphingolipids and ceramide to perturb insulin actions in
 (a)
(a)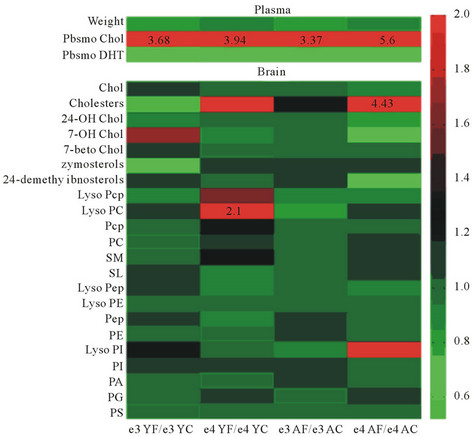 (b)
(b)
Figure 3. The brain lipid classes in apo E knockin mice (a) and AVASIMIBE drug treated C57BL/6J control mice (b) and effects of a HFHC diet.
various tissues [45-48]. Ceramides and programmed cell death [61] have been closely linked to inflammation, insulin resistance and amyloidogenesis and alterations in lipid mediators such as sphingolipids and ceramides have been shown to cause defective insulin action in obesity and diabetes. In studies related to lipid mediators and neurodegeneration, plasma samples from Alzheimer’s disease (AD) individuals have shown to have elevated levels of sphingolipids and ceramides in the blood plasma with indications of insulin resistance intimately involved in the convergence of risk factors for cardiovascular disease and AD [62-64].
3. Inflammatory Cytokines Are Linked to Brain Neuropeptides and Appetite Regulation
The world health organization has indicated that chronic diseases are the major risk for death and disability and include obesity, diabetes, cardiovascular disease, stroke, cancer and neurodegenerative diseases. The role of psychological and physiological stress and high fat, high glycaemic load diets are important risk factors since they cause inflammatory processes to accelerate depressive illnesses that are associated with abnormal cholesterol metabolism and NAFLD. Chronic diseases such as obesity and diabetes are associated with elevations in proinflammatory cytokines that corrupt insulin signalling with the promotion of the metabolic syndrome [24-27,31,65]. The synergistic role of stress and diets that produce adipose tissue inflammatory responses that effect neuroendocrine disease have become important for interventions in relation to lifestyle, drug therapy and diet [65-67]. The reversal of early inflammatory changes induced by these risk factors that accompany obesity and diabetes may improve the prognosis of individuals and prevent the risk for death and disability.
Inflammatory cytokines in chronic diseases may arise from the adipose tissue or from the brain and effects of inflammatory cytokines on the suprachiasmatic nucleus (SCN) in the hypothalamus may cause alteration in the SCN that leads to alterations in the circadian pacemaker that accelerate aging with the promotion of chronic diseases [65-67]. Diet and stress are sensitive to neuroendocrine disease and senescence with inflammatory changes that promote the alterations in the hypothalamic-pituitary hormones and relevance to the metabolic syndrome [65- 67]. Interest in cytokines and appetite has been clearly documented with the effect on food intake disorders related to peripheral production of inflammatory cytokines that effect peripheral hormones such as insulin, leptin and gastrointestinal hormones [68].
Interests in the understanding of cytokines to hypothalamic alterations involve various neuropeptides released from the brain such as corticotrophin releasing hormone (CRH) and neuropeptide Y (NPY) which have also been associated with stress, anxiety and depression [69-73]. Chronic diseases such as obesity and diabetes have led to major concerns in the Western communities and involve the role of abnormal release of brain CRH that is involved in appetite control and energy balance [74-78]. Interests in the role of leptin in the regulation of neuroinflammation possibly involve the effects of neuropeptides on the non amyloidogenic pathways of amyloid beta in brain cells. HFHC diets induce brain neuroinflammation with lipid peroxidation (Figures 3(a) and (b)) and the role of high fat diets that effect the release of leptin from adipose tissue is important to the control of neuroendocrine disease in various chronic diseases. In obesity, leptin resistance is proposed to be involved in appetite dysregulation by poor regulation of hypothalamic NPY; [a stimulator of food intake], and poor regulation of CRH; [an inhibitor of food intake] [74,79,80]. In mice and diabetic rats, high fat feeding reduced hypothalamic CRH expression with effects on NPY [81,82]. NPY has been shown to play a central role in the regulation of CRH expression and in mental disorders such as depression NPY levels are decreased [70,72-74,83]. In stress and anxiety the effects on appetite dysregulation possibly involve inadequate NPY and CRH release which can effect liver function and cholesterol homeostasis [84-86].
CRH has been shown to be important for protection of neurons and the release of CRH is associated with conditions related to stress and regulation of the non-amyloidogenic pathway by the actions of α-secretase [87-90] preventing the formation of inflammatory amyloid beta oligomers and plaque [91]. Amyloid beta oligomers induce inflammatory cytokines that have effects on various neuropeptides in the brain. CRH modulates the hypothalamic pituitary adrenal axis and in mouse models of AD chronic stressors lead to amyloid beta plaque development, suggesting a role for CRH in neuroendocrine disease and neurodegeneration Activators of α-secretase that stimulate the non-amyloidogenic pathways of amyloid beta have been studied and reduced food intake and promotion of healthy body composition helps prevent inflamemation and promotes insulin sensitivity, thereby reducing the risk of chronic disease (Figure 4). Activators include neuropeptides such as pituitary adenylate cyclase-activating polypeptide (PACAP), protein kinase C (PKC), statins and retinoids. [92-95]. In turn, PACAP, PKC activators and statins have been associated with reduced food intake [96-101].
4. Neuroendocrine Diseases and Effects on Appetite Regulation
The rise in chronic diseases in many countries have reached epidemic proportions due to diet and lifestyle, leading to an inability of the brain to regulate body weight and energy balance which can then lead to metabolic dysfunction and organ disease. In obesity and diabetes the central nervous system alterations in the brain can lead to an increased incidence of stroke and may be associated with other risk factors such as hypertension and hypercholesterolemia. The ability of the brain to regulate food intake, body weight and energy balance is dependent on the sensing of neurons in the parabrachial nucleus, thalamus, lateral hypothalamus, orbitofrontal complex, basolateral amygdala and insular cortex and has become important to the origins of cardiovascular disease which is one of the major global diseases.
Appetite regulation is dependent on neural activity increasing after fasting and decreasing postprandially as the brain senses biochemical changes in glucose, leptin and insulin levels in brain neurons. Studies have shown the effects of HFHC diets on brain lipid alterations in mice (Figures 3(a) and (b)) and suggest molecular mechanisms of endocrine disease involve lipid mediators and insulin resistance. Brain alterations in humans has been reported and induced by fat consumption with the extent of excess body fat in man associated with regional alterations in brain structure and a reduction in brain volume as assessed using voxel based morphometry (VBM) in obese individuals [102]. In these studies, body mass index (BMI) was negatively associated with gray matter (GM) density of the left post central gyrus in obese and lean subjects. In comparison to a group of lean subjects the group of obese individuals had significantly lower gray matter density in the post central gyrus, frontal operculum, putamen and middle frontal gyrus after
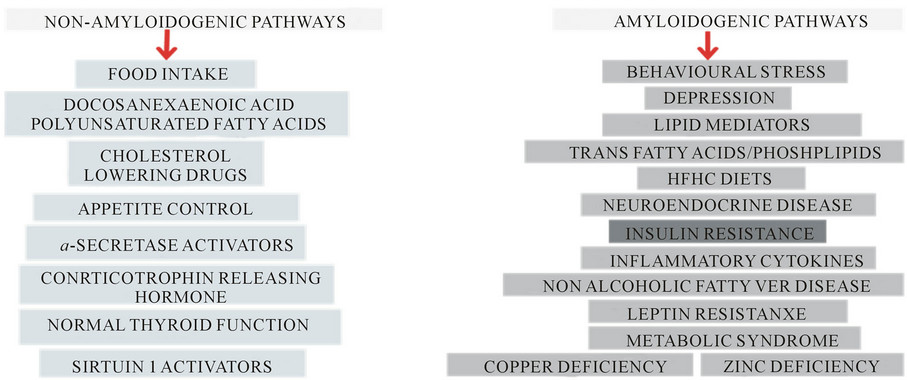
Figure 4. Amyloidogenic and non-amyloidogenic pathways are involved in the molecular mechanisms of chronic diseases.
adjustment for sex, age, handedness and global tissue density. This study identified structural brain differences in human obesity in several regions of the brain that are involved in the regulation of taste, reward and control of behaviour and suggest that the increase in weight gain as a result of obesity may affect brain structure and function [103]. In obese individuals high fat diet and inflammation of the hypothalamus has been reported and in particular neuronal alterations in the appetite centre the arcuate nucleus have been shown [104].
5. Lipid Mediators and Appetite Regulation
Brain lipid heat maps in both apo E KI mice and control mice fed HFHC diets (Figures 3(a) and (b)) have shown marked alteration in phosphatidylinositol (PI) and cholesterol metabolites with relevance to effects on food intake and appetite dysregulation as origins of metabolic disease associated with the NAFLD found in these mice. In other studies cholesterol metabolites such as 25 hydroxysterol and 7 hydroxysterol have been shown to be associated with appetite regulation and food intake [105]. PI intake was shown to have anti-obese effects with added PI to the diet associated with appetite control [106]. Effects of PI oxidation closely involve the loss of control of appetite regulation by neuropeptides leptin and insulin via the PI-3 kinase pathway [107-110]. Interests in various lipid mediators such as ceramides in inflammation and chronic diseases have increased since effects on ceramides on leptin actions and on insulin resistance indicate the role of lipids in the control of the hypothalamus and food intake in man [111]. Further interests in lipid mediators that effect food intake involve the production of malonyl-CoA that is relevant to high fat diets where malonyl-CoA levels are reduced [112,113]. The enzyme carnitine palmitoyltransferase 1 (CPT1) isoform is expressed in the brain and localized in neuron mitochondria [114]. The enzyme controls ceramide metabolism, food intake and energy homeostasis and effects of altered malonyl-CoA levels inhibit the enzyme [115-117]. The effects of inhibition of CPT1c by ceramide in the brain is related to neuropeptide dysregulation such as the orexigenic effects of ghrelin mediated by neuropeptide Y (NPY) and agouti-related protein (AgRP) in the hypothalamic arcuate nucleus [111,115,118,119]. MalonylCoA levels are involved with leptin control of feeding and levels are increased in chronic diseases such as obesity and diabetes [120-122]. Lowering levels of malonylCoA is closely related to reversal of NAFLD, reduction in cardiovascular disease, improving insulin resistance and promoting the non-amyloidogenic pathways [121, 122].
High fat and high calorie diets provide an oversupply of lipids to peripheral tissues and may contribute to the development of insulin resistance. Lipid accumulation (intra adominal fat) in obesity may indicate increase fatty acid intake or mitochondrial disease associated with diminished mitochondrial lipid oxidation and abnormal lipid metabolism in chronic diseases [123,124]. Interests in intervention with diet, lifestyle and drug therapy have accelerated with the increase in childhood obesity in Western countries. The metabolic syndrome disorder also shown in childhood obesity was associated with changes in brain volume and structure and alterations in appetite, hypertension and insulin resistance possibly associated with brain abnormalities in childhood obesity [125]. In obese individuals the loss of brain control is poorly understood and alterations in brain circuitry or feeding signals in obesity involve abnormal hormone regulation with poor control of appetite and body weight [12,126- 135].
6. Gut-Brain Interactions
Interest in chronic diseases such as obesity has led to the better understanding of the communication between the gastrointestinal tract and the CNS that involve the hypothalamus and brain stem. These regions of the brain integrate peripheral signals such as various factors released from the gut and adipose tissue that have effects on neuronal activity of the hypothalamus and brain stem that control appetite regulation [136-141]. In response to food intake various gut and adipose tissue hormones have effects on the hypothalamus, which affect central and peripheral circadian rhythms that release various neuropeptides that effect appetite, energy balance and body weight [141-146]. Signals from the gastrointestinal tract involved in appetite control communicate the need for food intake to the brain. There are chemical messengers from the upper GI tract e.g., cholecystokinin (CCK), secretin and glucose-dependent insulinotropic peptide or gastric inhibitory polypeptide, lower intestine glucagon-like peptide-1, from adipose tissue (leptin, adiponectin) and from the pancreas (insulin). These all communicate with the hypothalamus and allow food intake (orexic) or fast (anorexic). For example, ghrelin released from the intestine enters the brain and increases our appetite (is orexigenic), while insulin and leptin do the opposite, having an anorexigenic signal. The hypothalamus is the processing centre of the appetite regulating centre and integrates signals from the peripheral circulation, gastrointestinal tract and the brain. Hypothalamus neuronal cirucuits are involved in regulation of appetite, energy expenditure and control of major organ functions such as endocrine, gastrointestinal (GI), cardiovascular and reproductive systems. Neuropeptides and hormones produced by the hypothalamus and intestine stimulate appetite during fasting or inhibit appetite after feeding.
7. Hypothalamus and Neurodegenerative Diseases
The hypothalamus is involved with many biological functions including appetite and body weight control, feeding, emotion, memory, thermoregulation, fluid balance and insulin regulation. The three major systems that are involved with these functions include the autonomic nervous system, the neuroendocrine system and the limbic system. The hypothalamic nuclei that are involved in food intake include the arcuate nucleus (ARC), the paraventricular nucleus (PVN), the lateral hypothalamic area (LHA), the ventromedial nucleus and dorsomedial nucleus. ARC neurons at the bottom of the hypothalamus near the third ventricle have direct contact with peripheral satiety factors like leptin and insulin. Neurons in the hypothalamus are responsible for various connections to other brain regions and one of the important functions of the hypothalamus is control of the daily light dark cycle. The suprachiasmatic nucleus (SCN) that coordinates the neuronal and humoural systems and the circadian rhythms, activates the arcuate nucleus that releases neuropeptide Y (NPY) and agouti related protein (AgRP) that control physiological functions (body temperature, melatonin release, glucocorticoid secretion and behavioural functions (feeding and memory).
The SCN releases a number of hormones such as the corticosteroids and the SCN projects to the dorsal parvicellular paraventricular nucleus which projects to sympathetic preganglionic neurons which regulate melatonin output from the pineal gland. The dorsomedial nucleus connects to the ventrolateral preoptic nucleus (the sleep promoting region) and to the orexin neurons and melanin-concentrating hormone (MCH) neurons which regulate sleep and wakefulness. Appetite regulating hormones such as ghrelin, leptin and insulin can influence areas of the brain and are involved with resetting the circadian rhythms generated by the SCN. In response to the daily sleep/wake cycle corticosterone secretion increases during the night and related to food intake in rats with the release of melatonin from the pineal gland. The SCN may regulate the sleep-wake cycle and has effects on anxiety, stress and depression and food restriction effect the SCN and peripheral oscillators. In neuroendocrine diseases such as obesity, diabetes and neurodegenerative diseases the origins of these diseases involve the hypothalamus and SCN with alteration in appetite control in these individuals which is influenced by HFHC as consumed in Western countries.
Hypothalamic neurons have been clearly shown to be abnormal in chronic neuroendocrine diseases that involve obesity and diabetes [104,147]. In AD individuals many brain functions are lost as the disease destroys neurons and the hypothalamus has been shown to be involved with the early stages of the disease [147-152]. The disease advances to other regions of the brain and the progression of the disease leads to death of the AD individual; a period of time which varies from 10 years to 30 years after the onset of the disease. In Parkinson’s disease (PD) the examination of the hypothalamus have indicated abnormalities [151,152] with lewy body formation and marked nerve cell degeneration. Hypothalamic disorders in PD assist in the interpretation of the autonomic and endocrine abnormalities in these PD individuals. The abnormal crosstalk between the periphery and the hypothalamus involved with obesity, diabetes and cardiovascular disease are now closely linked to neurodegenerative diseases such as PD and AD.
8. Diets, Peripheral Endocrine Hormones and Brain Neuropeptides in Chronic Diseases
Future therapies that involve control of chronic diseases will involve diet, body size, adiposity and the role of the hypothalamus in the regulation of various neuropeptides involved in appetite regulation (Figure 5). Appetite disorders in chronic diseases are associated with abnormal inflammatory process, increased food intake, leptin resistance, hyperinsulinemia, neuropeptide dysregulation and GI hormone dysregulation. HFHC diets are closely linked to NAFLD and effect peripheral endocrine or central nervous systems that induce appetite dysregulation, obesity and NAFLD [153]. Influence on appetite level and feeding are related to neurons in the hypothalamus that express neuropeptides (Figure 5) that communicate with peripheral signals such as nutrients (glucose, amino acids, fatty acids) and gastrointestinal peptide hormones such as cholecystokinin and ghrelin.
8.1. Adipose Tissue Hormones
8.1.1. Leptin
Leptin is a 16 kda protein identified in 1994 and is synthesized by fat cells and acts as a satiety factor at the hypothalamus. The gene encoding leptin was identified by positional cloning as the ob mutation and ob/ob mice inherit the mutation on chromosome 6 as an autosomal recessive condition. Elevated leptin levels are associated with increased adipose tissue mass and leptin levels are proportional to the size of adipose tissue (Reviewed [33,154-161]. Food intake in obese mice (ob/ob) is not regulated and the mice become grossly obese, hyperinsulinemic and overweight. In db/db homozygous mice obesity is inherited as an autosomal recessive mutation and the db gene encodes the receptor for the ob gene product leptin. The effects of leptin on food intake and body weight regulation are mediated through the leptin receptor (ob-R) in the hypothalamus and binding to the ob-R
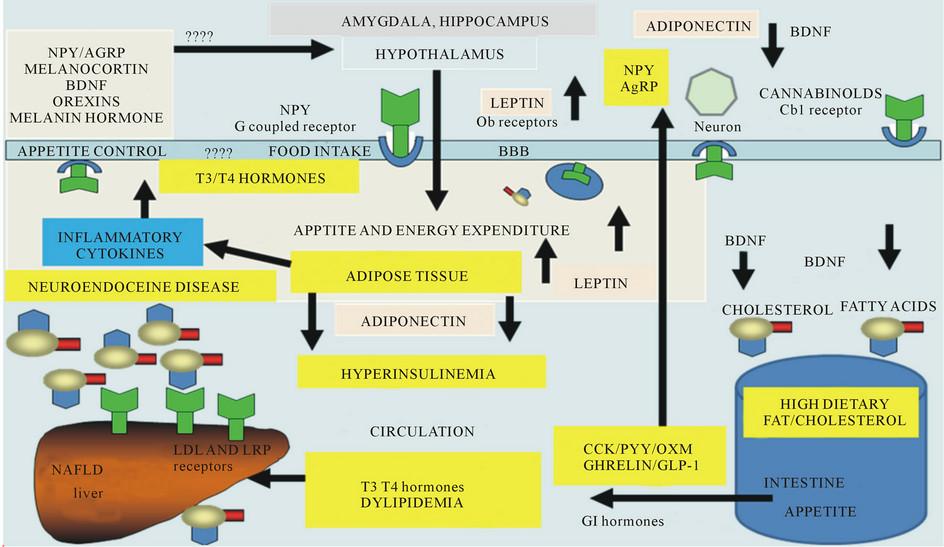
Figure 5. Appetite dysregulation with effects on inflammatoty cytolines, neuropeptides, adipokines, thyroid and GI hormones.
involves activation of PI 3K and other lipid intermediates. The arcuate nucleus (ARC) of the hypothalamus that contains the ob-R acts as the leptin signalling centre and the ARC acts via the orexigenic (appetite stimulating) pathway mediated by neuropeptide Y (NPY) and agouti related protein (AgRP) the anorexigenic (appetite-suppressing) via proopiomelanocortin (POMC) pathway.
8.1.2. Adiponectin
In chronic disease adiponectin levels are low as reviewed [162]. Adiponectin levels are high in plasma but are reduced in obese individuals with glucose intolerance, dyslipidemia and high risk of atherosclerosis. Interests in adiponectin and its interactions with insulin and leptin in the hypothalamus indicate its role in food intake and body energy homeostasis and injections with adiponectin reversed insulin resistance in obese mice [163]. Adiponectin is a collagen like protein secreted by adipocytes and has an important role in insulin sensitivity, energy homeostasis, glucose and fatty acid metabolism [164- 169]. Adiponectin has effects on peripheral tissues and the central nervous system with effects on the centre of the hypothalamus that controls hunger and satiety. Adiponectin receptors (Adip-R1 and Adip-R2) are under circadian rhythm and are abundantly expressed in the appetite centres of the hypothalamus and the POMC and NPY neurons of the arcuate nucleus indicating a role for central regulation of energy intake or expenditure [170- 172]. Adiponectin acts via phosphorylated AMP protein kinase (AMPK) which phosphorylates acetylCoA carboxylase indicating adiponectin regulates lipid metabolism by controlling metabolism of malonylCoA in tissues [173]; the metabolism of which is closely involved in food intake and energy expenditure in the hypothalamus [174-176]. Recent interests in inflammation, insulin resistance, dementia and AD implicate adiponectin as the adipokine involved in amyloidosis [177,178].
8.2. Neuropeptides and Appetite Regulation
Appetite regulating neuropeptides are involved in abnormal responses in brain-endocrine interactions in various chronic diseases and involve insulin resistance and neuropeptides such as melanocortin, NPY, melanin concentrating hormone (MCH), orexins (OR), endocannabinoids and brain derived neurotrophic factor (BDNF). In response to food, neuropeptides are released from the hypothalamus which effects energy expenditure and food intake; essential for control of body weight regulation which is abnormal in neuroendocrine diseases such as obesity, diabetes, Parkinson’s disease and Alzheimer’s disease [179-182].
8.2.1. Neuropeptide Y
High fat diets affect NPY [183-185] with effects in the brain that include effects on emotions such as stress, anxiety and depression [186-188]. The major physiological effects of NPY are to regulate food intake, suppress inflammation and regulate cholesterol metabolism [189-193]. NPY has neuroprotective effects and is present in the hypothalamus (orexigenic effects), hippocampus, amygdala (anxioloytic effects) and nucleus accumbens [193,194]. NPY synthesized in neurons such as the GABAergic neurons and transported to the synaptic nerve terminal where neurotransmitter modulation by NPY is at the pre and post synaptic terminals with the release of dopamine and glutamate. NPY acts with neurogenic and physiological effects at four G coupled receptors Y1-Y5 and Y6 with targeted brain specific receptor expression patterns [195].
8.2.2. Melanin Concentration Hormone
Melanin concentrating hormone (MCH) is a cyclic 19 amino acid peptide and is mainly expressed and synthesized in the lateral hypothalamus and zona incerta [196]. The neuropeptide binds to the melanin concentrating hormone 1 receptor (MCH1R) and obese (ob/ob) mice have overexpression of the MCH neuropeptide in the hypothalamus [196,197]. The MCH levels increase upon fasting and MCH is important in the regulation of various metabolic responses after consumption of diets rich in fat [198,199]. MCH1R is widely expressed in the brain with various physiological functions such as regulation of energy expenditure, food intake and body weight [200, 201]. Metabolic studies indicate that the hypothalamic MCH system is important in the regulation of energy homeostasis after consumption of high fat diets [202, 203]. Interests in MCH1R receptor and obesity have increased with the development of MCH1R antagonists that are involved in regulation of MCH regulation of metabolic processes [204,205].
8.2.3. Orexins
The orexins referred to as Orexin A and Orexin B recently isolated from the hypothalamus are derived from a 130 amino acid prepro-orexin. The orexins are synthesized in the lateral hypothalamus and preinforfornical area and the orexins bind to G protein coupled receptors referred to as Orexin receptor type 1 (OX1R) and Orexin receptor type 2 (OX2R). Binding of Orexins to receptors [206] allow neuronal firing with increased firing with increased intracellular calcium levels. Orexins have important roles in mediating spontaneous physical activity (SPA) and non exercise induced thermogenesis (NEAT). Orexins activate OX2R receptors in the ARC that stimulates Na+/Ca2+ exchange in GABAergic neurons associated with cell depolarization and these mechanisms stimulate feeding and appetite [207]. This may be important in obesity therapy with control of SPA and NEAT important to obesity resistance [207]. Diets rich in fat alter hypothalamic orexin release with effects on SPA and NEAT and may be relevant to the pathogenesis of obesity [202].
8.2.4. Endocannabinoids
Endocannabinoids such as endocannabinoids anandamide, 2-arachidonyl glycerol and 2-arachidonyl glyceryl ether have been detected in various mammals and produced by cleavage of membrane lipid precursors (Reviewed [208]. They act through G protein coupled receptors including CB1 which is found in the nervous system such as the hypothalamus, peripheral nervous system and peripheral organs [208]. The endocannabinoids are involved in the generation of ceramides and with the PI-3 kinase pathway involved in the control of neuronal survival by protection of ceramide induced apoptosis [209,210]. The endocannabinoids are involved in various physiological processes and include appetite regulation, emotional responses, learning and memory process, nociceptive transmission and motor activity. Leptin regulates endocannabinoids that act on the CB1 receptor, which modulates food intake and regulation of energy balance [211]. Interests in the actions of endocannabinoids has increased since drugs that are antagonists of CB1 receptors effect the expression of adiponectin and provide useful treatment for NAFLD in obesity and Type 2 diabetes [212-214].
8.2.5. Brain Derived Neurotrophic Factor
Brain derived neurotrophic factor (BDNF) is synthesized as a 32 kda precursor as pro BDNF from the BDNF gene at a locus at 11q13 that contains 11 exons and can be processed as a 14 kda or 28 kda protein intracellularly by furin/proconvertases or extracellularly by plasmin or metalloproteinase. The mature BDNF can be transported into vesicles from the golgi apparatus to the cell membrane and then secreted into the extracellular space. BDNF is involved in the regulation of food intake and the levels of BDNF are controlled by high fat diets [215, 216]. In mature neurons the BDNF peptide is involved with the regulation of synaptic plasticity and neurotransmission in the peripheral and central nervous system [215,216]. BDNF is involved in regulation of CB1 receptor expression and the proliferation, survival and maintenance of neurons [217]. In obese individuals with the metabolic syndrome, adiponectin levels are low and related to low BDNF levels [210,218-220] which is also a feature of neurodegeneration and Alzheimer’s disease.
8.3. Gastrointestinal Hormones and Effects on Hypothalamic Appetite Regulation
After a meal gut hormones are released into the blood that signals to the brain to inhibit food intake and control energy intake. The effects of these various hormones are short term with the short half-life of the hormones linking communication pathways to the brain. Ghrelin is characterized as an appetite stimulating hormone and levels rise after fasting indicating the onset of hunger and decrease postprandially after consumption of a meal (Reviewed [221,222]. It is secreted by the stomach as ghrelin and de-acylated (lack serine 3 acylation) with the acylated form essential for the activation of ghrelin receptor (GHS-R1a). Ghrelin effect on appetite control was related to hypothalamic NPY/AgRP neurones which express the ghrelin receptors [221,222]. Research in ghrelin based antiobesity studies have indicated a role of ghrelin in antiobesity therapy to date and possibilities remain for future research that targets other hormones for signals from the gut to the brain [223].
8.4. Miscellaneous Hormones and Peptides
Cholecystokinin (CCK) is an intestinal hormone and has clear effects on appetite and energy intake and after a meal CCK levels rise to inhibit food intake [224,225]. In response to ingested calories products of proglucagon cleavage such as glucagon like peptide (GLP-1) increases in the blood plasma as they are released from the L cells of the gastrointestinal tract. The increase in GLP-1 is associated with an improvement in body weight and the GLP-1 receptor is found in the CNS and GLP-1 has been used as a therapy for obesity [226-228]. Pancreatic islet beta cells release insulin and along with insulin release another peptide referred to as amylin. Amylin binds to a receptor complex that contains the calcitonin receptor. Anorectic effects of amylin include reduced gastric emptying and food intake. Analogues of amylin have been used in obese and diabetic man with modest weight reduction.
Other proglucagon cleavage peptides include oxyntomodulin (OXM) and peptide YY (PYY) that are secreted with GLP-1 in response to high calorie foods. OXM reduces energy intake and in obese individuals reduces body weight [229,230]. OXM acts via the GLP-1 receptor and has effects on the central nervous system with regulation of food intake. In several species PYY inhibits food intake and reduces body weight and administration of PYY to obese individuals is underway in various clinical trials [231,232]. Pancreatic polypeptide (PP) is secreted from the pancreatic islets and is similar in structure to PYY with reduction in food intake after administration to rodents and humans [233,234]. PP has effects on gastric ghrelin and gene expression of hypothalamic peptides such as NPY and AGRP that control food intake. Bombesin like peptides are found in the CNS and bind with high affinity to G protein coupled receptors with effects on hyperphagia and energy balance [235]. G coupled protein receptor Gpr17 is regulated by nutritional status and controls food intake by interaction with the AgRP neurons in the brain.
8.5. Thyroid Hormones and Food Intake
Hypothalamic control of appetite regulation and energy expenditure not only involves the hypothalamus but also the hypothalamic pituitary axis (HPT). Recent evidence indicates that the HPT axis can control food intake and effects on appetite and body weight is mediated by thyroid hormones [236]. Thyroid hormones may act directly on the hypothalamic appetite circuits and signalling factors such as thyroid stimulating hormone, triiodothyronine (T3) and thyroxine (T4) have recently shown to directly influence food intake and reverse NAFLD. Implications for thyroid hormone therapeutics to control neurodegeneration [237,238] with appetite dysregulation in obesity and other chronic diseases that involve thyroid hormones may also provide pharmacological treatments (Figure 5) for stress, anxiety and depression.
9. Zinc Deficiency and Neuroendocrine Effects on Appetite Regulation
Zinc deficiency has marked effects on brain zinc homeostasis and its deficiency has been associated with alterations in behaviour, learning and mental function [239- 241]. Under stress, anxiety and in depression disorders zinc levels alter with marked effects on chronic illness in Western countries [242]. Obesity and micronutrient deficiencies are related to metabolic defects in leptin and insulin metabolism [243,244]. In particular obese individuals in many studies have zinc deficiency which predisposes these individuals to glucose intolerance and appetite dysregulation [244-246]. Stress has been linked to body weight regulation and evidence suggests zinc’s involvement in inflammatory cytokine regulation as the molecular mechanism for brain dysfunction in chronic diseases [247].
Interests in the neuroendocrine system, energy metabolism and peripheral cholesterol metabolism have increased with the strong genetic identification and involvement NPY in plasma cholesterol regulation [190, 191]. The CNS and its control of lipid metabolism has identified hypothalamic NPY with evidence that NPY has effects on Y1 receptors to promote hepatic lipoprotein secretion to promote VLDL secretion via the sympathetic nervous system and on Y2 receptors to promote feeding [84]. BDNF has been shown to modulate NPY levels in the brain and several studies have indicated the involvement in neuronal plasticity, behaviour, appetite control and body weight regulation. Zinc is involved in the expression of brain BDNF and NPY synthesis and its effects on insulin, leptin and adiponectin [248-253] in the periphery indicates its role in the close relationship between appetite control and cholesterol homeostasis (Figure 6). In zinc deficiency, NPY is unable to bind to its receptors to initiate an orexigenic response.
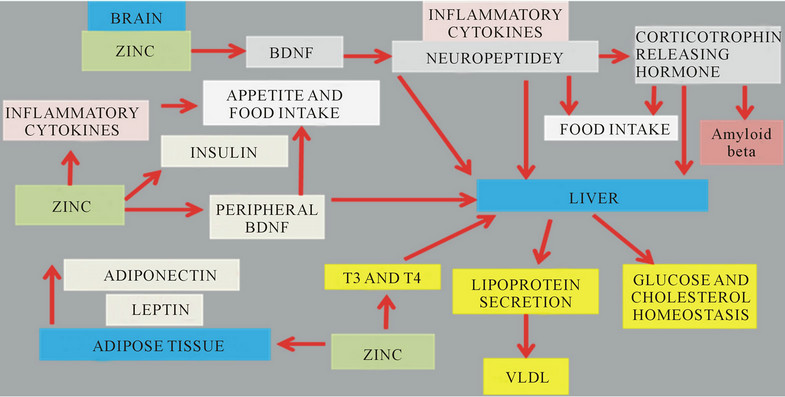
Figure 6. Zn deficiency in chronic diseases is associated with inflammation, appetite dysregulation andneuroendocrine disease.
In HFHC fed mice zinc deficiency was found and the effects on NPY dysregulation is possibly involved with the control of behaviour and stress responses in these mice as structures of the brain such as the amygdala and hippocampus are involved. Diets are rich in fat effect genes in the hypothalamus and are associated with plasma zinc dyshomeostasis and inflammation. The close relationship between zinc and lipid metabolism has been shown and zinc’s involvements with hundreds of enzymes includes effects on fatty acid metabolism that become abnormal in individuals with insulin resistance and chronic diseases. In HFHC diet-fed mice, zinc deficiency and ill effects on thyroid hormone metabolism possibly are closely involved in hypercholesterolemia and NAFLD.
10. Chronic Diseases and Overnutrition in Neurodegeneration
Overnutrition in chronic disease is in involved with central nervous system dysregulation of neuropeptides, peripheral hormone signalling from the pancreas (insulin), adipose tissue (leptin and adiponectin) and gastrointestinal tract. Excess free fatty acids inhibit insulin signalling and are involved in glucose dysregulation in the metabolic syndrome. Nuclear receptors include the sirtuin family and are NAD(+) dependent class III histone deacetylase (HDAC) proteins that target transcription factors to adapt gene expression to metabolic activity, insulin resistance and inflammation in chronic dieases has recently been reviewed [254-259]. Nutritional regulation by sirtuin 1 (Sirt1) that is involved with the hypothalamic control of food intake with regulation of the central melanocortin system via the fork head transcription factor has been reported [260-263]. Nutrients such as glucose, fatty acids, zinc and amino acids regulate hypothalamic Sirt1 involved with food intake regulation, insulin resistance, circadian clocks, lipid metabolism and energy expenditure (Figure 7). Regulation of neuronal Sirt1 by calorie restriction is involved with endocrine and somatotrophic disturbances that implicate growth hormone in insulin resistance and peripheral endocrine disease [264, 265]. Furthermore Sirt1 is downregulated in chronic obstructive pulmonary and kidney diseases associated with the metabolic syndrome and cardiovascular disease [266- 268]. The role of Sirt1 in cardiac function involves the peroxisome proliferator-activated receptor-α (PPAR α)- Sirt1 complex and downregulation of the complex is closely related to inflammation related cardiovascular disease [269,270]. Interests in PPAR α as a major regulator of cardiac lipid metabolism also involve the transcriptional control of malonyl-CoA decarboxylase [271].
Cellular SIRT1 expression/activity is important in the promotion of the non-amyloidogenic α-secretase processing of amyloid precursor protein which generates the AD peptide amyloid beta. The over-expression of SIRT1 in the hippocampus has been shown to provide protection against neurodegeneration in a mouse model of AD, and the over-expression of SIRT1 in the brains of AD-model transgenic mice has been shown to reduce brain Abeta production and amyloid deposition. The link between nutrition, food intake and amyloid beta production indicates that neuroendocrine disturbances in diabetes also possibly involve neurodegenerative diseases such as AD and PD.
11. Interventions with Drugs, Diet and Exercise to Prevent Chronic Diseases Such as Obesity and Diabetes
The global increase in chronic diseases such as obesity and diabetes has led to research in the understanding of the use of drugs in the stabilization of brain structures
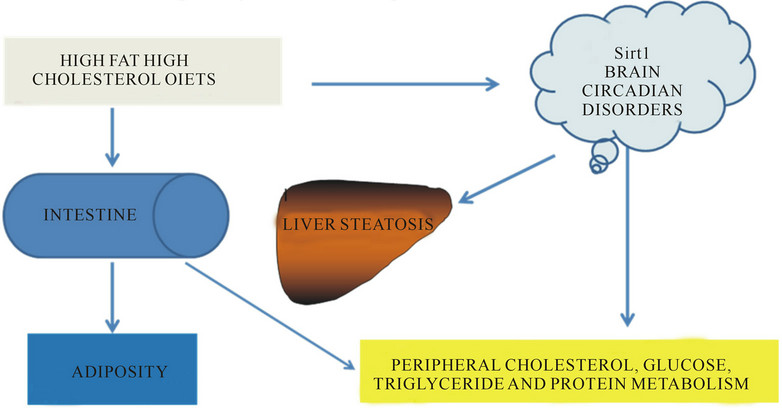
Figure 7. Diet and brain disease controls peripheral metabolism of glucose, lipids and proteins.
such as frontostriatal limbic circuits, hypothalamus brainstem circuits and parasympathetic nervous system. Interventions in chronic diseases that reverse early neuroendocrine disease are required in relation to neuropeptides and neurotransmitters that are regulated by these drugs and require further assessment. The promise of various drugs that target the CNS and peripheral tissues such as the adipose tissue, liver and muscle has indicated promise in the past 100 years however withdrawal of these drugs recently has resulted in the avoidance of harmful side effects to various individuals with neuroendocrine disease such as in obesity and diabetes. Interventions with anti-obese drugs that improve insulin resistance, dyslipidemia and the metabolic syndrome have not been appropriate to altered lifestyles and diet conditions.
Long term treatment of obesity with the use of antiobese drugs particularly agents that target the central nervous system have not been achieved due to safety concerns [272-274]. Anti-obesity drug discovery programmes have been ineffective with false starts, failures in clinical development, and withdrawals of drugs from the market and no improvement in NAFLD has been observed in these trials [275-278]. The discovery of novel CNS compounds that are under clinical development allow for assessment of agents that allow for appetite control, weight reduction and a reduction in the serious medical complications of obesity. In Western countries the ageing populations are afflicted with chronic diseases which are closely related to appetite dysregulation, zinc deficiency and endocrine abnormalities that are involved in the CNS and peripheral organ destruction. Exercise, drugs, diet and hormones that control calorie restriction activate nuclear sirtuins [279], prevent the metabolic syndromes and allow maintenance of the neuroendocrine system with reversal of NAFLD in these aged populations.
12. Conclusion
The acceleration in the rate of chronic diseases that involve insulin resistance and thyroid dysfunction has become of global concern in various countries including the United States. The rate of the most prevalent chronic disease; cardiovascular disease is linked to the metabolic syndrome and NAFLD and environmental factors such as stress, anxiety and depression are important contributors to the increasing rates of obesity related chronic diseases, such as diabetes and neurodegeneration. Intervention with drug therapy for obesity to prevent appetite dysregulation and neuroendocrine disturbances has not been successful. Hyperinsulinemia and hypercholesterolemia are connected to abnormal SCN rhythms and hypothalamic disturbances with loss of interactions between lipid mediators, endocrine hormones and neuropeptides released from the brain. Dietary and pharmacological therapies that are directed to reduce amyloidogenic pathways and accelerated aging and also connected to food intake will delay the onset of inflammation, mitochondrial disease and neurodegeneration that are involved in the pathogenesis of chronic diseases. Factors that reduce behavioural stress or depression and reduce the consumption of HFHC diets may delay the onset of neuroendocrine disturbances that are linked to abnormal gut-brain interactions and chronic diseases in Western countries. Insulin resistance and the development of chronic diseases may be related to deficiency in essential micronutrients in particular PI and zinc that are connected to diet and food intake. Nutritional programmes that improve lifespan require appropriate low fat/low glycaemic load diets and those that promote optimal function of the endocrine system, which may include thyroid hormone replacement therapy, to maintain mental health without the disorders of stress, anxiety and depression that are connected to biochemical changes such as insulin resistance and the metabolic syndrome in several chronic diseases.
REFERENCES
- B. Caballero, “The Global Epidemic of Obesity: An Overview,” Epidemiologic Reviews, Vol. 29, No. 1, 2007. pp. 1-5. doi:10.1093/epirev/mxm012
- K. L. Rennie and S. A. Jebb, “Prevalence of Obesity in Great Britain,” Obesity Reviews, Vol. 6, No. 1, 2005, pp. 11-12. doi:10.1111/j.1467-789X.2005.00164.x
- P. T. James, N. Rigby and R. Leach, “The Obesity Epidemic, Metabolic Syndrome and Future Prevention Strategies,” European Journal of Preventive Cardiology, Vol. 11, No. 1, 2004, pp. 3-8. doi:10.1097/01.hjr.0000114707.27531.48
- K. M. Flegal, et al., “Association of All-Cause Mortality with Overweight and Obesity Using Standard Body Mass Index Categories: A Systematic Review and Meta-Analysis,” The Journal of the American Medical Association, Vol. 309, No. 1, 2013, pp. 71-82. doi:10.1001/jama.2012.113905
- H. Greenberg, S. U. Raymond and S. R. Leeder, “Cardiovascular Disease and Global Health: Threat and Opportunity,” Health Affairs (Millwood), 2005.
- G. Bukhman and A. Kidder, “Cardiovascular Disease and Global Health Equity: Lessons from Tuberculosis Control Then and Now,” American Journal of Public Health, Vol. 98, No. 1, 2008, pp. 44-54. doi:10.2105/AJPH.2007.110841
- B. Mathew, et al., “Obesity: Effects on Cardiovascular Disease and Its Diagnosis,” Journal of the American Board of Family Medicine, Vol. 21, No. 6, 2008, pp. 562- 568. doi:10.3122/jabfm.2008.06.080080
- C. J. Lavie, M. R. Mehra and R. V. Milani, “Obesity and Heart Failure Prognosis: Paradox or Reverse Epidemiology?” European Heart Journal, Vol. 26, No. 1, 2005, pp. 5-7. doi:10.1093/eurheartj/ehi055
- R. L. C. Matteri, J. A. Carroll and C. J. Dyer, “Neuroendocrine Responses to Stress,” CAB International 2000, 2000.
- F. S. Luppino, et al., “Overweight, Obesity, and Depression: A Systematic Review and Meta-Analysis of Longitudinal Studies,” Archives of General Psychiatry, Vol. 67, No. 3, 2010, pp. 220-229. doi:10.1001/archgenpsychiatry.2010.2
- T. W. Strine, et al., “The Association of Depression and Anxiety with Obesity and Unhealthy Behaviors among Community-Dwelling US Adults,” General Hospital Psychiatry, Vol. 30, No. 2, 2008, pp. 127-137. doi:10.1016/j.genhosppsych.2007.12.008
- B. Jeanrenaud, S. Halimi and G. Van der Werve, “Neuroendocrine Disorders Seen as Triggers of the Triad: Obesity-Insulin Resistance-Abnormal Glucose Tolerance,” Diabetes/Metabolism Reviews, Vol. 1, No. 3, 2009, pp. 261-291. doi:10.1002/dmr.5610010303
- E. Susaki and K. I. Nakayama, “An Animal Model Manifesting Neurodegeneration and Obesity,” Aging, Vol. 2, No. 7, 2010, pp. 453-456.
- P. Bjorntorp, “Neuroendocrine Perturbations as a Cause of Insulin Resistance,” Diabetes/Metabolism Research and Reviews, Vol. 15, No. 6, 1999, pp. 427-441. doi:10.1002/(SICI)1520-7560(199911/12)15:6<427::AID-DMRR68>3.0.CO;2-C
- P. Bjorntorp, “Insulin Resistance: The Consequence of a Neuroendocrine Disturbance?” International Journal of Obesity and Related Metabolic Disorders, Vol. 19, Suppl. 1, 1995, pp. S6-S10.
- M. Sjostrand, and J. W. Eriksson, “Neuroendocrine Mechanisms in Insulin Resistance,” Molecular and Cellular Endocrinology, Vol. 297, No. 1-2, 2009, pp. 104-111. doi:10.1016/j.mce.2008.05.010
- J. Gimble, A. Ptitsyn and Z. Floyd, “Circadian Rhythms in Adipose Tissue,” Endocrine Abstracts, Vol. 29, Suppl. 50, 2012, p. 3.
- J. M. Gimble, et al., “Circadian Rhythms in Adipose Tissue: An Update,” Current Opinion in Clinical Nutrition & Metabolic Care, Vol. 14, No. 6, 2011, pp. 554-561. doi:10.1097/MCO.0b013e32834ad94b
- P. Loria, et al., “Endocrine and Liver Interaction: The Role of Endocrine Pathways in NASH,” Nature Reviews. Gastroenterology & Hepatology, Vol. 6, No. 4, 2009, pp. 236-247.
- P. Schrauwen, “Lipid-Induced Cell Stress and Insulin Resistance,” Scandinavian Journal of Food and Nutrition, Vol. 50, Suppl. 2, 2006, pp. 62-67. doi:10.1080/17482970601066132
- J. P. Felber, et al., “Role of Lipid Oxidation in Pathogenesis of Insulin Resistance of Obesity and Type II Diabetes,” Diabetes, Vol. 36, No. 11, 1987, pp. 1341-1350.
- M. F. Gregor and G. S. Hotamisligil, “Inflammatory Mechanisms in Obesity,” Annual Review of Immunology, Vol. 29, 2011, pp. 415-445. doi:10.1146/annurev-immunol-031210-101322
- C. de Luca and J. M. Olefsky, “Inflammation and Insulin Resistance,” FEBS Letters, Vol. 582, No. 1, 2008, pp. 97- 105. doi:10.1016/j.febslet.2007.11.057
- J. K. Suagee, B. A. Corl and R. J. Geor, “A Potential Role for Pro-Inflammatory Cytokines in the Development of Insulin Resistance in Horses,” Animals, Vol. 2, No. 2, 2012, pp. 243-260.doi:10.3390/ani2020243
- B. E. Wisse, “The Inflammatory Syndrome: The Role of Adipose Tissue Cytokines in Metabolic Disorders Linked to Obesity,” Journal of the American Society of Nephrology, Vol. 15, No. 11, 2004, pp. 2792-2800. doi:10.1097/01.ASN.0000141966.69934.21
- J. K. Kiecolt-Glaser, et al., “Chronic Stress and Age-Related Increases in the Proinflammatory Cytokine IL-6,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 100, No. 15, 2003, pp. 9090-9095. doi:10.1073/pnas.1531903100
- H. Anisman and Z. Merali, “Cytokines, Stress, and Depressive Illness,” Brain, Behavior and Immunity, Vol. 16, No. 5, 2002, pp. 513-524.
- A. Marette, “Mediators of Cytokine-Induced Insulin Resistance in Obesity and Other Inflammatory Settings,” Current Opinion in Clinical Nutrition & Metabolic Care, Vol. 5, No. 4, 2002, pp. 377-383. doi:10.1097/00075197-200207000-00005
- N. Ouchi, et al., “Adipokines in Inflammation and Metabolic Disease,” Nature Reviews. Immunology, Vol. 11, No. 2, 2011, pp. 85-97.
- V. Mohamed-Ali, J. H. Pinkney and S. W. Coppack, “Adipose Tissue as an Endocrine and Paracrine Organ,” International Journal of Obesity and Related Metabolic Disorders, Vol. 22, No. 12, 1998, pp. 1145-1158.
- F. D. Popescu, “Cytokines and Neuropeptides in the Relationship Stress-Immunity,” INFO Medica, Vol. 1, No. 143, 2008, pp. 8-10.
- M. G. Myers Jr., et al., “Obesity and Leptin Resistance: Distinguishing Cause from Effect,” Trends in Endocrinology & Metabolism, Vol. 21, No. 11, 2010, pp. 643-651. doi:10.1016/j.tem.2010.08.002
- C. S. Mantzoros, “The Role of Leptin in Human Obesity and Disease: A Review of Current Evidence,” Annals of Internal Medicine, Vol. 130, No. 8, 1999, pp. 671-680. doi:10.7326/0003-4819-130-8-199904200-00014
- S. M. de la Monte, et al., “Insulin Resistance and Neurodegeneration: Roles of Obesity, Type 2 Diabetes Mellitus and Non-Alcoholic Steatohepatitis,” Current Opinion in Investigational Drugs, Vol. 10, No. 10, 2009, pp. 1049-1060.
- E. Stice, et al., “Psychological and Behavioral Risk Factors for Obesity Onset in Adolescent Girls: A Prospective Study,” Journal of Consulting and Clinical Psychology, Vol. 73, No. 2, 2005, pp. 195-202. doi:10.1037/0022-006X.73.2.195
- J. Brody, “The Global Epidemic of Childhood Obesity: Poverty, Urbanization and Nutrition,” Nutrition Bytes, Vol. 8, No. 2, 2002, pp. 1-7.
- B. Biondi, “Thyroid and Obesity: An Intriguing Relationship,” The Journal of Clinical Endocrinology & Metabolism, Vol. 95, No. 8, 2010, pp. 3614-3617. doi:10.1210/jc.2010-1245
- E. N. Pearce, “Thyroid Hormone and Obesity,” Current Opinion in Endocrinology, Diabetes, and Obesity, Vol. 19, No. 5, 2012, pp. 408-413.
- A. Verma, et al., “Hypothyroidism and Obesity. Cause or Effect?” Saudi Medical Journal, Vol. 29, No. 8, 2008, pp. 1135-1138.
- M. Rotondi, F. Magri and L. Chiovato, “Thyroid and Obesity: Not a One-Way Interaction,” The Journal of Clinical Endocrinology & Metabolism, Vol. 96, No. 2, 2011, pp. 344-346. doi:10.1210/jc.2010-2515
- T. Reinehr, “Obesity and Thyroid Function,” Molecular and Cellular Endocrinology, Vol. 316, No. 2, 2010, pp. 165-171. doi:10.1016/j.mce.2009.06.005
- G. Boden, “Free Fatty Acids as Target for Therapy. Diabetes and the Endocrine Pancreas II,” Current Opinion in Endocrinology & Diabetes, Vol. 11, No. 5, 2004, pp. 258-263. doi:10.1097/01.med.0000141928.95173.63
- R. Belfort, et al., “Dose-Response Effect of Elevated Plasma Free Fatty Acid on Insulin Signalling,” Diabetes, Vol. 54, No. 6, 2005, pp. 1640-1648. doi:10.2337/diabetes.54.6.1640
- X. L. Wang, et al., “Free Fatty Acids Inhibit Insulin Signaling-Stimulated Endothelial Nitric Oxide Synthase Activation through Upregulating PTEN or Inhibiting Akt Kinase,” Diabetes, Vol. 55, No. 8, 2006, pp. 2301-2310. doi:10.2337/db05-1574
- W. L. Holland and S. A. Summers, “Sphingolipids, Insulin Resistance, and Metabolic Disease: New Insights from in Vivo Manipulation of Sphingolipid Metabolism,” Endocrine Reviews, Vol. 29, No. 4, 2008, pp. 381-402. doi:10.1210/er.2007-0025
- C. Lipina and H. S. Hundal, “Sphingolipids: Agents Provocateurs in the Pathogenesis of Insulin Resistance,” Diabetologia, Vol. 54, No. 7, 2011, pp. 1596-1607. doi:10.1007/s00125-011-2127-3
- C. Schmitz-Peiffer, “Targeting Ceramide Synthesis to Reverse Insulin Resistance,” Diabetes, Vol. 59, No. 10, 2010, pp. 2351-2353. doi:10.2337/db10-0912
- J. M. Gill and N. Sattar, “Ceramides: A New Player in the Inflammation-Insulin Resistance Paradigm?” Diabetologia, Vol. 52, No. 12, 2009, pp. 2475-2477. doi:10.1007/s00125-009-1546-x
- M. Kolak, et al., “Adipose Tissue Inflammation and Increased Ceramide Content Characterize Subjects with High Liver Fat Content Independent of Obesity,” Diabetes, Vol. 56, No. 8, 2007, pp. 1960-1968. doi:10.2337/db07-0111
- F. Samad, et al., “Adipose Tissue and Ceramide Biosynthesis in the Pathogenesis of Obesity,” Advances in Experimental Medicine and Biology, Vol. 721, 2011, pp. 67- 86. doi:10.1007/978-1-4614-0650-1_5
- S. A. Young, et al., “Sphingolipid and Ceramide Homeostasis: Potential Therapeutic Targets,” Biochemistry Research International, Vol. 2012, 2012, Article ID: 248135.
- R. N. Bergman and M. Ader, “Free Fatty Acids and Pathogenesis of Type 2 Diabetes Mellitus,” Trends in Endocrinology & Metabolism, Vol. 11, No. 9, 2000, pp. 351-356. doi:10.1016/S1043-2760(00)00323-4
- P. J. Meikle and M. J. Christopher, “Lipidomics Is Providing New Insight into the Metabolic Syndrome and Its Sequelae,” Current Opinion in Lipidology, Vol. 22, No. 3, 2011, pp. 210-215. doi:10.1097/MOL.0b013e3283453dbe
- K. H. Pietilainen, et al., “Acquired Obesity Is Associated with Changes in the Serum Lipidomic Profile Independent of Genetic Effects—A Monozygotic Twin Study,” PLoS One, Vol. 2, No. 2, 2007, p. e218. doi:10.1371/journal.pone.0000218
- A. Kontush and M. J. Chapman, “Lipidomics as a Tool for the Study of Lipoprotein Metabolism,” Current Atherosclerosis Reports, Vol. 12, No. 3, 2010, pp. 194- 201. doi:10.1007/s11883-010-0100-0
- P. Puri, et al., “A Lipidomic Analysis of Nonalcoholic Fatty Liver Disease,” Hepatology, Vol. 46, No. 4, 2007, pp. 1081-1090. doi:10.1002/hep.21763
- W. L. Lim, et al., “Effects of a High-Fat, High-Cholesterol Diet on Brain Lipid Profiles in Apolipoprotein E Epsilon3 and Epsilon4 Knock-In Mice,” Neurobiology of Aging, Vol. 34, No. 9, 2013, pp. 2217-2224. doi:10.1016/j.neurobiolaging.2013.03.012
- I. J. Martins, K. M. Wood, A. Z. Fernandez, K. Taddei, R. N. Martins, “Anti-Oxidative Acyl CoA Cholesterol Acyltransferase Inhibitor AVASIMIBE Reduces the Impact of a High Cholesterol Diet on Brain Lipid Peroxidation in Mice,” ADPD, Florence, 2013. www.kenes.com.
- M. O. Grimm, et al., “Trans Fatty Acids Enhance Amyloidogenic Processing of the Alzheimer Amyloid Precursor Protein (APP),” The Journal of Nutritional Biochemistry, Vol. 23, No. 10, 2012, pp. 1214-1223. doi:10.1016/j.jnutbio.2011.06.015
- [61] M. O. Grimm, et al., “Effect of Different Phospholipids on alpha-Secretase Activity in the Non-Amyloidogenic Pathway of Alzheimer’s Disease,” International Journal of Molecular Sciences, Vol. 14, No. 3, 2013, pp. 5879- 5898. doi:10.3390/ijms14035879
- [62] L. Arana, et al., “Ceramide and Ceramide 1-Phosphate in Health and Disease,” Lipids in Health and Disease, Vol. 9, 2010, p. 15. doi:10.1186/1476-511X-9-15
- [63] R. L. Creegan, W. L. F. Lin, I. J. Martins, S. M. Laws, V. Gupta, S. Rainey-Smith, et al., “Plasma Lipidomics in Alzheimer’s disease (AD); Preliminary Data from the Australian Imaging, Biomarkers and Lifestyle Study (AIBL) of Ageing,” ADPD, Florence, 2013. www.kenes.com
- [64] J. T. Lee, et al., “Amyloid-Beta Peptide Induces Oligodendrocyte Death by Activating the Neutral Sphingomyelinase-Ceramide Pathway,” The Journal of Cell Biology, Vol. 164, No. 1, 2004, pp. 123-131. doi:10.1083/jcb.200307017
- [65] L. Puglielli, et al., “Ceramide Stabilizes Beta-Site Amyloid Precursor Protein-Cleaving Enzyme 1 and Promotes Amyloid Beta-Peptide Biogenesis,” The Journal of Biological Chemistry, Vol. 278, No. 22, 2003, pp. 19777- 19783. doi:10.1074/jbc.M300466200
- [66] T. H. Jones and R. L. Kennedy, “Cytokines and Hypothalamic-Pituitary Function,” Cytokine, Vol. 5, No. 6, 1993, pp. 531-538. doi:10.1016/S1043-4666(05)80001-8
- [67] M. Bentivoglio, et al., “The Aging Suprachiasmatic Nucleus and Cytokines: Functional, Molecular, and Cellular Changes in Rodents,” Chronobiology International, Vol. 23, No. 1-2, 2006, pp. 437-449. doi:10.1080/07420520500545797
- [68] M. Bentivoglio, M. Nygård, M. Palomba and K. Kristensson, “The Biological Clock in Inflammation and Sleep Switch Alterations,” Neuroimmunology of Sleep, Vol. 1, 2007, pp. 87-96. doi:10.1007/978-0-387-69146-6_4
- [69] J. B. Buchanan and R. W. Johnson, “Regulation of Food Intake by Inflammatory Cytokines in the Brain,” Neuroendocrinology, Vol. 86, No. 3, 2007, pp. 183-190. doi:10.1159/000108280
- [70] B. J. Mickey, et al., “Emotion Processing, Major Depression, and Functional Genetic Variation of Neuropeptide Y,” Archives of General Psychiatry, Vol. 68, No. 2, 2011, pp. 158-166. doi:10.1001/archgenpsychiatry.2010.197
- [71] A. Thorsell, “Brain Neuropeptide Y and CorticotropinReleasing Hormone in Mediating Stress and Anxiety,” Experimental Biology and Medicine, Vol. 235, No. 10, 2010, pp. 1163-1167. doi:10.1258/ebm.2010.009331
- [72] Y. Koutmani, et al., “Corticotropin-Releasing Hormone Exerts Direct Effects on Neuronal Progenitor Cells: Implications for Neuroprotection,” Molecular Psychiatry, Vol. 18, No. 3, 2013, pp. 300-307. doi:10.1038/mp.2012.198
- [73] G. Nikisch, et al., “Neuropeptide Y and CorticotropinReleasing Hormone in CSF Mark Response to Antidepressive Treatment with Citalopram,” The International Journal of Neuropsychopharmacology, Vol. 8, No. 3, 2005, pp. 403-410. doi:10.1017/S1461145705005158
- [74] D. A. Haas and S. R. George, “Neuropeptide Y-Induced Effects on Hypothalamic Corticotropin-Releasing Factor Content and Release Are Dependent on Noradrenergic/ Adrenergic Neurotransmission,” Brain Research, Vol. 498, No. 2, 1989, pp. 333-338. doi:10.1016/0006-8993(89)91112-8
- [75] M. Jang and D. R. Romsos, “Neuropeptide Y and Corticotropin-Releasing Hormone Concentrations within Specific Hypothalamic Regions of Lean but not ob/ob Mice Respond to Food-Deprivation and Refeeding,” The Journal of Nutrition, Vol. 128, No. 12, 1998, pp. 2520-2525.
- [76] J. R. Glowa, et al., “Effects of Corticotropin Releasing Hormone on Appetitive Behaviours,” Peptides, Vol. 13, No. 3, 1992, pp. 609-621. doi:10.1016/0196-9781(92)90097-M
- [77] T. Katsuyoshi, “Hormonal Mechanism of Appetite Control and Clinical Application. Basis: Hormone and Appetite Control. Role of CRH, Urocortin and Glucocorticoid in the Regulation of Food Intake,” Clinical Endocrinology, Vol. 53, 2005, pp. 361-370.
- [78] R. Denis, “The Role of Corticotropin-Releasing Hormone in the Regulation of Energy Balance,” Current Opinion in Endocrinology & Diabetes, Vol. 6, No. 1, 1999, p. 10. doi:10.1097/00060793-199902000-00003
- [79] D. Richard, Q. Huang and E. Timofeeva, “The Corticotropin-Releasing Hormone System in the Regulation of Energy Balance in Obesity,” International Journal of Obesity, Vol. 24, Suppl. 2, 2000, pp. S36-S39. doi:10.1038/sj.ijo.0801275
- [80] K. Roguski, et al., “Leptin Modulates the CorticotropinReleasing Hormone (CRH) Action on Progesterone Release from Cultured Rat Granulosa Cells,” Neuro Endocrinology Letters, Vol. 21, No. 5, 2000, pp. 383-389.
- [81] K. Arvaniti, Q. Huang and D. Richard, “Effects of Leptin and Corticosterone on the Expression of CorticotropinReleasing Hormone, Agouti-Related Protein, and Proopiomelanocortin in the Brain of ob/ob Mouse,” Neuroendocrinology, Vol. 73, No. 4, 2001, pp. 227-236. doi:10.1159/000054639
- [82] H. E. Auvinena, J. A. Romijn, N. R. Biermasza, et al., “The Effects of High Fat Diet on the Basal Activity of the Hypothalmus-Pituitary-Adrenal Axis in Mice,” Journal of Endocrinology, Vol. 214, No. 2, 2012, pp. 191-197.
- [83] M. Chavez, et al., “Effect of a High-Fat Diet on Food Intake and Hypothalamic Neuropeptide Gene Expression in Streptozotocin Diabetes,” The Journal of Clinical Investigation, Vol. 102, No. 2, 1998, pp. 340-346. doi:10.1172/JCI603
- [84] H. Husum and A. A. Mathe, “Early Life Stress Changes Concentrations of Neuropeptide Y and Corticotropin-Releasing Hormone in Adult Rat Brain. Lithium Treatment Modifies These Changes,” Neuropsychopharmacology, Vol. 27, No. 5, 2002, pp. 756-764. doi:10.1016/S0893-133X(02)00363-9
- [85] J. M. Stafford, et al., “Central Nervous System Neuropeptide Y Signaling Modulates VLDL Triglyceride Secretion,” Diabetes, Vol. 57, No. 6, 2008, pp. 1482-1490. doi:10.2337/db07-1702
- [86] J. J. Geerling, et al., “Acute Central Neuropeptide Y Administration Increases Food Intake but Does Not Affect Hepatic Very Low-Density Lipoprotein (VLDL) Production in Mice,” PLoS One, Vol. 8, No. 2, 2013, p. e55217. doi:10.1371/journal.pone.0055217
- [87] K. A. Paschos, et al., “Corticotropin-Releasing Hormone Receptors Mediate Opposing Effects in Cholestasis-Induced Liver Cell Apoptosis,” Endocrinology, Vol. 151, No. 4, 2010, pp. 1704-1712. doi:10.1210/en.2009-1208
- [88] N. Bayatti and C. Behl, “The Neuroprotective Actions of Corticotropin Releasing Hormone,” Ageing Research Reviews, Vol. 4, No. 2, 2005, pp. 258-270. doi:10.1016/j.arr.2005.02.004
- [89] J. E. Kang, et al., “Acute Stress Increases Interstitial Fluid Amyloid-Beta via Corticotropin-Releasing Factor and Neuronal Activity,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 104, No. 25, 2007, pp. 10673-10678. doi:10.1073/pnas.0700148104
- [90] W. A. Pedersen, et al., “Corticotropin-Releasing Hormone Protects Neurons against Insults Relevant to the Pathogenesis of Alzheimer’s Disease,” Neurobiology of Disease, Vol. 8, No. 3, 2001, pp. 492-503. doi:10.1006/nbdi.2001.0395
- [91] F. Lezoualc’h, et al., “Corticotropin-Releasing HormoneMediated Neuroprotection against Oxidative Stress Is Associated with the Increased Release of Non-Amyloidogenic Amyloid Beta Precursor Protein and with the Suppression of Nuclear Factor-kappaB,” Molecular Endocrinology, Vol. 14, No. 1, 2000, pp. 147-159. doi:10.1210/me.14.1.147
- [92] D. Rat, et al., “Neuropeptide Pituitary Adenylate CyclaseActivating Polypeptide (PACAP) Slows Down Alzheimer’s Disease-Like Pathology in Amyloid Precursor Protein-Transgenic Mice,” The FASEB Journal, Vol. 25, No. 9, 2011, pp. 3208-3218. doi:10.1096/fj.10-180133
- [93] R. Postina, “Activation of Alpha-Secretase Cleavage,” Journal of Neurochemistry, Vol. 120, Suppl. 1, 2012, pp. 46-54. doi:10.1111/j.1471-4159.2011.07459.x
- [94] S. Parvathy, et al., “Atorvastatin-Induced Activation of Alzheimer’s Alpha Secretase Is Resistant to Standard Inhibitors of Protein Phosphorylation-Regulated Ectodomain Shedding,” Journal of Neurochemistry, Vol. 90, No. 4, 2004, pp. 1005-1010. doi:10.1111/j.1471-4159.2004.02521.x
- [95] D. M. Skovronsky, et al., “Protein Kinase C-Dependent Alpha-Secretase Competes with Beta-Secretase for Cleavage of Amyloid-Beta Precursor Protein in the TransGolgi Network,” The Journal of Biological Chemistry, Vol. 275, No. 4, 2000, pp. 2568-2575. doi:10.1074/jbc.275.4.2568
- [96] S. E. Hammack, et al., “Roles for Pituitary Adenylate Cyclase-Activating Peptide (PACAP) Expression and Signaling in the Bed Nucleus of the Stria Terminalis (BNST) in Mediating the Behavioral Consequences of Chronic Stress,” Journal of Molecular Neuroscience, Vol. 4, No. 3, 2010, pp. 327-340. doi:10.1007/s12031-010-9364-7
- [97] L. Mounien, et al., “Pituitary Adenylate Cyclase-Activating Polypeptide Inhibits Food Intake in Mice through Activation of the Hypothalamic Melanocortin System,” Neuropsychopharmacology, Vol. 34, No. 2, 2009, pp. 424-435. doi:10.1038/npp.2008.73
- [98] J. E. Morley, et al., “Pituitary Adenylate Cyclase Activating Polypeptide (PACAP) Reduces Food Intake in Mice,” Peptides, Vol. 13, No. 6, 1992, pp. 1133-1135. doi:10.1016/0196-9781(92)90019-Y
- [99] X. Qu, J. P. Seale and R. Donnelly, “Tissue and IsoformSelective Activation of Protein Kinase C in Insulin-Resistant Obese Zucker Rats-Effects of Feeding,” Journal of Endocrinology, Vol. 162, No. 2, 1999, pp. 207-214. doi:10.1677/joe.0.1620207
- [100] M. Sastre, T. Klockgether and M. T. Heneka, “Contribution of Inflammatory Processes to Alzheimer’s Disease: Molecular Mechanisms,” International Journal of Developmental Neuroscience, Vol. 24, No. 2-3, 2006, pp. 167-176. doi:10.1016/j.ijdevneu.2005.11.014
- [101] N. S. Patel, et al., “Inflammatory Cytokine Levels Correlate with Amyloid Load in Transgenic Mouse Models of Alzheimer’s Disease,” Journal of Neuroinflammation, Vol. 2, No. 1, 2005, p. 9. doi:10.1186/1742-2094-2-9
- [102] S. Askarova, X. Yang and J. C. Lee, “Impacts of Membrane Biophysics in Alzheimer’s Disease: From Amyloid Precursor Protein Processing to Abeta Peptide-Induced Membrane Changes,” International Journal of Alzheimer’s Disease, Vol. 2011, 2011, Article ID: 134971.
- [103] N. Pannacciulli, et al., “Brain Abnormalities in Human Obesity: A Voxel-Based Morphometric Study,” Neuroimage, Vol. 31, No. 4, 2006, pp. 1419-1425. doi:10.1016/j.neuroimage.2006.01.047
- [104] C. A. Raji, et al., “Brain Structure and Obesity,” Human Brain Mapping, Vol. 31, No. 3, 2010, pp. 353-364.
- [105] J. P. Thaler and M. W. Schwartz, “Minireview: Inflammation and Obesity Pathogenesis: The Hypothalamus Heats Up,” Endocrinology, Vol. 151, No. 9, 2010, pp. 4109-4115. doi:10.1210/en.2010-0336
- [106] A. A. Kandutsch, H. J. Heiniger and H. W. Chen, “Effects of 25-Hydroxycholesterol and 7-Ketocholesterol, Inhibitors of Sterol Synthesis, Administered Orally to Mice,” Biochimica et Biophysica Acta, Vol. 486, No. 2, 1977, pp. 260-272. doi:10.1016/0005-2760(77)90022-4
- [107] K. Shimizu, et al., “Anti-Obesity Effect of Phosphatidylinositol on Diet-Induced Obesity in Mice,” Journal of Agricultural and Food Chemistry, Vol. 58, No. 21, 2010, pp. 11218-11225. doi:10.1021/jf102075j
- [108] A. S. Metlakunta, M. Sahu and A. Sahu, “Hypothalamic Phosphatidylinositol 3-Kinase Pathway of Leptin Signaling Is Impaired during the Development of Diet-Induced Obesity in FVB/N Mice,” Endocrinology, Vol. 149, No. 3, 2008, pp. 1121-1128. doi:10.1210/en.2007-1307
- [109] J. P. Warne, et al., “Impairment of Central Leptin-Mediated PI3K Signaling Manifested as Hepatic Steatosis Independent of Hyperphagia and Obesity,” Cell Metabolism, Vol. 14, No. 6, 2011, pp. 791-803. doi:10.1016/j.cmet.2011.11.001
- [110] J. W. Hill, et al., “Acute Effects of Leptin Require PI3K Signaling in Hypothalamic Proopiomelanocortin Neurons in Mice,” The Journal of Clinical Investigation, Vol. 118, No. 5, 2008, pp. 1796-1805. doi:10.1172/JCI32964
- [111] K. D. Niswender, et al., “Insulin Activation of Phosphatidylinositol 3-Kinase in the Hypothalamic Arcuate Nucleus: A Key Mediator of Insulin-Induced Anorexia,” Diabetes, Vol. 52, No. 2, 2003, pp. 227-231. doi:10.2337/diabetes.52.2.227
- [112] S. Gao, et al., “Important Roles of Brain-Specific Carnitine Palmitoyltransferase and Ceramide Metabolism in Leptin Hypothalamic Control of Feeding,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 108, No. 23, 2011, pp. 9691-9696. doi:10.1073/pnas.1103267108
- [113] X. F. Gao, W. Chen, X. P. Kong, A. M. Xu, Z. G. Wang, G. Sweeney and D. Wu, “Enhanced Susceptibility of Cpt1c Knockout Mice to Glucose Intolerance Induced by a High-Fat Diet Involves Elevated Hepatic Gluconeogenesis and Decreased Skeletal Muscle Glucose Uptake,” The Journal of Biological Chemistry, Vol. 287, No. 25, 2012, pp. 212-243.
- [114] M. D. Lane, et al., “Regulation of Food Intake and Energy Expenditure by Hypothalamic Malonyl-CoA,” International Journal of Obesity, Vol. 32, Suppl. 4, 2008, pp. S49-S54. doi:10.1038/ijo.2008.123
- [115] S. Ramirez, et al., “Hypothalamic Ceramide Levels Regulated by CPT1C Mediate the Orexigenic Effect of Ghrelin,” Diabetes, Vol. 62, No. 7, 2013, pp. 2329-2337. doi:10.2337/db12-1451
- [116] P. Carrasco, et al., “Ceramide Levels Regulated by Carnitine Palmitoyltransferase 1C Control Dendritic Spine Maturation and Cognition,” The Journal of Biological Chemistry, Vol. 287, No. 25, 2012, pp. 21224-21232. doi:10.1074/jbc.M111.337493
- [117] Z. Hu, et al., “A Role for Hypothalamic Malonyl-CoA in the Control of Food Intake,” The Journal of Biological Chemistry, Vol. 280, No. 48, 2005, pp. 39681-39683. doi:10.1074/jbc.C500398200
- [118] S. Gao, et al., “Malonyl-CoA Mediates Leptin Hypothalamic Control of Feeding Independent of Inhibition of CPT-1a,” American Journal of Physiology: Regulatory, Integrative and Comparative Physiology, Vol. 301, No. 1, 2011, pp. R209-R217. doi:10.1152/ajpregu.00092.2011
- [119] M. J. Wolfgang, et al., “The Brain-Specific Carnitine Palmitoyltransferase-1c Regulates Energy Homeostasis,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 103, No. 19, 2006, pp. 7282-7287. doi:10.1073/pnas.0602205103
- [120] R. Lage, et al., “Ghrelin Effects on Neuropeptides in the Rat Hypothalamus Depend on Fatty Acid Metabolism Actions on BSX but Not on Gender,” The FASEB Journal, Vol. 24, No. 8, 2010, pp. 2670-2679. doi:10.1096/fj.09-150672
- [121] L. Herrero, et al., “Alteration of the Malonyl-CoA/Carnitine Palmitoyltransferase I Interaction in the Beta-Cell Impairs Glucose-Induced Insulin Secretion,” Diabetes, Vol. 54, No. 2, 2005, pp. 462-471. doi:10.2337/diabetes.54.2.462
- [122] M. D. Lane and S. H. Cha, “Effect of Glucose and Fructose on Food Intake via Malonyl-CoA Signaling in the Brain,” Biochemical and Biophysical Research Communications, Vol. 382, No. 1, 2009, pp. 1-5. doi:10.1016/j.bbrc.2009.02.145
- [123] M. J. Wolfgang, et al., “Regulation of Hypothalamic Malonyl-CoA by Central Glucose and Leptin,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 104, No. 49, 2007, pp. 19285- 19290. doi:10.1073/pnas.0709778104
- [124] F. Pintus, G. Floris and A. Rufini, “Nutrient Availability Links Mitochondria, Apoptosis, and Obesity,” Aging, Vol. 4, No. 11, 2012, pp. 734-741.
- [125] W. I. Sivitz, “Mitochondria Dysfunction in Obesity and Diabetes,” Endocrinology, Vol. 6, No. 1, 2010, pp. 20-27.
- [126] P. L. Yau, et al., “Obesity and Metabolic Syndrome and Functional and Structural Brain Impairments in Adolescence,” Pediatrics, Vol. 130, No. 4, 2012, pp. e856-e864. doi:10.1542/peds.2012-0324
- [127] J. J. Hillebrand, D. de Wied and R. A. Adan, “Neuropeptides, Food Intake and Body Weight Regulation: A Hypothalamic Focus,” Peptides, Vol. 23, No. 12, 2002, pp. 2283-2306. doi:10.1016/S0196-9781(02)00269-3
- [128] M. W. Schwartz, “Central Nervous System Control of Food Intake,” Nature, Vol. 404, 2007, pp. 661-671.
- [129] B. C. Field, O. B. Chaudhri and S. R. Bloom, “Obesity Treatment: Novel Peripheral Targets,” British Journal of Clinical Pharmacology, Vol. 68, No. 6, 2009, pp. 830- 843. doi:10.1111/j.1365-2125.2009.03522.x
- [130] T. J. Bartness and C. K. Song, “Brain-Adipose Tissue Neural Crosstalk,” Physiology & Behavior, Vol. 91, No. 4, 2007, pp. 343-351. doi:10.1016/j.physbeh.2007.04.002
- [131] R. S. Ahima and D. A. Antwi, “Brain Regulation of Appetite and Satiety,” Endocrinology and Metabolism Clinics of North America, Vol. 37, No. 4, 2008, pp. 811-823. doi:10.1016/j.ecl.2008.08.005
- [132] H. Zheng, et al., “Appetite Control and Energy Balance Regulation in the Modern World: Reward-Driven Brain Overrides Repletion Signals,” International Journal of Obesity, Vol. 33, Suppl. 2, 2009, pp. S8-S13. doi:10.1038/ijo.2009.65
- [133] P. J. Havel, “Peripheral Signals Conveying Metabolic Information to the Brain: Short-Term and Long-Term Regulation of Food Intake and Energy Homeostasis,” Experimental Biology and Medicine, Vol. 226, No. 11, 2001, pp. 963-977.
- [134] A. J. Bruce-Keller, J. N. Keller and C. D. Morrison, “Obesity and Vulnerability of the CNS,” Biochimica et Biophysica Acta, Vol. 1792, No. 5, 2009, pp. 395-400. doi:10.1016/j.bbadis.2008.10.004
- [135] K. Suzuki, C. N. Jayasena and S. R. Bloom, “Obesity and Appetite Control,” Experimental Diabetes Research, Vol. 2012, 2012, Article ID: 824305.
- [136] C. X. Yi, et al., “Ventromedial Arcuate Nucleus Communicates Peripheral Metabolic Information to the Suprachiasmatic Nucleus,” Endocrinology, Vol. 147, No. 1, 2006, pp. 283-294. doi:10.1210/en.2005-1051
- [137] O. B. Chaudhri, K. Wynne and S. R. Bloom, “Can Gut Hormones Control Appetite and Prevent Obesity?” Diabetes Care, Vol. 31, Suppl. 2, 2008, pp. S284-S289. doi:10.2337/dc08-s269
- [138] S. Chearskul, et al., “Obesity and Appetite-Related Hormones,” Journal of the Medical Association of Thailand, Vol. 95, No. 11, 2012, pp. 1472-1479.
- [139] K. Suzuki, C. N. Jayasena and S. R. Bloom, “The Gut Hormones in Appetite Regulation,” Journal of Obesity, Vol. 2011, 2011, Article ID: 528401.
- [140] K. A. Simpson and S. R. Bloom, “Appetite and Hedonism: Gut Hormones and the Brain,” Endocrinology and Metabolism Clinics of North America, Vol. 39, No. 4, 2010, pp. 729-743. doi:10.1016/j.ecl.2010.08.001
- [141] C. X. Yi and M. H. Tschop, “Brain-Gut-Adipose-Tissue Communication Pathways at a Glance,” Diseases Models & Mechanisms, Vol. 5, No. 5, 2012, pp. 583-587. doi:10.1242/dmm.009902
- [142] P. C. Konturek, T. Brzozowski and S. J. Konturek, “Gut Clock: Implication of Circadian Rhythms in the Gastrointestinal Tract,” Journal of Physiology and Pharmacology, Vol. 62, No. 2, 2011, pp. 139-150.
- [143] C. Dibner, U. Schibler and U. Albrecht, “The Mammalian Circadian Timing System: Organization and Coordination of Central and Peripheral Clocks,” Annual Review of Physiology, Vol. 72, 2010, pp. 517-549. doi:10.1146/annurev-physiol-021909-135821
- [144] O. Froy, “Metabolism and Circadian Rhythms—Implications for Obesity,” Endocrine Reviews, Vol. 31, No. 1, 2010, pp. 1-24. doi:10.1210/er.2009-0014
- [145] O. Froy, “Circadian Rhythms and Obesity in Mammals,” ISRN Obesity, Vol. 2012, 2012, Article ID: 437198. doi:10.5402/2012/437198
- [146] J. Mendoza and E. Challet, “Brain Clocks: From the Suprachiasmatic Nuclei to a Cerebral Network,” Neuroscientist, Vol. 15, No. 5, 2009, pp. 477-488. doi:10.1177/1073858408327808
- [147] C. B. Saper, et al., “The Hypothalamic Integrator for Circadian Rhythms,” Trends in Neurosciences, Vol. 28, No. 3, 2005, pp. 152-157. doi:10.1016/j.tins.2004.12.009
- [148] M. Lee and J. Korner, “Review of Physiology, Clinical Manifestations, and Management of Hypothalamic Obesity in Humans,” Pituitary, Vol. 12, No. 2, 2009, pp. 87- 95. doi:10.1007/s11102-008-0096-4
- [149] S. Baloyannis, “The Hypothalamus in Alzheimer’s Disease: A Study with Silver Impregnation Techniques and Electron Microscope,” Alzheimer’s and Dementia, Vol. 5, No. 4, 2009, p. 40. doi:10.1016/j.jalz.2009.05.644
- [150] D. F. Swaab, et al., “The Human Hypothalamus in Development, Sexual Differentiation, Aging and Alzheimer’s Disease,” Progress in Brain Research, Vol. 91, 1992, pp. 465-472. doi:10.1016/S0079-6123(08)62369-9
- [151] J. A. van de Nes, et al., “The Distribution of Alz-50 Immunoreactivity in the Hypothalamus and Adjoining Areas of Alzheimer’s Disease Patients,” Brain, Vol. 116, No. 1, 1993, pp. 103-115. doi:10.1093/brain/116.1.103
- [152] J. W. Langston and L. S. Forno, “The Hypothalamus in Parkinson Disease,” Annals of Neurology, Vol. 3, No. 2, 1978, pp. 129-133. doi:10.1002/ana.410030207
- [153] K. Otake, et al., “Hypothalamic Dysfunction in Parkinson’s Disease Patients,” Acta Medica Hungarica, Vol. 50, No. 1-2, 1994, pp. 3-13.
- [154] E. Fabbrini, S. Sullivan and S. Klein, “Obesity and Nonalcoholic Fatty Liver Disease: Biochemical, Metabolic, and Clinical Implications,” Hepatology, Vol. 51, No. 2, 2010, pp. 679-689. doi:10.1002/hep.23280
- [155] J. G. Langendonk, et al., “Circadian Rhythm of Plasma Leptin Levels in Upper and Lower Body Obese Women: Influence of Body Fat Distribution and Weight Loss,” The Journal of Clinical Endocrinology & Metabolism, Vol. 83, No. 5, 1998, pp. 1706-1712. doi:10.1210/jc.83.5.1706
- [156] D. P. Figlewicz, “Adiposity Signals and Food Reward: Expanding the CNS Roles of Insulin and Leptin,” American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, Vol. 284, No. 4, 2003, pp. R882-R892.
- [157] J. Harvey and M. L. Ashford, “Leptin in the CNS: Much More than a Satiety Signal,” Neuropharmacology, Vol. 44, No. 7, 2003, pp. 845-854. doi:10.1016/S0028-3908(03)00076-5
- [158] S. P. Kalra, et al., “Rhythmic, Reciprocal Ghrelin and Leptin Signaling: New Insight in the Development of Obesity,” Regulatory Peptides, Vol. 111, No. 1-3, 2003, pp. 1-11. doi:10.1016/S0167-0115(02)00305-1
- [159] M. D. Klok, S. Jakobsdottir and M. L. Drent, “The Role of Leptin and Ghrelin in the Regulation of Food Intake and Body Weight in Humans: A Review,” Obesity Reviews, Vol. 8, No. 1, 2007, pp. 21-34. doi:10.1111/j.1467-789X.2006.00270.x
- [160] B. Bodosi, et al., “Rhythms of Ghrelin, Leptin, and Sleep in Rats: Effects of the Normal Diurnal Cycle, Restricted Feeding, and Sleep Deprivation,” American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, Vol. 287, No. 5, 2004, pp. R1071-R1079. doi:10.1152/ajpregu.00294.2004
- [161] J. Sanchez, et al., “Diurnal Rhythms of Leptin and Ghrelin in the Systemic Circulation and in the Gastric Mucosa Are Related to Food Intake in Rats,” Pflügers Archiv, Vol. 448, No. 5, 2004, pp. 500-506. doi:10.1007/s00424-004-1283-4
- [162] A. Kalsbeek, et al., “The Suprachiasmatic Nucleus Generates the Diurnal Changes in Plasma Leptin Levels,” Endocrinology, Vol. 142, No. 6, 2001, pp. 2677-2685. doi:10.1210/en.142.6.2677
- [163] D. K. Oh, T. Ciaraldi and R. R. Henry, “Adiponectin in Health and Disease,” Diabetes, Obesity and Metabolism, Vol. 9, No. 3, 2007, pp. 282-289. doi:10.1111/j.1463-1326.2006.00610.x
- [164] Y. Qi, et al., “Adiponectin Acts in the Brain to Decrease Body Weight,” Nature Medicine, Vol. 10, No. 5, 2004, pp. 524-529. doi:10.1038/nm1029
- [165] S. Dridi and M. Taouis, “Adiponectin and Energy Homeostasis: Consensus and Controversy,” The Journal of Nutritional Biochemistry, Vol. 20, No. 11, 2009, pp. 831- 839. doi:10.1016/j.jnutbio.2009.06.003
- [166] J. Kawano and R. Arora, “The Role of Adiponectin in Obesity, Diabetes, and Cardiovascular Disease,” Journal of the CardioMetabolic Syndrome, Vol. 4, No. 1, 2009, pp. 44-49. doi:10.1111/j.1559-4572.2008.00030.x
- [167] S. J. Chen, et al., “Relationships between Inflammation, Adiponectin, and Oxidative Stress in Metabolic Syndrome,” PLoS One, Vol. 7, No. 9, 2012, p. e45693. doi:10.1371/journal.pone.0045693
- [168] J. P. Whitehead, et al., “Adiponectin—A Key Adipokine in the Metabolic Syndrome,” Diabetes, Obesity and Metabolism, Vol. 8, No. 3, 2006, pp. 264-280. doi:10.1111/j.1463-1326.2005.00510.x
- [169] K. Rabe, et al., “Adipokines and Insulin Resistance,” Molecular Medicine, Vol. 14, No. 11-12, 2008, pp. 741- 751. doi:10.2119/2008-00058.Rabe
- [170] S. Yaturu, S. Prado and S. R. Grimes, “Changes in Adipocyte Hormones Leptin, Resistin, and Adiponectin in Thyroid Dysfunction,” Journal of Cellular Biochemistry, Vol. 93, No. 3, 2004, pp. 491-496. doi:10.1002/jcb.20188
- [171] M. Barnea, Z. Madar and O. Froy, “High-Fat Diet Delays and Fasting Advances the Circadian Expression of Adiponectin Signaling Components in Mouse Liver,” Endocrinology, Vol. 150, No. 1, 2009, pp. 161-168. doi:10.1210/en.2008-0944
- [172] E. Guillod-Maximin, et al., “Adiponectin Receptors Are Expressed in Hypothalamus and Colocalized with Proopiomelanocortin and Neuropeptide Y in Rodent Arcuate Neurons,” Journal of Endocrinology, Vol. 200, No. 1, 2009, pp. 93-105. doi:10.1677/JOE-08-0348
- [173] J. Thundyil, et al., “Adiponectin receptor signalling in the brain,” British Journal of Pharmacology, Vol. 165, No. 2, 2012, pp. 313-327. doi:10.1111/j.1476-5381.2011.01560.x
- [174] L. Qiao, et al., “Adiponectin Reduces Plasma Triglyceride by Increasing VLDL Triglyceride Catabolism,” Diabetes, Vol. 57, No. 7, 2008, pp. 1824-1833. doi:10.2337/db07-0435
- [175] X. Wu, et al., “Involvement of AMP-Activated Protein Kinase in Glucose Uptake Stimulated by the Globular Domain of Adiponectin in Primary Rat Adipocytes,” Diabetes, Vol. 52, No. 6, 2003, pp. 1355-1363. doi:10.2337/diabetes.52.6.1355
- [176] N. B. Ruderman, A. K. Saha and E. W. Kraegen, “Minireview: Malonyl CoA, AMP-Activated Protein Kinase, and Adiposity,” Endocrinology, Vol. 144, No. 12, 2003, pp. 5166-5171. doi:10.1210/en.2003-0849
- [177] J. Karbowska and Z. Kochan, “Role of Adiponectin in the Regulation of Carbohydrate and Lipid Metabolism,” Journal of Physiology and Pharmacology, Vol. 57, Suppl. 6, 2006, pp. 103-113.
- [178] L. Cantarini, et al., “Serum Leptin, Resistin, Visfatin and Adiponectin Levels in Tumor Necrosis Factor ReceptorAssociated Periodic Syndrome (TRAPS),” Clinical and Experimental Rheumatology, Vol. 30, No. 3, 2012, pp. S108-S114.
- [179] T. M. van Himbergen, et al., “Biomarkers for Insulin Resistance and Inflammation and the Risk for All-Cause Dementia and Alzheimer Disease: Results from the Framingham Heart Study,” Archives of Neurology, Vol. 69, No. 5, 2012, pp. 594-600. doi:10.1001/archneurol.2011.670
- [180] N. R. Cutler, “Neuropeptides in Alzheimer’s Disease: Somatostatin, Neuropeptide Y and Galanin,” American Journal of Alzheimer’s Care and Related Disorders and Reseacrh, Vol. 5, No. 1, 1990, pp. 7-12. doi:10.1177/153331759000500103
- [181] H. Umegaki, “Neurodegeneration in Diabetes Mellitus,” Advances in Experimental Medicine and Biology, Vol. 724, 2012, pp. 258-265. doi:10.1007/978-1-4614-0653-2_19
- [182] M. F. Beal, et al., “Neuropeptide Y Immunoreactivity Is Reduced in Cerebral Cortex in Alzheimer’s Disease,” Annals of Neurology, Vol. 20, No. 3, 1986, pp. 282-288. doi:10.1002/ana.410200303
- [183] N. Croce, et al., “Neuropeptide Y Protects Rat Cortical Neurons against Beta-Amyloid Toxicity and Re-Establishes Synthesis and Release of Nerve Growth Factor,” ACS Chemical Neuroscience, Vol. 3, No. 4, 2012, pp. 312-318. doi:10.1021/cn200127e
- [184] B. Beck, “Neuropeptide Y in Normal Eating and in Genetic and Dietary-Induced Obesity,” Philosophical Transactions of the Royal Society B Biological Science, Vol. 361, No. 1471, 2006, pp. 1159-1185. doi:10.1098/rstb.2006.1855
- [185] M. Stachon, E. Furstenberg and J. Gromadzka-Ostrowska, “Effects of High-Fat Diets on Body Composition, Hypothalamus NPY, and Plasma Leptin and Corticosterone Levels in Rats,” Endocrine, Vol. 30, No. 1, 2006, pp. 69- 74. doi:10.1385/ENDO:30:1:69
- [186] H. Wang, L. H. Storlien and X. F. Huang, “Effects of Dietary Fat Types on Body Fatness, Leptin, and ARC Leptin Receptor, NPY, and AgRP mRNA Expression,” American Journal of Physiology-Endocrinology and Metabolism, Vol. 282, No. 6, 2002, pp. E1352-E1359.
- [187] M. Heilig, “The NPY System in Stress, Anxiety and Depression,” Neuropeptides, Vol. 38, No. 4, 2004, pp. 213- 224. doi:10.1016/j.npep.2004.05.002
- [188] S. D. Primeaux, et al., “Effects of Altered Amygdalar Neuropeptide Y Expression on Anxiety-Related Behaviours,” Neuropsychopharmacology, Vol. 30, No. 9, 2005, pp. 1589-1597. doi:10.1038/sj.npp.1300705
- [189] R. O. Tasan, et al., “The Central and Basolateral Amygdala Are Critical Sites of Neuropeptide Y/Y2 ReceptorMediated Regulation of Anxiety and Depression,” The Journal of Neuroscience, Vol. 30, No. 18, 2010, pp. 6282-6290. doi:10.1523/JNEUROSCI.0430-10.2010
- [190] C. Veyrat-Durebex, et al., “Aging and Long-Term Caloric Restriction Regulate Neuropeptide Y Receptor Subtype Densities in the Rat Brain,” Neuropeptides, Vol. 47, No. 3, 2013, pp. 163-169. doi:10.1016/j.npep.2013.01.001
- [191] M. I. Uusitupa, et al., “Neuropeptide Y: A Novel Link between the Neuroendocrine System and Cholesterol Metabolism,” Annals of Medicine, Vol. 30, No. 6, 1998, pp. 508-510. doi:10.3109/07853899809002597
- [192] J. M. Rojas, et al., “Central Nervous System Neuropeptide Y Signaling via the Y1 Receptor Partially Dissociates Feeding Behavior from Lipoprotein Metabolism in Lean Rats,” American Journal of Physiology-Endocrinology and Metabolism, Vol. 303, No. 12, 2012, pp. E1479- E1488. doi:10.1152/ajpendo.00351.2012
- [193] T. M. McShane, M. E. Wilson and P. M. Wise, “Effects of Lifelong Moderate Caloric Restriction on Levels of Neuropeptide Y, Proopiomelanocortin, and Galanin mRNA,” The Journals of Gerontology: Series A, Vol. 54, No. 1, 1999, pp. B14-B21. doi:10.1093/gerona/54.1.B14
- [194] S. D. Primeaux, D. A. York and G. A. Bray, “Neuropeptide Y Administration into the Amygdala Alters High Fat Food Intake,” Peptides, Vol. 27, No. 7, 2006, pp. 1644- 1651. doi:10.1016/j.peptides.2005.12.009
- [195] M. Decressac and R. A. Barker, “Neuropeptide Y and Its Role in CNS Disease and Repair,” Experimental Neurology, Vol. 238, No. 2, 2012, pp. 265-272. doi:10.1016/j.expneurol.2012.09.004
- [196] L. E. Kuo, et al., “Neuropeptide Y Acts Directly in the Periphery on Fat Tissue and Mediates Stress-Induced Obesity and Metabolic Syndrome,” Nature Medicine, Vol. 13, No. 7, 2007, pp. 803-811.
- [197] R. D. Cone, “Anatomy and Regulation of the Central Melanocortin System,” Nature Neuroscience, Vol. 8, No. 5, 2005, pp. 571-578.
- [198] G. Segal-Lieberman, et al., “Melanin-Concentrating Hormone Is a Critical Mediator of the Leptin-Deficient Phenotype,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 100, No. 17, 2003, pp. 10085-10090. doi:10.1073/pnas.1633636100
- [199] N. Balthasar, et al., “Divergence of Melanocortin Pathways in the Control of Food Intake and Energy Expenditure,” Cell, Vol. 123, No. 3, 2005, pp. 493-505. doi:10.1016/j.cell.2005.08.035
- [200] S. Boghossian, M. Park and D. A. York, “Melanocortin Activity in the Amygdala Controls Appetite for Dietary Fat,” American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, Vol. 298, No. 2, 2010, pp. R385-R393. doi:10.1152/ajpregu.00591.2009
- [201] S. Chung, et al., “Acute Homeostatic Responses to Increased Fat Consumption in MCH1R Knockout Mice,” Journal of Molecular Neuroscience, Vol. 42, No. 3, 2010, pp. 459-463. doi:10.1007/s12031-010-9358-5
- [202] D. J. Marsh, et al., “Melanin-Concentrating Hormone 1 Receptor-Deficient Mice Are Lean, Hyperactive, and Hyperphagic and Have Altered Metabolism,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 99, No. 5, 2002, pp. 3240-3245. doi:10.1073/pnas.052706899
- [203] I. Morgansterna, G.-Q. Chang, O. Karatayeva and S. F Liebowitza, “Increased Orexin and Melanin-Concentrating Hormone Expression in the Perifornical Lateral Hypothalamus of Rats Prone to Overconsuming a FatRich Diet,” Pharmacology Biochemistry and Behavior, Vol. 96, No. 4, 2010, pp. 413-422. doi:10.1016/j.pbb.2010.06.013
- [204] A. A. Butler, et al., “A Unique Metabolic Syndrome Causes Obesity in the Melanocortin-3 Receptor-Deficient Mouse,” Endocrinology, Vol. 141, No. 9, 2000, pp. 3518- 3521. doi:10.1210/en.141.9.3518
- [205] T. J. Kowalski and T. Sasikumar, “Melanin-Concentrating Hormone Receptor-1 Antagonists as Antiobesity Therapeutics: Current Status,” BioDrugs, Vol. 21, No. 5, 2007, pp. 311-321. doi:10.2165/00063030-200721050-00003
- [206] S. C. Benoit, et al., “A Novel Selective Melanocortin-4 Receptor Agonist Reduces Food Intake in Rats and Mice without Producing Aversive Consequences,” Journal of Neuroscience, Vol. 20, No. 9, 2000, pp. 3442-3448.
- [207] C. E. Perez-Leighton, et al., “Role of Orexin Receptors in Obesity: From Cellular to Behavioral Evidence,” International Journal of Obesity, Vol. 37, No. 2, 2013, pp. 167- 174.
- [208] C. Kotz, et al., “Brain Orexin Promotes Obesity Resistance,” Annals of the New York Academy of Sciences, Vol. 1264, No. 1, 2012, pp. 72-86. doi:10.1111/j.1749-6632.2012.06585.x
- [209] E. Fride, “Endocannabinoids in the Central Nervous System—An Overview,” Prostaglandins, Leukotrienes and Essential Fatty Acids, Vol. 66, No. 2-3, 2002, pp. 221- 233. doi:10.1054/plef.2001.0360
- [210] T. Gomez Del Pulgar, et al., “Cannabinoids Protect Astrocytes from Ceramide-Induced Apoptosis through the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway,” The Journal of Biological Chemistry, Vol. 277, No. 39, 2002, pp. 36527-36533. doi:10.1074/jbc.M205797200
- [211] G. Velasco, et al., “Cannabinoids and Ceramide: Two Lipids Acting Hand-by-Hand,” Life Sciences, Vol. 77, No. 14, 2005, pp. 1723-1731. doi:10.1016/j.lfs.2005.05.015
- [212] V. Di Marzo, et al., “Leptin-Regulated Endocannabinoids Are Involved in Maintaining Food Intake,” Nature, Vol. 410, No. 6830, 2001, pp. 822-825.
- [213] A. Mallat and S. Lotersztajn, “Cannabinoid Receptors as Novel Therapeutic Targets for the Management of Non-Alcoholic Steatohepatitis,” Diabetes & Metabolism, Vol. 34, No. 6, 2008, pp. 680-684. doi:10.1016/S1262-3636(08)74604-4
- [214] D. H. Nam, et al., “Blockade of Cannabinoid Receptor 1 Improves Insulin Resistance, Lipid Metabolism, and Diabetic Nephropathy in db/db Mice,” Endocrinology, Vol. 153, No. 3, 2012, pp. 1387-1396. doi:10.1210/en.2011-1423
- [215] Z. D. Thornton-Jones, et al., “The Cannabinoid CB1 Receptor Inverse Agonist, Rimonabant, Modifies Body Weight and Adiponectin Function in Diet-Induced Obese Rats as a Consequence of Reduced Food Intake,” Pharmacology Biochemistry and Behavior, Vol. 84, No. 2, 2006, pp. 353-359. doi:10.1016/j.pbb.2006.06.001
- [216] S. Suzuki, et al., “Brain-Derived Neurotrophic Factor Regulates Cholesterol Metabolism for Synapse Development,” The Journal of Neuroscience, Vol. 27, No. 24, 2007, pp. 6417-6427. doi:10.1523/JNEUROSCI.0690-07.2007
- [217] R. Molteni, et al., “A High-Fat, Refined Sugar Diet Reduces Hippocampal Brain-Derived Neurotrophic Factor, Neuronal Plasticity, and Learning,” Neuroscience, Vol. 112, No. 4, 2002, pp. 803-814. doi:10.1016/S0306-4522(02)00123-9
- [218] V. De Chiara, et al., “Brain-Derived Neurotrophic Factor Controls Cannabinoid CB1 Receptor Function in the Striatum,” The Journal of Neuroscience, Vol. 30, No. 24, 2010, pp. 8127-8137. doi:10.1523/JNEUROSCI.1683-10.2010
- [219] I. Tasci, H. K. Kabul and A. Aydogdu, “Brain Derived Neurotrophic Factor (BDNF) in Cardiometabolic Physiology and Diseases,” Anadolu Kardiyoloji Dergisi, Vol. 12, No. 8, 2012, pp. 684-688.
- [220] M. Suwa, et al., “Serum Brain-Derived Neurotrophic Factor Level Is Increased and Associated with Obesity in Newly Diagnosed Female Patients with Type 2 Diabetes Mellitus,” Metabolism, Vol. 55, No. 7, 2006, pp. 852-857. doi:10.1016/j.metabol.2006.02.012
- [221] H. Rosas-Vargas, J. D. Martinez-Ezquerro and T. Bienvenu, “Brain-Derived Neurotrophic Factor, Food Intake Regulation, and Obesity,” Archives of Medical Research, Vol. 42, No. 6, 2011, pp. 482-494. doi:10.1016/j.arcmed.2011.09.005
- [222] M. Patterson, S. R. Bloom and J. V. Gardiner, “Ghrelin and Appetite Control in Humans—Potential Application in the Treatment of Obesity,” Peptides, Vol. 32, No. 11, 2011, pp. 2290-2294. doi:10.1016/j.peptides.2011.07.021
- [223] B. Holst and T. W. Schwartz, “Constitutive Ghrelin Receptor Activity as a Signaling Set-Point in Appetite Regulation,” Trends in Pharmacological Sciences, Vol. 25, No. 3, 2004, pp. 113-117. doi:10.1016/j.tips.2004.01.010
- [224] M. Scerif, A. P. Goldstone and M. Korbonits, “Ghrelin in Obesity and Endocrine Diseases,” Molecular and Cellular Endocrinology, Vol. 340, No. 1, 2011, pp. 15-25. doi:10.1016/j.mce.2011.02.011
- [225] J. Gibbs, R. C. Young and G. P. Smith, “Cholecystokinin Decreases Food Intake in Rats,” Obesity Research, Vol. 5, No. 3, 1997, pp. 284-290. doi:10.1002/j.1550-8528.1997.tb00305.x
- [226] T. J. Little, M. Horowitz and C. Feinle-Bisset, “Role of Cholecystokinin in Appetite Control and Body Weight Regulation,” Obesity Reviews, Vol. 6, No. 4, 2005, pp. 297-306. doi:10.1111/j.1467-789X.2005.00212.x
- [227] L. M. Neff and R. F. Kushner, “Emerging Role of GLP-1 Receptor Agonists in the Treatment of Obesity,” Diabetes, Metabolic Syndrome and Obesity, Vol. 2010, 2010, pp. 263-273. doi:10.2147/DMSOTT.S6816
- [228] J. J. Carlson, et al., “Preand Post-Prandial Appetite Hormone Levels in Normal Weight and Severely Obese Women,” Nutrition & Metabolism, Vol. 6, 2009, p. 32. doi:10.1186/1743-7075-6-32
- [229] S. S. Torekov, S. Madsbad and J. J. Holst, “Obesity—An Indication for GLP-1 Treatment? Obesity Pathophysiology and GLP-1 Treatment Potential,” Obesity Reviews, Vol. 12, No. 8, 2011, pp. 593-601. doi:10.1111/j.1467-789X.2011.00860.x
- [230] K. Wynne, B. C. Field and S. R. Bloom, “The Mechanism of Action for Oxyntomodulin in the Regulation of Obesity,” Current Opinion in Investigational Drugs, Vol. 11, No. 10, 2010, pp. 1151-1157.
- [231] M. R. Druce and S. R. Bloom, “Oxyntomodulin: A Novel Potential Treatment for Obesity,” Treatments in Endocrinology, Vol. 5, No. 5, 2006, pp. 265-272.
- [232] A. Asakawa, et al., “Characterization of the Effects of Pancreatic Polypeptide in the Regulation of Energy Balance,” Gastroenterology, Vol. 124, No. 5, 2003, pp. 1325-1336. doi:10.1016/S0016-5085(03)00216-6
- [233] V. Lassmann, et al., “Low Plasma Levels of Pancreatic Polypeptide in Obesity,” Diabetes, Vol. 29, No. 6, 1980, pp. 428-430.
- [234] D. Renshaw and R. L. Batterham, “Peptide YY: A Potential Therapy for Obesity,” Current Drug Targets, Vol. 6, No. 2, 2005, pp. 171-179. doi:10.2174/1389450053174523
- [235] M. H. Tschop and E. Ravussin, “Peptide YY: Obesity’s Cause and Cure?” American Journal of Physiology-Endocrinology and Metabolism, Vol. 293, No. 5, 2007, pp. E1131-E1133. doi:10.1152/ajpendo.00568.2007
- [236] K. Wada, et al., “Bombesin, Obesity, and Social Behaviour,” Molecular Psychiatry, Vol. 3, No. 3, 1998, pp. 204- 206. doi:10.1038/sj.mp.4000387
- [237] A. Amin, W. S. Dhillo and K. G. Murphy, “The Central Effects of Thyroid Hormones on Appetite,” Journal of Thyroid Research, Vol. 2011, 2011, Article ID: 306510.
- [238] M. Pilhatsch, et al., “Hypothyroidism and Mood Disorders: Integrating Novel Insights from Brain Imaging Techniques,” Thyroid Research, Vol. 4, Suppl. 1, 2011, p. S3. doi:10.1186/1756-6614-4-S1-S3
- [239] E. Cano-Europa, et al., “Hypothyroidism Induces Selective Oxidative Stress in Amygdala and Hippocampus of Rat,” Metabolic Brain Disease, Vol. 23, No. 3, 2008, pp. 275-287. doi:10.1007/s11011-008-9099-0
- [240] C. J. Frederickson, et al., “Importance of Zinc in the Central Nervous System: The Zinc-Containing Neuron,” Journal of Nutrition, Vol. 130, Suppl. 5, 2000, pp. 1471S- 1483S.
- [241] N. Whittle, et al., “Rescue of Impaired Fear Extinction and Normalization of Cortico-Amygdala Circuit Dysfunction in a Genetic Mouse Model by Dietary Zinc Restriction,” The Journal of Neuroscience, Vol. 30, No. 41, 2010, pp. 13586-13596. doi:10.1523/JNEUROSCI.0849-10.2010
- [242] A. Takeda, et al., “Increases in Extracellular Zinc in the Amygdala in Acquisition and Recall of Fear Experience and Their Roles in Response to Fear,” Neuroscience, Vol. 168, No. 3, 2010, pp. 715-722. doi:10.1016/j.neuroscience.2010.04.017
- [243] R. Tudor, P. D. Zalewski and R. Ratnaike, “Zinc in Health and Chronic Disease,” The Journal of Nutrition Health and Aging, Vol. 9, No. 1, 2005, pp. 45-51.
- [244] O. P. Garcia, K. Z. Long and J. L. Rosado, “Impact of Micronutrient Deficiencies on Obesity,” Nutrition Reviews, Vol. 67, No. 10, 2009, pp. 559-572. doi:10.1111/j.1753-4887.2009.00228.x
- [245] J. Kim and S. Lee, “Effect of Zinc Supplementation on Insulin Resistance and Metabolic Risk Factors in Obese Korean Women,” Nutrition Research and Practice, Vol. 6, No. 3, 2012, pp. 221-225. doi:10.4162/nrp.2012.6.3.221
- [246] C. W. Levenson, “Zinc Regulation of Food Intake: New Insights on the Role of Neuropeptide Y,” Nutrition Reviews, Vol. 61, No. 7, 2003, pp. 247-249.
- [247] K. Ohinata, et al., “Orally Administered Zinc Increases Food Intake via Vagal Stimulation in Rats,” The Journal of Nutrition, Vol. 139, No. 3, 2009, pp. 611-616. doi:10.3945/jn.108.096370
- [248] M. Foster and S. Samman, “Zinc and Regulation of Inflammatory Cytokines: Implications for Cardiometabolic Disease,” Nutrients, Vol. 4, No. 7, 2012, pp. 676-694. doi:10.3390/nu4070676
- [249] M. Sowa-Kucma, et al., “Antidepressant-Like Activity of Zinc: Further Behavioral and Molecular Evidence,” Journal of Neural Transmission, Vol. 115, No. 12, 2008, pp. 1621-1628. doi:10.1007/s00702-008-0115-7
- [250] C. S. Mantzoros, et al., “Zinc May Regulate Serum Leptin Concentrations in Humans,” Journal of the American College of Nutrition, Vol. 17, No. 3, 1998, pp. 270-275. doi:10.1080/07315724.1998.10718758
- [251] E. S. Ott and N. F. Shay, “Zinc Deficiency Reduces Leptin Gene Expression and Leptin Secretion in Rat Adipocytes,” Experimental Biology and Medicine, Vol. 226, No. 9, 2001, pp. 841-846.
- [252] D. B. Briggs, et al., “Zinc Enhances Adiponectin Oligomerization to Octadecamers but Decreases the Rate of Disulfide Bond Formation,” BioMetals, Vol. 25, No. 2, 2012, pp. 469-486. doi:10.1007/s10534-012-9519-9
- [253] S. D. Soheilykhan, M. R. Mohammed, S. M. Mohammadi, M. Afkhami-Ardekani, S. A. Eghbali and F. Dehghan, “The Effect of Zinc Supplementation on Serum Adiponectin Concentration and Insulin Resistance in First Degree Relatives of Diabetic Patients,” Iranian Journal of Diabetes and Obesity, Vol. 4, No. 2, 2012, pp. 57-62.
- [254] R. G. Lee, et al., “Zinc Deficiency Increases Hypothalamic Neuropeptide Y and Neuropeptide Y mRNA Levels and Does Not Block Neuropeptide Y-Induced Feeding in Rats,” The Journal of Nutrition, Vol. 128, No. 7, 1998, pp. 1218-1223.
- [255] M. K. Hansen and T. M. Connolly, “Nuclear Receptors as Drug Targets in Obesity, Dyslipidemia and Atherosclerosis,” Current Opinion in Investigational Drugs, Vol. 9, No. 3, 2008, pp. 247-255.
- [256] L. Guarente, “Sirtuins, Aging, and Metabolism,” Cold Spring Harbor Symposia on Quantitative Biology, Vol. 76, 2011, pp. 81-90. doi:10.1101/sqb.2011.76.010629
- [257] C. Harrison, “Neurodegenerative Disorders: A Neuroprotective Role for Sirtuin 1,” Nature Reviews Drug Discovery, Vol. 11, 2012, p. 108.
- [258] T. Kawada, et al., “Dietary Regulation of Nuclear Receptors in Obesity-Related Metabolic Syndrome,” Asia Pacific Journal of Clinical Nutrition, Vol. 17, Suppl. 1, 2008, pp. 126-130.
- [259] H. I. Swanson, et al., “Role of Nuclear Receptors in Lipid Dysfunction and Obesity-Related Diseases,” Drug Metabolism & Disposition, Vol. 41, No. 1, 2013, pp. 1-11. doi:10.1124/dmd.112.048694
- [260] I. Cakir, et al., “Hypothalamic Sirt1 Regulates Food Intake in a Rodent Model System,” PLoS One, Vol. 4, No. 12, 2009, p. e8322. doi:10.1371/journal.pone.0008322
- [261] T. Kitamura and T. Sasaki, “Hypothalamic Sirt1 and Regualtion of Food Intake,” Diabetology International, Vol. 3, No. 3, 2012, pp. 109-112. doi:10.1007/s13340-012-0088-5
- [262] M. O. Dietrich, et al., “Agrp Neurons Mediate Sirt1’s Action on the Melanocortin System and Energy Balance: roles for Sirt1 in Neuronal Firing and Synaptic Plasticity,” The Journal of Neuroscience, Vol. 30, No. 35, 2010, pp. 11815-11825. doi:10.1523/JNEUROSCI.2234-10.2010
- [263] A. O. Schaffhauser, et al., “Effects of a High-Fat Diet and Strain on Hypothalamic Gene Expression in Rats,” Obesity Research, Vol. 10, No. 11, 2002, pp. 1188-1196. doi:10.1038/oby.2002.161
- [264] A. K. Lee, et al., “Effect of High-Fat Feeding on Expression of Genes Controlling Availability of Dopamine in Mouse Hypothalamus,” Nutrition, Vol. 26, No. 4, 2010, pp. 411-422. doi:10.1016/j.nut.2009.05.007
- [265] D. E. Cohen, et al., “Neuronal SIRT1 Regulates Endocrine and Behavioral Responses to Calorie Restriction,” Genes & Development, Vol. 23, No. 24, 2009, pp. 2812- 2817. doi:10.1101/gad.1839209
- [266] A. Takano, et al., “Growth Hormone Induces Cellular Insulin Resistance by Uncoupling Phosphatidylinositol 3-Kinase and Its Downstream Signals in 3T3-L1 Adipocytes,” Diabetes, Vol. 50, No. 8, 2001, pp. 1891-1900. doi:10.2337/diabetes.50.8.1891
- [267] M. Kitada, et al., “Sirtuins and Renal Diseases: Relationship with Aging and Diabetic Nephropathy,” Clinical Science, Vol. 124, No. 3, 2013, pp. 153-164. doi:10.1042/CS20120190
- [268] S. Rajendrasozhan, et al., “SIRT1, an Antiinflammatory and Antiaging Protein, Is Decreased in Lungs of Patients with Chronic Obstructive Pulmonary Disease,” American Journal of Respiratory and Critical Care Medicine, Vol. 177, No. 8, 2008, pp. 861-870. doi:10.1164/rccm.200708-1269OC
- [269] J. R. Archer and E. H. Baker, “Diabetes and Metabolic Dysfunction in COPD,” Respiratory Medicine, Vol. 5, No. 3-4, 2009, pp. 67-74.
- [270] A. Planavila, et al., “Sirt1 Acts in Association with PPARalpha to Protect the Heart from Hypertrophy, Metabolic Dysregulation, and Inflammation,” Cardiovascular Research, Vol. 90, No. 2, 2011, pp. 276-284. doi:10.1093/cvr/cvq376
- [271] S. Oka, et al., “PPARalpha-Sirt1 Complex Mediates Cardiac Hypertrophy and Failure through Suppression of the ERR Transcriptional Pathway,” Cell Metabolism, Vol. 14, No. 5, 2011, pp. 598-611. doi:10.1016/j.cmet.2011.10.001
- [272] F. M. Campbell, et al., “A Role for Peroxisome Proliferator-Activated Receptor Alpha (PPARalpha) in the Control of Cardiac Malonyl-CoA Levels: Reduced Fatty Acid Oxidation Rates and Increased Glucose Oxidation Rates in the Hearts of Mice Lacking PPARalpha Are Associated with Higher Concentrations of Malonyl-CoA and Reduced Expression of Malonyl-CoA Decarboxylase,” The Journal of Biological Chemistry, Vol. 277, No. 6, 2002, pp. 4098-4103. doi:10.1074/jbc.M106054200
- [273] R. J. Rodgers, M. H. Tschop and J. P. Wilding, “AntiObesity Drugs: Past, Present and Future,” Disease Models & Mechanisms, Vol. 5, No. 5, 2012, pp. 621-626. doi:10.1242/dmm.009621
- [274] V. Hainer and I. A. Hainerova, “Do We Need Anti-Obesity Drugs?” Diabetes/Metabolism Research and Reviews, Vol. 28, Suppl. 2, 2012, pp. 8-20. doi:10.1002/dmrr.2349
- [275] D. J. Heal, J. Gosden and S. L. Smith, “What Is the Prognosis for New Centrally-Acting Anti-Obesity Drugs?” Neuropharmacology, Vol. 63, No. 1, 2012, pp. 132-146. doi:10.1016/j.neuropharm.2012.01.017
- [276] G. Derosa and P. Maffioli, “Anti-Obesity Drugs: A Review about Their Effects and Their Safety,” Expert Opinion on Drug Safety, Vol. 11, No. 3, 2012, pp. 459-471. doi:10.1517/14740338.2012.675326
- [277] M. F. Li and B. M. Cheung, “Rise and Fall of Anti-Obesity Drugs,” World Journal of Diabetes, Vol. 2, No. 2, 2011, pp. 19-23. doi:10.4239/wjd.v2.i2.19
- [278] E. Yulyaningsih, et al., “NPY Receptors as Potential Targets for Anti-Obesity Drug Development,” British Journal of Pharmacology, Vol. 163, No. 6, 2011, pp. 1170- 1202. doi:10.1111/j.1476-5381.2011.01363.x
- [279] T. M. Fong, “Development of Anti-Obesity Agents: Drugs that Target Neuropeptide and Neurotransmitter Systems,” Expert Opinion on Investigational Drugs, Vol. 17, No. 3, 2008, pp. 321-325. doi:10.1517/13543784.17.3.321