Open Journal of Gastroenterology
Vol.2 No.4(2012), Article ID:25047,8 pages DOI:10.4236/ojgas.2012.24030
Clinical management of gastrointestinal amyloidosis
![]()
Department of Gastroenterology and Hepatology, Nagasaki University Hospital, Nagasaki, Japan
Email: *hajimei2002@yahoo.co.jp
Received 13 August 2012; revised 15 September 2012; accepted 16 October 2012
Keywords: Gastrointestinal Amyloidosis; Secondary Amyloidosis; Epidermolysis Bullosa; Colchicine
ABSTRACT
Amyloidosis is characterized by extracellular deposition of abnormal protein, consisting of primary, secondary, hemodialysis-related, hereditary, senile and localized type. Primary amyloidosis is associated with monoclonal light chains. Secondary amyloidosis is associated with inflammatory, infectious, and neoplastic diseases. Amyloid deposition in the gastrointestinal tract can manifest the symptoms including diarrhea, steatorrhea, or constipation. For diagnosis, one should obtain an immunofixation of serum or urine as well as biopsy sampling of gastrointestinal mucosa stained specifically. While most gastrointestinal complications are managed symptomatically, treatment depends upon the type of amyloidosis. Causal therapy is reserved for a select few from various subtypes of this disorder.
1. INTRODUCTION
Amyloidosis is defined as an extracellular deposit nonbranching fibrils with a β-pleated sheet configuration [1,2]. It can be acquired or hereditary and systemic or localized [3]. The common forms of systemic amyloidosis are: primary, secondary, dialysis-related, and senile and familial amyloidotic polyneuropathy [3]. Primary amyloidosis is associated with monoclonal light chains in serum and/or urine with 15% of patients having multiple myeloma [4]. Secondary amyloidosis is caused by the deposition of amyloid A (AA) protein, which results from proteolytic cleavage of the circulating acute-phase reactant serum amyloid A [5]. Secondary amyloidosis is associated with a variety of chronic inflammatory disorders, chronic microbial infections or less commonly, malignant neoplasms [6]. Clinical manifestations of amyloidosis may vary from asymptomatic to fatal forms [7]. Amyloidosis is one of the unusual diseases about which a physician may not think when it is affecting the patient. There has been a progress in the understanding of the pathogenesis, clinical features, diagnostic measures and therapy of this disorder. Herein, we will discuss clinical management of gastrointestinal (GI) amyloidosis.
2. PATHOGENESIS
Amyloidosis is defined as an extracellular deposit of protein fibrils with a β-sheet fibrillar structure and characteristic properties typically after staining with Congo Red dye [1,2]. The current nomenclature consists of the first letter, A (for amyloid), followed by a description of the precursor protein [6]. This β-pleated configuration provides relative resistance to physiological solvents and to normal proteolytic digestion [1,2]. There are six types: primary, secondary, hemodialysis-related, hereditary, senile, and localized.
Primary amyloidosis (amyloid light chain, AL) is the most common form of amyloidosis [3,6]. Automated protein analysis of the amino acid sequences reveals that these peptides represent the N-terminal of the variable region of the κ or λ light-chains of the immunoglobulins [8]. The circulating light-chain proteins are called BenceJones proteins [8], which are secreted by a plasma-cell clone. AL has been associated with certain plasma-cell dyscrasias, such as multiple myeloma and Waldenstrom’s macroglobulinaemia [8].
Secondary amyloidosis (AA) is caused by the deposition of AA protein, which results from proteolytic cleavage of the circulating acute-phase reactant, serumamyloid A [9]. AA amyloidosis is associated with infectious, inflammatory, or, less commonly, neoplastic disorders. Rheumatoid arthritis is found in 48% of such patients [10]. In contrast, the incidence of amyloidosis in patients with rheumatoid arthritis ranges from 7% to 21% [11-14]. Other inflammatory disorders such as Crohn’s disease, ankylosing spondylitis, Reiter’s syndrome, psoriasis, progressive systemic sclerosis, primary biliary cirrhosis, and serum lupus erythematosus are listed in the underlying diseases of AA amyloidosis [3,6]. Other associations include infectious processes such as leprosy, tuberculosis, osteomyelitis, and bronchiectasis; tumors such as renal cell carcinoma, GI stromal tumors [15], and intestinal carcinomas; and other disorders, such as idiopathic retroperitoneal fibrosis [16]. Familial-Mediterranean fever (FMF) is a rare cause of AA amyloidosis, characterized by recurrent episodes of fever, pleuritis and peritonitis [17]. As for the dialysis-related amyloidosis, the fibrils are composed of β2-microglobulins, possibly retained in the body because of decreased renal clearance [18].
Senile amyloidosis is mainly found in patients over 80 years old and involves the heart, but can also be seen throughout the GI tract [19]. GI amyloid was observed in the subserosal veins in the large and small bowel [19]. The plaques in the brains of patients with Alzheimer’s disease are critical in the pathogenesis of this disorder.
Hereditary amyloidosis, an autosomal dominant inheritance of amyloidogenic proteins, is rare. The most common form is variant transthyretin; the deposition results in familial amyloidotic polyneuropathy [6]. Otherwise, there are hereditary nonneuropathic systemic amyloidoses associated with mutations in genes for apolipoprotein AI, lysozyme, and fibrinogen α-chain [6]. Local amyloidosis is found in the esophagus, stomach, small bowel, and colon [6].
3. CLINICAL MANIFESTATIONS
Clinical symptoms of amyloidosis may depend on the amount and location of the amyloid deposits irrespective of whether it is primary or secondary amyloidosis, and vary from asymptomatic to fatal forms. Patients with GI amyloidosis can present with abdominal pain, dysmotility, diarrhea, bleeding, pseudo-obstruction and malabsorption [3,6,7]. The common presenting symptoms are fatigue, light-headedness and weight loss [8]. Edema is the most common finding at presentation in all types of amyloidosis. The involvement of the kidneys, heart and peripheral nerves leads to the common presenting symptoms [20]. Renal involvement is characterized initially by proteinuria, with azotaemia developing late in the course of the disease. Cardiac deposition results in restrictive cardiomyopathy with ensuing congestive-heart failure and various arrhythmias [3,6-8,20].
The most common oral manifestation of amyloidosis is macroglossia, which may cause sleep apnea, speech difficulties, oral dysphagia, difficulty in chewing, malocclusion of the teeth, dental indentations of the tongue, and eventually, airway obstruction [6]. Papules, vesicles, and ulcers may be seen [21]. There may be swelling of the floor of the mouth, hardening of the soft tissues in the perioral region, loss of facial expression, and difficulty in opening the mouth [22]. The submandibular glands may be involved, resulting in xerostomia, resembling Sjogren’s syndrome [23].
Amyloidosis of the gastrointestinal tract, with biopsyproven disease, is rare [24]. In the latest study on gastrointestinal amyloidosis, among the 2334 patients with all types of amyloidosis, 76 patients (3.2%) had biopsyproven amyloid involvement of the gastrointestinal tract. The median age was 61 years. Systemic amyloidosis with dominant gastrointestinal involvement was present in 60 (79%) patients. Of the 60 systemic cases, 50 (83%) had immunoglobulin light-chain, 5 (8%) familial lysozyme, 5 (8%) transthyretin amyloidosis. The most frequent symptoms for all patients were weight loss in 33 (45%) and gastrointestinal bleeding in 27 (36%). Incidental identification of amyloidosis on routine endoscopic surveillance played a role in the diagnosis of 11 patients. Although a rare manifestation of amyloidosis, staining for amyloid should be considered in patients undergoing gastrointestinal biopsy who have unexplained chronic gastrointestinal symptoms [24].
Esophageal disease in amyloidosis includes symptoms such as dysphagia, chest pain, heartburn, and hematemesis. The most common radiographic feature is the dilated, atonic esophagus with decreased peristalsis, sometimes with distal narrowing and proximal dilatation, associated with tracheobronchial aspiration [3,6]. Less common findings are ulcerations or masses [25]. A variety of manometric abnormalities may be seen in the esophagus. Generally, the lower esophageal sphincter pressure is normal or low, often with associated heartburn [26]. The amplitude of contractions is decreased. Occasionally, the picture may mirror that seen in achalasia. Unlike idiopathic achalasia, patients experience a rapid onset of symptoms and weight loss [27]. Neural involvement in esophageal amyloidosis is suggested by lack of the expected secondary peristalsis and LES relaxation caused by esophageal distension [28]. The prevalence of esophageal disease in amyloidosis ranges from 13% in a radiologic study to 22% in an autopsy series [29].
In patients with amyloidosis, gastric involvement occurs in 8% of patients by biopsy and 12% by autopsy, with only 1% being symptomatic [29]. Symptoms via gastric involvement include nausea, vomiting, hematemesis, and epigastric pain [28]. Gastric outlet obstruction, thickened gastric folds, gastric ulcers, hematomas, arteriovenous malformations, granular-appearing mucosa, plaque-like lesions, and gastroparesis may be rarely occurred [30-36].
The degree of amyloid deposition is greatest in the small intestine [37], with 31% of patients with amyloidosis being affected at autopsy [29]. The vasculature is frequently involved usually in the submucosa. Patients with amyloidosis of the small intestine present with diarrhea, steatorrhea, protein loss, hemorrhage, obstruction, infarction, perforation, intussusception, pneumatosis intestinalis, and constipation [38-42]. Malabsorption is also found in certain amyloid patients [43].
The clinical manifestations of amyloid deposit into the colon may mimic other diseases including inflammatory bowel disease, malignancy, ischemic colitis, and collagenous colitis [44-47]. Complications include colonic dilatation, pseudo-obstruction, stricture formation, rectal bleeding, submucosal hemorrhage, volvulus, infarction, and perforation [41,48-52].
Hepatic amyloidosis has no clinical significance in the majority of patients, although symptomatic patients may exhibit weight loss, fatigue, abdominal discomfort and anorexia (26%) [53]. Familial amyloidotic polyneuropathy affects the liver predominantly; other organs involved are the spleen, heart, thyroid, adrenal glands and vitreous corpus of the eyes. Clinical splenomegaly might be found in patients with amyloidosis [5,54]. Gallbladder involvement in amyloidosis is very rare in only a few case series [55].
4. DIAGNOSIS
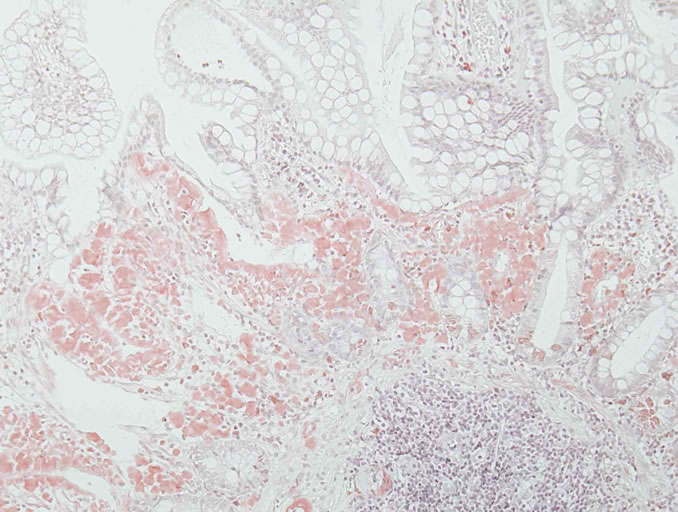
Diagnosis is often made long after the onset of symptoms because of its non-specific and varied presentation, which can mimic many other conditions. It requires confirmation of the presence of amyloid deposition by histology and is achieved by characteristic properties after specific staining with Congo red [3,4,6]. Direct fast scarlet staining [56] can also produce the characteristic red appearance in normal light and apple-green birefringence in polarized light indicating prominent mucosal amyloid deposition (Figure 1).
The prevalence of radiologic abnormalities is greatest in the small intestine [57]. There may be innumerable fine granular densities or a coarse mucosal appearance, punctuate lucent defects, multiple, sometimes ulcerated nodular polyps, erosions, thickened folds and impaired motor activity [14,58-60].
Screening endoscopic biopsies of the gastrointestinal tract are diagnostic in most cases of systemic amyloidosis. A variety of mucosal abnormalities are observed in the GI tract by endoscopy; involvement of the small intestine is especially common (Figure 2). The lesions may appear as a fine granular, granular and nodular mucosa, polypoid protrusions, erosions, ulcerations, mucosal friability, and bowel wall thickening [3,6,57,60]. Tada et al. reported endoscopic appearances of gastrointestinal amyloidosis at different location [61]. In their study, endoscopic findings of the esophagus, stomach, duodenum, and colorectum were assessed in 37 patients with amyloidosis involving the gastrointestinal tract. Endoscopic examinations revealed fine granular appearance, polypoid protrusions, erosions, ulcerations, and mucosal friability in many cases. These findings were most marked and noticed most often in the second portion of the duodenum. The frequency of amyloid deposition in the bi-
 (a)
(a)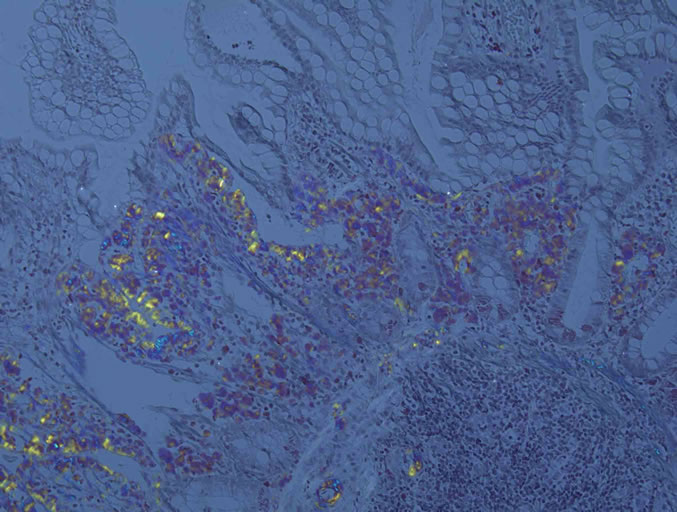 (b)
(b)
Figure 1. Direct fast scarlet staining produced the characteristic orange-red appearance in normal light (a) and apple-green birefringence in polarized light indicating prominent mucosal amyloid deposition (b). Direct fast scarlet staining showed less nonspecific background than Congo red.
opsy specimens was as follows; 100% in the duodenum, 95% in the stomach, 91% in the colorectum, and 72% in the esophagus. The degree of amyloid deposition in the duodenum, which was the highest of the entire gastrointestinal tract, significantly correlated with the frequency of endoscopic findings such as fine granular appearance and polypoid protrusions. Therefore, the two endoscopic findings described above are characteristic of this disease and may reflect amyloid deposition in the mucosa or submucosa of the alimentary tract. The results indicate that for a diagnosis of amyloidosis, it is important to examine the upper gastrointestinal tract, especially the duodenum, using endoscopy and biopsy techniques. Macroscopically, AL amyloidosis typically forms polypoid protrusions, while secondary amyloidosis is principally characterized by granular-appearing mucosa, as seen in the intestine of current case [3,6,61].
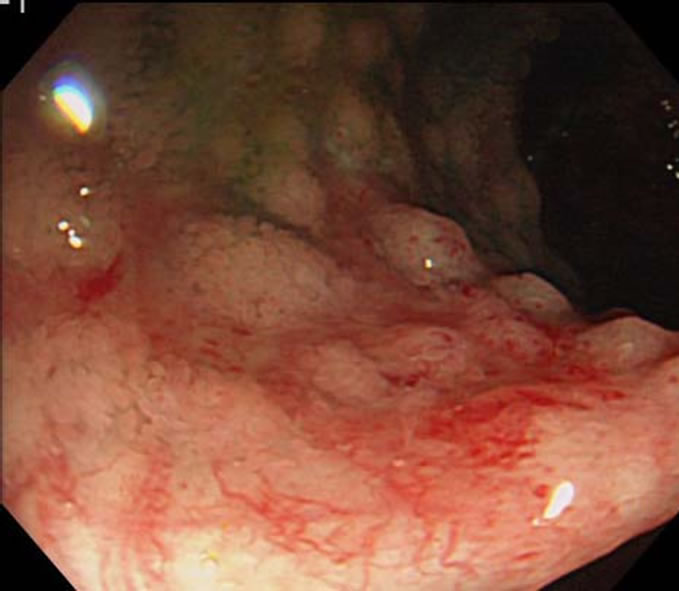
Figure 2. The small-bowel lesions appeared as granular and nodular mucosa with mucosal friability, and hemorrhagic erosions.
In order to image total body amyloid deposits, 123Ilabeled serum amyloid P component is injected into the circulation [62]. It binds to amyloid fibrils in the body. The diagnostic sensitivity of this scintigraphy is 90% for AA and AL amyloidosis [63].
The liver on ultrasound or computed tomography scan is enlarged with homogeneous echogenicity or density [54]. Computed tomography scan may show diffuse or focal regions of decreased attenuation and lack of parenchymal enhancement of the spleen. On magnetic resonance imaging, there is also homogeneous intensity with T1 increased and T2 not significantly altered [54,64].
5. TREATMENT
The principle of amyloidosis treatment should be directed at the underlying cause, and aimed to arrest amyloid accumulation and formation by reducing the abundance of the fibril precursor protein. This approach can result in stabilization or improvement of the affected organ function [3,6,65]. The goal of treating primary amyloidosis is to suppress the synthesis of immunoglobulin light chains by controlling the underlying plasma cell disorder with chemotherapy [65]. Recently, high-dose chemotherapy combined with hematopoietic stem cell transplantation is being performed for AL amyloidosis with encouraging results [66]. More recently, a patient suffering from severe intestinal amyloidosis secondary to aggressive rheumatoid arthritis was treated with tocilizumab, a humanized anti-interleukin 6 receptor antibody [67]. The treatment leaded to amelioration of gastrointestinal manifestations as well as systemic joint pain. Moreover, her serum AA protein levels returned to the normal ranges and repeat endoscopic biopsy revealed no colonic amyloid accumulation. Tocilizumab can be a treatment of choice for AA amyloidosis associated with the inflammatory arthritis, potentially interfering in the amyloidogenic precursor biosynthesis induced by the representative proinflammatory cytokine, intereukin 6 [68]. Again, a novel small molecule inhibitor, R-1-[6- [(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexa-noyl]pyrrolidine-2 carboxylic acid (CPHPC) has been developed to specifically target serum amyloid P component which is a universal constituent of amyloid deposits and contributes to their formation [69]. CPHPC depletes serum amyloid P levels and arrests amyloid deposition in an open label proof of principle study in systemic amyloidosis [70]. However, such promising specific agents hitherto have not been available in the clinical settings of systemic amyloidosis. Practically, treatment of AA amyloidosis includes control of the primary disease [3,6,65]. Prolonged anti-tuberculous therapy, for example, was reported to reduce established amyloidosis [71]. Chlorambucil and tumor necrosis factor-α inhibitors against inflammatory arthritis cause regression of the amyloid deposition [72,73]. However, the therapeutic effects are anecdotal and not conclusive as shown in sporadic cases or limited series.
Colchicine, an alkaloid extracted from Colchicum autumnale of the lily family, is prescribed for treatment of diverse illnesses such as gout, Behçet’s disease and primary biliary cirrhosis [74]. This drug exerts inhibitory actions on microtubule formation, thus affecting cellular mitosis, and can interfere with phagocytic properties of polymorphonuclear cells [73]. First, there was a case observation that it was an effective remedy for prophylaxis of attacks of FMF a genetic disease characterized by short febrile bouts of serositis, and this was confirmed by controlled studies [74,75]. Colchicine is nowadays an affective medication in the prevention and treatment of amyloidosis secondary to FMF [73]. Cases of nephritic syndrome that resolved after prolonged use of colchicine were reported with not only FMF but also ulcerative colitis and ankylosing spondylitis [76,77]. In a preliminary trial, colchicine (1.5 mg/day) was given for amyloid nephropathy in patients with dystrophic type of inherited epidermolysis bullosa (EB) [78]. More recently, we reported that gastrointestinal amyloidosis secondary to EB was cured by colchicine at the same dosage 2 months later [79].
GI complications are managed with symptomatic control. It is important to assure adequate nutrition and hydration. (3) For instance, symptoms of gastroparesis have effectively been treated with metoclopramide or domperidone [80,81]. Neostigmine may be helpful in alleviating acute colonic pseudo-obstruction; it could be used as an alternative to colonic decompression in patients with various acute medical conditions [82]. Diarrhoea has been successfully treated with loperamide or octreotide [83]. Total parenteral nutrition is used if dysmotility-related symptoms are disabling, and the patient becomes malnourished.3 Antibiotics can be used for bacterial overgrowth. Patients should receive supplementation, especially with fat-soluble vitamins [84]. The treatment of GI bleeding may be similar to that in other diseases.
6. CONCLUSION
Amyloidosis can manifest a variety of GI symptoms with investigational studies showing a range of abnormalities. One should obtain an immunofixation of serum or urine as well as biopsy sampling of GI mucosa stained with Congo Red. While most gastrointestinal complications are managed symptomatically, treatment depends upon the type of amyloidosis. Causal therapy is reserved for a select few from various subtypes of this disorder.
![]()
![]()
REFERENCES
- Stermark, P. (2005) Aspects on human amyloid forms and their fibril polypeptides. FEBS Journal, 272, 5942- 5949. doi:10.1111/j.1742-4658.2005.05024.x
- Glenner, G.G. (1980) Amyloid deposits and amyloidosis. The β-fibrilloses. New England Journal of Medicine, 302, 1283-1292. doi:10.1056/NEJM198006053022305
- Petre, S., Shah, I.A. and Gilani, N. (2008) Review article: Gastrointestinal amyloidosis-clinical features, diagnosis and therapy. Alimentary Pharmacology & Therapeutics, 27, 1006-1016. doi:10.1111/j.1365-2036.2008.03682.x
- Kyle, R.A. and Gertz, M.A. (1995) Primary systemic amyloidosis: Clinical and laboratory features in 474 cases. Seminars in Hematology, 32, 45-59.
- Gillmore, J.D., Lovat, L.B. and Hawkins, P.N. (1999) Amyloidosis and the liver. Journal of Hepatology, 30, 17- 33.
- Ebert, E.C. and Nagar, M. (2008) Gastrointestinal manifestations of amyloidosis. American Journal of Gastroenterology, 103, 776-787. doi:10.1111/j.1572-0241.2007.01669.x
- Kuang, L., Sun, W., Gibson, M.F., et al. (2003) Gastrointestinal amyloidosis with ulceration, hemorrhage, small bowel diverticula, and perforation. Digestive Diseases and Sciences, 48, 2023-2026. doi:10.1023/A:1026190809074
- Gertz, M.A., Lacy, M.Q. and Dispenzieri, A. (1999) Amyloidosis. Hematology/Oncology Clinics of North America, 13, 1211-1233. doi:10.1016/S0889-8588(05)70122-2
- Browning, M.J., Banks, R.A., Tribe, C.R., et al. (1985) Ten years experience in an amyloid clinic: A clinicopathologic survery. Oxford Journals Medicine, 54, 213-227.
- Gertz, M.A. and Kyle, R.A. (1991) Secondary systemic amyloidosis: Response and survival in 64 patients. Medicine, 70, 246-256.
- Tiitinen, S., Kaarela, K., Helin, H., et al. (1993) Amyloidosis—Incidence and early risk factors in patients with rheumatoid arthritis. Scandinavian Journal of Rheumatology, 22, 158-161. doi:10.3109/03009749309099264
- Kobayashi, H., Tada, S., Fuchigami, T., et al. (1996) Secondary amyloidosis in patients with rheumatoid arthritis: Diagnostic and prognostic value of gastroduodenal biopsy. British Journal of Rheumatology, 35, 44-49. doi:10.1093/rheumatology/35.1.44
- Suzuki, A., Ohosone, Y., Obana, M., et al. (1994) Cause of death in 81 autopsied patients with rheumatoid arthritis. The Journal of Rheumatology, 21, 33-36.
- Kuroda, T., Tanabe, N., Sakatsume, M., et al. (2002) Comparison of gastroduodenal, renal and abdominal fat biopsies for diagnosing amyloidosis in rheumatoid arthritis. Clinical Rheumatology, 21, 123-128. doi:10.1007/PL00011217
- Overstreet, K., Barone, R.M. and Robin, H.S. (2003) Secondary amyloidosis and gastrointestinal stromal tumors. A case report and discussion of pathogenesis. Archives of Pathology & Laboratory Medicine, 127, 470- 473.
- Hosaka, N., Ito, M., Taki, Y., et al. (2003) Amyloid A gastrointestinal amyloidosis associated with idiopathic retroperitoneal fibrosis. Report of a rare autopsy case and review of the literature. Archives of Pathology & Laboratory Medicine, 127, 735-738.
- Friedman, S. and Janowitz, H. (1998) Systemic amyloidosis and the gastrointestinal tract. Gastroenterology Clinics of North America, 27, 595-614. doi:10.1016/S0889-8553(05)70022-4
- Floege, J. and Ketteler, M. (2001) Beta2-microglobulinderived amyloidosis: An update. Kidney International, 78, S164-S171. doi:10.1046/j.1523-1755.2001.07823.x
- Rocken, C., Saeger, W. and Linke, R.P. (1994) Gastrointestinal amyloid deposits in old age. Report on 110 consecutive autopsical patients and 98 retrospective bioptic specimens. Pathology-Research and Practice, 190, 641- 649.
- Gertz, M.A. and Kyle, R.A. (1997) Hepatic amyloidosis: clinical appraisal in 77 patients. Hepatology, 25, 118-121. doi:10.1002/hep.510250122
- Jacobs, P., Sellars, S. and King, H.S. (1988) Massive macroglossia, amyloidosis and myeloma. Journal of Postgraduate Medicine, 64, 696-698. doi:10.1136/pgmj.64.755.696
- Pereira, C.M., Gasparetto, P.F., Correa, M.E., et al. (2005) Primary oral and perioral amyloidosis associated with multiple myeloma. General Dentistry, 53, 340-341.
- Gogel, H.K., Searles, R.P., Volpicelli, N.A., et al. (1983) Primary amyloidosis presenting as Sjogren’s syndrome. Archives of Internal Medicine, 143, 2325-2326. doi:10.1001/archinte.1983.00350120123027
- Cowan, A.J., Skinner, M., Seldin, D.C., et al. (2012) Amyloidosis of the gastrointestinal tract: A 13-year single center referral experience. Haematologica, in press. doi:10.3324/haematol.2012.068155
- Heitzman, E.J., Heitzman, G.C. and Elliott, C.F. (1962) Primary esophageal amyloidosis. Archives of Internal Medicine, 109, 595-600. doi:10.1001/archinte.1962.03620170093015
- Rubinow, A., Burakoff, R., Cohen, A.S., et al. (1983) Esophageal manometry in systemic amyloidosis. A study of 30 patients. American Journal of Medicine, 75, 951- 956. doi:10.1016/0002-9343(83)90874-4
- Suris, X., Moya, F., Panes, J., et al. (1993) Achalasia of the esophagus in secondary amyloidosis. American Journal of Gastroenterology, 88, 1959-1960.
- Battle, W.M., Rubin, M.R., Cohen, S., et al. (1979) Gastrointestinal motility dysfunction in amyloidosis. New England Journal of Medicine, 301, 24-25. doi:10.1056/NEJM197907053010105
- Briggs, G.W. (1961) Amyloidosis. Annals of Internal Medicine, 55, 943-957.
- Menke, D.M., Kyle, R.A., Fleming, R., et al. (1993) Symptomatic gastric amyloidosis in patients with primary systemic amyloidosis. Mayo Clinic Proceedings, 68, 763- 767.
- Bjornsson, S., Johannsson, J.H. and Sigurjonsson, F. (1987) Localizedprimary amyloidosis of the stomach presenting with gastric hemorrhage. Acta Medica Scandinavica, 221, 115-119. doi:10.1111/j.0954-6820.1987.tb01252.x
- Lau, C.F., Fok, K.O., Hui, P.K., et al. (1999) Intestinal obstruction and gastrointestinal bleeding due to systemic amyloidosis in a woman with occult plasma cell dyscrasia. European Journal of Gastroenterology and Hepatology, 11, 681-685. doi:10.1097/00042737-199906000-00017
- Iijima-Dohi, N., Shinji, A., Shimizu, T., et al. (2004) Recurrent gastric hemorrhaging with large submucosal hematomas in a patient with primary AL systemic amyloidosis: Endoscopic and histopathological findings. Internal Medicine, 43, 468-472.
- Walley, V.M. (1986) Amyloid deposition in a gastric arteriovenous malformation. Archives of Pathology and Laboratory Medicine, 110, 69-71.
- Chang, H.S., Myung, S.J., Yang, S.K., et al. (2004) Massive small bowel bleeding in a patient with amyloidosis. Gastrointestestinal Endoscopy, 59, 126-129. doi:10.1016/S0016-5107(03)02352-6
- MacManus, Q. and Okies, J.E. (1978) Amyloidosis of the stomach: Report of an unusual case and review of the literature. American Journal of Surgery, 42, 607-610.
- Reddy, A.B., Wright, R.A., Wheeler, G.E., et al. (1983) Nonobstructive gastroparesis in amyloidosis improved with metoclopramide. Archives of Internal Medicine, 143, 247-248. doi:10.1001/archinte.1983.00350020069014
- Legge, D.A., Carlson, H.C. and Wollaeger, E.E. (1970) Roentgenologic appearance of systemic amyloidosis involving the gastrointestinal tract. American Journal of Roentgenology, 110, 406-412.
- French, J.M., Gall, G., Parish, D.J., et al. (1965) Peripheral and autonomic nerve involvement in primary amyloidosis associated with uncontrollable diarrhea and steatorrhoea. American Journal of Medicine, 39, 277-284. doi:10.1016/0002-9343(65)90052-5
- Jeong, Y.S., Jun, J.B., Kim, T.H., et al. (2000) Successful treatment of protein-losing enteropathy due to AA amyloidosis with somatostatin analogue and high dose steroid in ankylosing spondylitis. Clinical and Experimental Rheumatology, 18, 619-621.
- Kuan, L., Sun, W., Gibson, M.F., et al. (2003) Gastrointestinal amyloidosis with ulceration, hemorrhage, small bowel diverticula, and perforation. Digestive Diseases and Sciences, 48, 2023-2026. doi:10.1023/A:1026190809074
- Choi, H.S., Heller, D., Picken, M.M., et al. (1989) Infarction of intestine with massive amyloid deposition in two patients on long-term hemodialysis. Gastroenterology, 96, 230-234.
- Pearson, D.C., Price, L.M. and Urbanski, S. (1996) Pneumatosis cystoides intestinalis: An unusual complication of systemic amyloidosis. Journal of Clinical Gastroenterology, 22, 74-76. doi:10.1097/00004836-199601000-00022
- Herskovic, T., Bartholomew, L.G. and Green, P.A. (1964) Amyloidosis and malabsorption syndrome. Archives of Internal Medicine, 114, 629-633. doi:10.1001/archinte.1964.03860110099009
- Vernon, S.E. (1982) Amyloid colitis. Diseases of the Colon and Rectum, 25, 728-730. doi:10.1007/BF02629550
- Chen, J.H., Lai, S.J., Tsai, P.P., et al. (2002) Localized amyloidosis mimicking carcinoma of the colon. American Journal of Roentgenology, 179, 536-537.
- Biggers, J.A., Remmers, A.R., Lindley, J.D., et al. (1975) Femoral neuropathy and ischemic colitis associated with amyloidosis in hemodialysis patients. Annals of Surgery, 182, 161-162. doi:10.1097/00000658-197508000-00014
- Garcia-Gonzalez, R., Fernandez, F.A., Garijo, M.F., et al. (1998) Amyloidosis of the rectum mimicking collagenous colitis. Pathology-Research and Practice, 194, 731-735. doi:10.1016/S0344-0338(98)80134-9
- Kumar, S.S., Appavu, S.S., Abcarian, H., et al. (1983) Amyloidosis of the colon. Report of a case and review of the literature. Diseases of the Colon and Rectum, 26, 541- 544. doi:10.1007/BF02563751
- Fraser, A.G., Arthur, J.F. and Hamilton, I. (1991) Intestinal pseudoobstruction secondary to amyloidosis responsive to cisapride. Digestive Diseases and Sciences, 36, 532-535. doi:10.1007/BF01298889
- Rives, S., Pera, M., Rosinol, L., et al. (2002) Primary systemic amyloidosis presenting as a colonic stricture: Successful treatment with left hemicolectomy followed by autologous hematopoietic stem-cell transplantation. Diseases of the Colon and Rectum, 45, 1263-1266. doi:10.1007/s10350-004-6403-x
- Gonzalez-Sanchez, J.A., Molinero, R.M., Sayans, J.D., et al. (1989) Colonic perforation by amyloidosis. Report of a case. Diseases of the Colon and Rectum, 32, 437-440. doi:10.1007/BF02563700
- Buck, F.S. and Koss, M.N. (1991) Hepatic amyloidosis: Morphologic differences between systemic AL and AA types. Human Pathology, 22, 904-907. doi:10.1016/0046-8177(91)90180-W
- Levy, M., Polliack, A., Lender, M., et al. (1974) The liver in amyloidosis. Digestion, 10, 40-51. doi:10.1159/000197521
- Kim, S.H., Han, J.K., Lee, K.H., et al. (2003) Abdominal amyloidosis: Spectrum of radiological findings. Clinical Radiology, 58, 610-620. doi:10.1016/S0009-9260(03)00142-9
- Takahashi, T., Miura, H., Matsu-ura, Y., et al. (2005) Urine cytology of localized primary amyloidosis of the ureter: A case report. Acta Cytologica, 49, 319-322. doi:10.1159/000326156
- Braunstein, J.M., Aman, A. and Warman, J. (2007) Colonic amyloidosis. Clincal Gastroenterology and Hepatology, 5, A30.
- Smith, T.R. and Cho, K.C. (1986) Small intestine amyloidosis producing a stippled punctuate mucosal pattern: Radiological-pathological correlation. American Journal of Gastroenterology, 81, 477-479.
- Pandarinath, G.S., Levine, S.M., Sorokin, J.J., et al. (1978) Selective massive amyloidosis of the small intestine mimicking multiple tumors. Radiology, 129, 609-610.
- [61] Tada, S., Iida, M., Fuchigami, T., et al. (1990) Barium meal study for amyloidosis of the small intestine: Measurements on radiograph. Gastrointestinal Radiology, 15, 320-324. doi:10.1007/BF01888809
- [62] Tada, S., Iida, M., Iwashita, A., et al. (1990) Endoscopic and biopsy findings of the upper digestive tract in patients with amyloidosis. Gastrointestinal Endoscopy, 36, 10-14. doi:10.1016/S0016-5107(90)70913-3
- [63] Lovat, L.B., Persey, M.R., Madhoo, S., et al. (1988) The liver in systemic amyloidosis: Insights from 123I serum amyloid P component scintigraphy in 484 patients. Gut, 42, 727-734. doi:10.1136/gut.42.5.727
- [64] Hazenberg, B.P.C., van Rijswijk, M.H., Piers, A., et al. (2006) Diagnostic performance of 123I-labeled serum amyloid P component scintigraphy in patients with amyloidosis. American Journal of Medicine, 119, E15-E24. doi:10.1016/j.amjmed.2005.08.043
- [65] Ohta, H., Kokuryu, H., Ota, T., et al. (1997) CT, MR imaging and radionuclide imaging in a patient with hepatic involvement in primary amyloidosis. Clinical Nuclear Medicine, 22, 778-780. doi:10.1097/00003072-199711000-00012
- [66] Rajkumar, S.V. and Gertz, M.A. (2007) Advances in the treatment of amyloidosis. New England Journal of Medicine, 356, 2413-2415. doi:10.1056/NEJMe078027
- [67] Gertz, M.A., Lacy, M.Q., Dispenzieri, A., et al. (2002) Stem cell transplantation for the management of primary systemic amyloidosis. American Journal of Medicine, 113, 549-555. doi:10.1016/S0002-9343(02)01208-1
- [68] Inoue, D., Arima, H., Kawanami, C., et al. (2010) Excellent therapeutic effect of tocilizumab on intestinal amyloid a deposition secondary to active rheumatoid arthritis. Clinical Rheumatology, 29, 1195-1197. doi:10.1007/s10067-010-1422-6
- [69] Perry, M.E., Stirling, A. and Hunter, J.A. (2007) Effect of etanercept on serum amyloid A protein (SAA) levels in patients with AA amyloidosis complicating inflammatory arthritis. Clinical Rheumatology, 27, 923-925. doi:10.1007/s10067-008-0875-3
- [70] Bodin, K., Ellmerich, S., Kahan, M.C., et al. (2010) Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature, 468, 93-97. doi:10.1038/nature09494
- [71] Gillmore, J.D., Tennent, G.A., Hutchinson, W.L., et al. (2010) Sustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosis. British Journal of Haematology, 148, 760-767. doi:10.1111/j.1365-2141.2009.08036.x
- [72] Sunga, M.N., Reyes, C.V., Zvetina, J., et al. (1989) Resolution of secondary amyloidosis 14 years after adequate chemotherapy for skeletal tuberculosis. Southern Medical Journal, 82, 92-93. doi:10.1097/00007611-198901000-00024
- [73] Gillmore, J.D., Lovat, L.B., Persey, M.R., et al. (2001) Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet, 358, 24-29. doi:10.1016/S0140-6736(00)05252-1
- [74] Fernandez-Nebro, A., Tomero, E., Ortiz-Santamaria, V., et al. (2005) Treatment of rheumatic inflammatory disease in 25 patients with secondary amyloidosis using tumor necrosis factor alpha antagonists. American Journal of Medicine, 118, 552-556. doi:10.1016/j.amjmed.2005.01.028
- [75] Bhat, A., Naguwa, S.M., Cheema, G.S., et al. (2009) Colchicine revisited. Annals of the New York Academy of Sciences, 1173, 766-773. doi:10.1111/j.1749-6632.2009.04674.x
- [76] Livneh, A., Zemer, D., Langevitz, P., et al. (1993) Colchicine in the treatment of AA and AL amyloidosis. Seminars in Arthritis and Rheumatism, 23, 206-214. doi:10.1016/S0049-0172(05)80042-3
- [77] Meyers, S., Janowitz, H.D., Gumaste, V.V., et al. (1988) Colchicine therapy of the renal amyloidosis of ulcerative colitis. Gastroenterology, 94, 1503-1507.
- [78] Kagan, A., Husza’r, M., Frumkin, A., et al. (1999) Reversal of nephrotic syndrome due to AA amyloidosis in psoriatic patients on long-term colchicine treatment. Case report and review of the literature. Nephron, 82, 348-353. doi:10.1159/000045450
- [79] Kaneko, K., Someya, T., Ohtaki, R., et al. (2003) Colchicine therapy in amyloid nephropathy due to recessive dystrophic epidermolysis bullosa. Pediatric Nephrology, 18, 1311-1312. doi:10.1007/s00467-003-1310-2
- [80] Chen, C.C., Isomoto, H. and Hayashi, T. (2012) Gastrointestinal amyloidosis secondary to inherited skin disorder. Gastroenterology, 142, e9-e10. doi:10.1053/j.gastro.2011.08.052
- [81] Reddy, A.B., Wright, R.A., Wheeler, G.E., et al. (1983) Nonobstructive gastroparesis in amyloidosis improved with metoclopramide. Archives of Internal Medicine, 143, 247-248. doi:10.1001/archinte.1983.00350020069014
- [82] Gil, R., Debiais, F., Lefevre, F.P., et al. (1984) Amyloidosis with gastroparesis. Improvement with domperidone. Presse Médicale, 13, 564.
- [83] Ponec, R.J., Saunders, M.D. and Kimmey, M.B. (1999) Neostigmine for the treatment of acute colonic pseudoobstruction. New England Journal of Medicine, 341, 137- 141. doi:10.1056/NEJM199907153410301
- [84] Hayman, S.R., Lacy, M.Q., Kyle, R.A., et al. (2001) Primary systemic amyloidosis: A cause of malabsorption syndrome. American Journal of Medicine, 111, 535-540. doi:10.1016/S0002-9343(01)00919-6
- [85] Poullos, P.D. and Stollman, N. (2003) Gastrointestinal amyloidosis: Approach to treatment. Current Treatment Options in Gastroenterology, 6, 17-25. doi:10.1007/s11938-003-0029-2
ABBREVIATIONS
GI, gastrointestinal;
AA, secondary amyloidosis;
FMF, familial-Mediterranean fever;
CPHPC, R-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxohexa-noyl]pyrrolidine-2 carboxylic acid;
EB, epidermolysis bullosa.
NOTES
*Corresponding author.