Open Journal of Polymer Chemistry
Vol.3 No.1(2013), Article ID:28470,5 pages DOI:10.4236/ojpchem.2013.31002
Immobilization of His-Tagged Proteins on Various Solid Surfaces Using NTA-Modified Chitosan
Department of Chemical and Environmental Engineering, Graduated School of Engineering, Gunma University, Kiryu-shi, Japan
Email: *katsura@cee.gunma-u.ac.jp
Received December 6, 2012; revised January 7, 2013; accepted January 20, 2013
Keywords: Protein Immobilization; Chitosan; Ni-NTA (Nickel-Nitrilotriacetic Acid); Solid Surface
ABSTRACT
Continued advancement of protein array, bioelectrode, and biosensor technologies will necessitate development of methods that allow for increased protein immobilization capacity and more control over protein orientation. Toward these ends, we developed a method involving modification of chitosan with nitrilotriacetic acid (NTA) to achieve immobilization of a larger amount of His-tagged protein than is possible with current methods. The immobilization capacity of our method was evaluated using His-tagged GFP (Green Fluorescent Protein) as a model protein. The average immobilization density on modified glass was about 32 ng/mm2. Our method is suitable for use on a variety of solid surfaces, including glassy carbon, silicon wafers, polycarbonate, and beaten gold.
1. Introduction
The immobilization of proteins on the surface of solid materials is a key technique in the production of protein arrays, biosensors, and bioelectrodes for use in the analytical and bioelectronics fields. For these applications, it is necessary to arrange the orientation of immobilized protein molecules so that their function will be maintained. A number of different methods have been reported for immobilizing proteins on a variety of surfaces [1]. Proteins may be immobilized by physical absorption (e.g., dot blot [2,3] and polyacrylamide gel [4] methods), through covalent coupling or cross-linking [1,5-10], as a self-assembled monolayer (SAM) [1,6,11], or through affinity interactions (e.g., avidin/biotin [1,12], Ni-NTA (nickel-nitrilotriacetic acid)/His-tagged protein [1], and intein methods [1,6,13,14]).
A dot blot is a technique used in molecular biology to detect biomolecules on nitrocellulose or PVDF (polyvinylidene difluoride) membranes [2,3]. Dot blotting is based on physical adsorption and allows for rapid immobilization of substrates and protein molecules. Deactivation of immobilized proteins can be suppressed by spotting them onto polymers such as polyacrylamide gels or polyethyleneglycol applied to a solid surface by inhibiting protease attack and minimizing shear, interfacial temperature or solvent denaturation [4]. Because protein molecules are fixed within the three-dimensional space of the gel, this method allows for the immobilization of at least 10 times more protein than is possible with simple surface absorption methods. However, most methods, including those described above, do not allow for controlling the orientation of protein molecules; therefore, a portion of each fixed protein molecule may be inaccessible to substrates.
MacBeath and Schreiber immobilized proteins on BSA-NHS (bovine serum albumin-N-hydroxysuccinimide)-coated slides fabricated by attaching a molecular layer of BSA to the surface of a glass slide and then activating the BSA with N,N,disuccinimidyl carbonate [13]. Protein molecules are then “printed” on the slide and immobilized through covalent urea or amide linkages with the activated lysine, aspartate, and glutamate residues on BSA molecules. Another method based on covalent coupling or cross-linking formation was also reported [5-10]. In this method, protein molecules are immobilized to a surface through a cross-linking reaction in which epoxy groups on a modified glass surface react with amino groups on the surface of the protein (epoxy coupling reaction). Major drawbacks associated with methods based on covalent bonds however are the high possibility that protein activity will be lost due to the lack of control over protein orientation.
A self-assembled monolayer is an organized layer of amphiphilic molecules in which one end of the molecule, the “head group”, shows a specific, reversible affinity for a given substrate [1,6,11]. The hydrophilic head groups assemble together on the substrate, while the hydrophobic tail (alkyl chain) groups assemble far from the substrate surface. Because immobilization by this method is based on adsorption, it is difficult to control protein orientation.
Affinity methods such as those based on streptavidin/ biotin or His-tag/Ni-NTA interactions are commonly used in biochemical and histomorphology studies [1]. Affinity techniques exploit specific binding between streptavidin and biotin-labeled proteins or between Histagged proteins and Ni-NTA [1]. The interaction between biotin and streptavidin is the strongest noncovalent biological interaction known, with a dissociation constant (KD) on the order of 4 × 10−14 M [1,12]. However, it is necessary to modify the substrate with streptavidin or NTA and the protein of interest with biotin or a His-tag prior to immobilization.
For intein-mediated immobilization, target genes are inserted in-frame and the mRNA is translated together with that encoding a target protein. This precursor protein (tag-intein-target protein) undergoes autocatalytic protein splicing, resulting in 2 products (tag-target protein and intein). By using this method, the N-terminal cysteine-containing peptides were specific immobilized onto a thioester-functionalized glass slide, which was subsequently used for screening of epitope mapping of kinase/phosphatase assays [15]. This strategy was extended to the immobilization of proteins in a microarray format using intein [6,13,14]. Intein is a segment of a whole protein that can be used to splice proteins with a peptide bond. However, this method involving intein may not be applicable to every protein, and its use requires high protein labeling efficiency.
These protein absorption/immobilization techniques were designed for immobilizing large amounts of highly active proteins at a high density. However, there is still a need for techniques that will enable immobilization of greater amounts of protein while preserving protein activity. Immobilizing a sufficient quantity of a protein of interest over a minimal surface area remains challenging for many applications, as does maintaining the orientation of immobilized proteins so as to preserve their activity. To solve the problem of maximizing the amount and density of protein immobilized, we increased the effecttive surface area available for immobilization by modifying the surface of various solid substrates with a polymer (chitosan). To address the problem of maintaining the proper orientation of an immobilized protein, we modified chitosan bound to the surface with NTA to allow for specific binding between NTA and the Histagged protein. The method we describe here is simple with respect to both preparation of the substrate surface and protein immobilization. In addition, the method is suitable for use with a variety of substrates, and thus should have a wide range of applications.
2. Experimental
2.1. Materials
We purchased 3-aminopropyltriethoxysilane from ShinEtsu Chemical (Tokyo, Japan). Shrimp shell chitosan (>75%, deacetylated) was purchased from Sigma-Aldrich (St. Louis, MO, USA). N-(5-amino-1-carboxypentyl) iminodiacetic monohydrate disodium salt (AB-NTA) was obtained from Dojindo (Kumamoto, Japan). Glutaraldehyde, NiCl2, imidazole, and all other reagents were purchased from Wako (Osaka, Japan).
2.2. Purification of Recombinant His-Tagged GFP Protein
The His-tag modified GFP expression vector (pGGFPH) was a kind gift from Professor H. Nakano (Graduate School of Bioagricultural Science, Nagoya University, Japan) [15]. His-tagged GFP was overexpressed in E. coli Rosetta(DE3) (Novagen; Madison, WI, USA) and purified using Ni-NTA Superflow chromatography (Qiagen; Germantown, MD, USA) at 4˚C according to the manufacturer’s protocol. The protein concentration was determined according to the method of Bradford using a protein assay kit (Bio-Rad; Benicia, CA, USA) with BSA as the standard.
2.3. Preparation of Glass Slides
Glass slides printed with highly water-repellent mark (Matsunami; Tokyo, Japan) were washed with acetone and milli-Q water. The slides were soaked in 1 M NaOH overnight at room temperature, and then washed with milli-Q water and 99.5% ethanol. Next, 3-aminopropyltriethoxysilane was vacuum-deposited onto the cleaned glass (CG) surface for 2 h at room temperature, washed with milli-Q water, and then the slides were baked for 2 h at 100˚C after blowing off residual water with dry air. This process produced amino silane-treated CG slides (AGs). The Ags were soaked in 1% (v/v) glutaraldehyde overnight at 37˚C to produce glutaraldehyde-treated AGs (GAGs). The GAGs were incubated overnight at 37˚C in chitosan (0.05% (w/v)) dissolved in 0.1 M acetic acid buffer (pH 5.0) supplemented with 0.25 mM sodium azide, producing chitosan immobilized glass slides (CIGs). The CIGs were rinsed twice with 0.1 M acetic acid buffer (pH 5) and then 3 times with milli-Q water. The CIGs were soaked in 1% (v/v) glutaraldehyde overnight at 37˚C to produce glutaraldehyde-treated CIGs (GCIGs). The GCIGs were incubated overnight at 37˚C in a 0.05% (w/v) solution of N-(5-Amino-1-carboxypentyl) iminodiacetic acid (AB-NTA) in 0.1 M HEPES buffer (pH 8.0), then rinsed 3 times with milli-Q water. The slides were then soaked in blocking solution (1% (v/v) glycine) for 1 h at 37˚C, then rinsed 3 times with milli-Q water, 3 times with 0.5 M NiCl2, and 3 times with milli-Q water to produce Ni-NTA immobilized GCIGs (Ni-NTAGCIGs).
2.4. Preparation of Other Modified Solid Substrates
Preparation of glassy carbon plates, silicon wafer plates, and polycarbonate plates was exactly same way to the protocol used to prepare the modified glass slides. Beaten gold for use in immobilization was prepared as follows. Cysteine was used instead of 3-aminopropyltriethoxysilane to modify gold surfaces with amino groups. Beaten gold was soaked in 1 M HNO3 for 2 h, rinsed with milliQ water, then pasted to the slide glass with double-faced tape. Cysteine thiol groups were adsorbed onto the cleaned gold surface by soaking the glass in 1 M cysteine solution (pH 9.4) for 2 h with stirring. The amino groups of cysteine residues bound to the surface were then crosslinked with amino groups of chitosan using glutaraldehyde. The subsequent preparation steps for beaten gold were almost the same as those used in the preparation of modified glass from the step of modified CIGs.
2.5. Immobilization and Quantification of His-Tagged GFP
Immobilization of His-tagged GFP was examined on various substrates (CG, AG, GAG, CIG, GCIG, NiNTAGCIG, modified glassy carbon, modified silicon wafers, modified polycarbonate, and a modified gold surface). His-tagged GFP was adjusted to 200 μg/ml with TG buffer (50 mM Tris-HCl (pH 8.0), 10% (v/v) glycerol). A total of 100 ng of His-tagged GFP was spotted on each substrate and incubated for 60 min in a moist, dark chamber. Substrates were then washed with TG buffer, and immobilized His-tagged GFP was observed with a UV illuminator (TFML-20E, Funakoshi; Tokyo, Japan) or a fluorescence microscope (Nikon ECLIPSE TE2000-U; Tokyo, Japan). Images were captured with a digital camera (Nikon DIGITAL CAMERA D80; Tokyo, Japan). The amount of His-tagged GFP immobilized on each surface was determined by comparing the images with those for known amounts of His-tagged GFP (0, 0.25, 0.5, and 2 μg/spot) using Image J version 1.44 (http://rsbweb.nih.gov/ij/). Immobilized His-tagged GFP was also observed under a fluorescence microscope equipped with a Nikon CF Plan 10×/0.30 EPI Infinity lens. The scale was determined by comparison with the appropriate scale on the captured images.
3. Results and Discussion
Figure 1(a) diagrams the preparation of surfaces for immobilization of His-tagged protein. The washed glass was treated with 3-aminopropyltriethoxysilane, followed by glutaraldehyde. Soaking the glutaraldehyde-treated glass in chitosan solution resulted in cross-linking of the amino groups bound to the modified glass with the amino groups of chitosan. The chitosan-immobilized glass was treated with glutaraldehyde again to activate free chitosan amino groups. Subsequent soaking of the chitosanimmobilized glass in AB-NTA solution resulted in crosslinking of the activated amino groups in chitosan with the amino groups of AB-NTA. His-tagged GFP could then be immobilized onto the modified glass by charging the NTA with Ni2+ (Figure 1(b)).
The fluorescence associated with immobilized Histagged GFP at each modification step is shown in Figure 2. Fluorescence was very low for the washed glass (WG), aminosilane-treated glass (AG), glutaraldehyde-treated AG (GAG), and chitosan-immobilized glass (CIG). Higher fluorescence signals were observed with the glu-

Figure 1. (a) Preparation of the modified glass substrate using chitosan and Ni-NTA; (b) Schematic of completed modified glass substrate with immobilized His-tagged GFP.
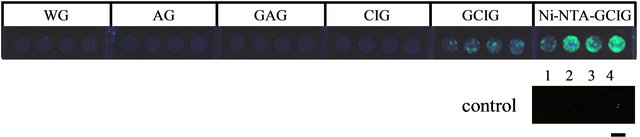
Figure 2. Fluorescence of immobilized His-tagged GFP at each modification step. WG: washed glass, AG: aminosilane-treated glass, GAG: glutaraldehyde-treated AG, CIG: chitosan-immobilized glass, GCIG: glutaraldehyde-treated CIG, Ni-NTAGCIG: Ni-NTA-immobilized GCIG. Control spots were used to estimate the amount of His-tagged GFP immobilized. Spot 1: 0 μg His-tagged GFP, Spot 2: 0.25 μg His-tagged GFP, Spot 3: 0.5 μg His-tagged GFP, and Spot 4: 2.0 μg His-tagged GFP. Scale bar = 4 mm.
taraldehyde-treated CIG (GCIG) and Ni-NTA-immobilized GCIG (Ni-NTAGCIG). Comparing the fluorescence obtained with GAG and GCIG surfaces demonstrated that His-tagged GFP was immobilized more efficiently on GCIG than on GAG. This result indicated that chitosan modification increased the number of sites available for cross-linking His-tagged GFP. More Histagged GFP was immobilized onto GCIG than NiNTAGCIG surfaces, indicating that the cross-linking of AB-NTA and chitosan was more efficient than the crosslinking of His-tagged GFP and AB-NTA.
The molecular weight of AB-NTA (about 263 Da) is much lower than that of His-tagged GFP (about 27 kDa). Therefore, without orientation control through AB-NTA, considerable steric hindrance associated with His-tagged GFP would be expected. Our results demonstrated that large amounts of His-tagged GFP can be immobilized by increasing the number of cross-linking sites with chitosan and providing orientation control with NTA. Based upon comparison with His-tagged GFP controls, we estimated that 0.26, 0.52, 0.41, and 0.43 μg His-tagged GFP/diameter 4 mm spot was immobilized onto the Ni-NTAGCIG surface (with an average of about 0.45 μg/the spot of diameter 4 mm, or about 32 ng/mm2).
To test the specificity of immobilization, His-tagged GFP immobilized on Ni-NTAGCIG surfaces was striped by soaking the slides in 1 M imidazole solution. Histagged GFP was removed from Ni-NTAGCIG slides by treatment with either imidazole (Figure 3) or 0.5 M EDTA (data not shown). This result strongly suggested that the immobilization of His-tagged GFP onto NiNTAGCIG-modified surfaces is due to formation of a complex between the His-tag and Ni-NTA. When Histagged GFP was re-spotted onto Ni-NTAGCIG slides from which it had been removed, almost the same amount of His-tagged GFP was immobilized (data not shown).
We were also able to successfully immobilize Histagged GFP onto other solid surfaces, such as glassy carbon, silicon wafers (polished and unpolished), polycarbonate, and beaten gold (Figure 4). The immobiliza-

Figure 3. Removal of immobilized His-tagged GFP from NiNTAGCIG slides. (a) Fluorescence of His-tagged GFP immobilized onto a Ni-NTAGCIG surface; (b) Fluorescence of the same surface after soaking in 1 M imidazole solution for 60 min at 4˚C. Scale bar = 4 mm.
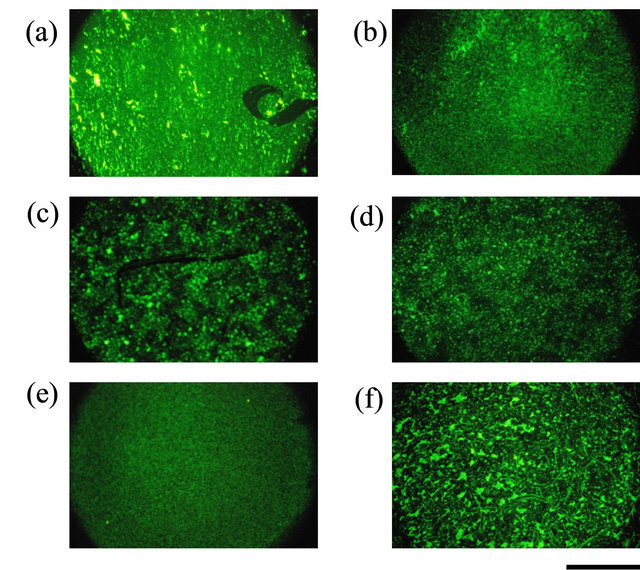
Figure 4. Immobilization of His-tagged GFP on various solid substrates using chitosan and Ni-NTA modification. (a) Glass; (b) Glassy carbon; (c) Silicon wafer (polished surface); (d) Silicon wafer (unpolished surface); (e) Polycarbonate; (f) Beaten gold. Immobilized His-tagged GFP was observed with a fluorescence microscope. Scale bar = 500 μm.
tion efficiency was higher on the glassy carbon than on the other materials.
4. Conclusion
The immobilization capacity of our method was evaluated using His-tagged GFP as a model protein. The average immobilization density on modified glass was about 32 ng/mm2. We found that incorporation of chitosan increases the amount of His-tagged GFP that can be immobilized onto a variety of solid surfaces, including glassy carbon, silicon wafers, polycarbonate, and beaten gold. Our method could thus be applied to the immobilization of large amounts of highly active protein to surfaces such as enzyme electrodes or sensors. Experiments aimed at improving the method by eliminating the irregularities associated with polymer modification are ongoing.
REFERENCES
- F. Rusmini, Z. Zhong and J. Feijen, “Protein Immobilization Strategies for Protein Biochips,” Biomacromolecules, Vol. 8, No. 6, 2007, pp. 1775-1789. doi:10.1021/bm061197b
- H. Ge, “UPA, a Universal Protein Array System for Quantitative Detection of Protein-Protein, Protein-DNA, Protein-RNA and Protein-Ligand Interactions,” Nucleic Acid Research, Vol. 28, No. 2, 2000, p. e3. doi:10.1093/nar/28.2.e3
- L. J. Holt, K. Bussow, G. Walter and L. M. Tomlinson, “By-Passing Selection: Direct Screening for AntibodyAntigen Interactions Using Protein Arrays,” Nucleic Acid Research, Vol. 28, No. 2, 2000, p. e72. doi:10.1093/nar/28.15.e72
- A. Pollak, H. Blumenfeld, M. Wax, R. L. Baughn and G. M. Whitesides, “Enzyme Immobilization by Condensation Copolymerization into Cross-Linked Polyacrylamide Gels,” Journal of the American Chemical Society, Vol. 102, No. 20, 1980, pp. 6324-6336. doi:10.1021/ja00540a026
- G. MacBeath and S. L. Schreiber, “Printing Proteins as Microarrays for High-Throughput Function Determination,” Science, Vol. 289, No. 5485, 2000, pp. 1760-1763.
- J. A. Camarero, “Recent Developments in the Site-Specific Immobilization of Proteins onto Solid Supports,” Biopolymers, Vol. 90, No. 3, 2008, pp. 450-458. doi:10.1002/bip.20803
- M. A. Gauthier and H. A. Klok, “Peptide/Protein-Polymer Conjugates: Synthetic Strategies and Design Concepts,” Chemical Communications, No. 23, 2008, pp. 2591-2611. doi:10.1039/b719689j
- M. Köhn, “Immobilization Strategies for Small Molecule, Peptide and Protein Microarrays,” Journal of Peptide Science, Vol. 15, No. 6, 2009, pp. 393-397. doi:10.1002/psc.1130
- M. Frasconi, F. Mazzei and T. Ferri, “Protein Immobilization at Gold-Thiol Surfaces and Potential for Biosensing,” Analytical and Bioanalytical Chemistry, Vol. 398, No. 4, 2010, pp. 1545-1564. doi:10.1007/s00216-010-3708-6
- S. Cosnier and M. Holzinger, “Electrosynthesized Polymers for Biosensing,” Chemical Society Reviews, Vol. 40, No. 5, 2011, pp. 2146-2156. doi:10.1039/c0cs00090f
- D. Samanta and A. Sarkar, “Immobilization of Bio-Macromolecules on Self-Assembled Monolayers: Methods and Sensor Applications,” Chemical Society Reviews, Vol. 40, No. 5, 2011, pp. 2567-2592. doi:10.1039/c0cs00056f
- A. Holmberg, A. Blomstergren, O. Nord, M. Lukacs, J. Lundeberg and M. Uhlén, “The Biotin-Streptavidin Interaction Can Be Reversibly Broken Using Water at Elevated Temperatures,” Electrophoresis, Vol. 26, No. 3, 2005, pp. 501-510. doi:10.1002/elps.200410070
- M. L. Lesaicherre, R. Y. Lue, G. Y. Chen, Q. Zhu and S. Q. Yao, “Intein-Mediated Biotinylation of Proteins and Its Application in a Protein Microarray,” Journal of the American Chemical Society, Vol. 124, No. 30, 2002, pp. 8768-8769. doi:10.1021/ja0265963
- A. Girish, H. Sun, D. S. Yeo, G. Y. Chen, T. K. Chua and S. Q. Yao, “Site-Specific Immobilization of Proteins in a Microarray Using Intein-Mediated Protein Splicing,” Bioorganic & Medicinal Chemistry Letters, Vol. 15, No. 10, 2005, pp. 2447-2451. doi:10.1016/j.bmcl.2005.03.079
- T. Kinpara, R. Mizuno, Y. Murakami, M. Kobayashi, S. Yamaura, Q. Hasan, Y. Morita, H. Nakano, T. Yamane, and E. Tamiya, “A Picoliter Chamber Array for Cell-Free Protein Synthesis,” Journal of Biochemistry, Vol. 136, No. 2, 2004, pp. 149-154. doi:10.1093/jb/mvh102
NOTES
*Corresponding author.