Open Journal of Polymer Chemistry
Vol.2 No.4(2012), Article ID:25160,4 pages DOI:10.4236/ojpchem.2012.24018
Teaching Polymer Chemistry: Revisiting the Syllabus
1Université de Lyon, Université Claude Bernard Lyon, Ingénierie des Matériaux Polymères, Villeurbanne, France
2Institut Charles Sadron, CNRS 23, Rue du Loess, Strasbourg Cedex, France
3Université du Maine, Unité de Chimie Organique Moléculaire et Macromoléculaire, Le Mans cedex, France
4Lorraine Université, Laboratoire de Chimie-Physique Macromoléculaire, Nancy, France
5Centre Québécois des Matériaux Fonctionnels, Département de Chimie, U. Sherbrooke, J1K2R1, Qc, Canada
6Groupement Français d’Etudes et d’Applications des Polymères (GFP), Institut Charles Sadron, 23, Rue du Loess, Strasbourg Cedex, France
Email: *thierry.hamaide@univ-lyon1.fr
Received August 24, 2012; revised September 25, 2012; accepted October 5, 2012
Keywords: Teaching Polymer Chemistry; Chain Polymerization; Step-Growth Polymerization; Polycondensation; Polyaddition
ABSTRACT
As anyone who has taught polymer chemistry to undergraduate students might already appreciate, we are faced with inherent difficulties in clearly communicating the subject matter due its interdisciplinary character. This paper is thus aimed at proposing a new educational plan to teach this fundamental course in order to overcome some inherent obstacles. It is especially devoted to professors in charge of teaching a first course in polymer chemistry. We intend to emphasize polymerization by beginning with the simplest chemical pathway, namely the living anionic polymerization. In addition, some pedagogical difficulties are outlined all along this paper. This novel and distinct way to teach polymer chemistry makes the students with a background in organic chemistry more equipped to face difficulties arising from the introduction of new concepts.
1. Introduction
This article is based on a series of meetings among members of the Groupe Français d’Etudes et d’Applications des Polymères (GFP-Polymer French Association) [1]. By sharing our experience in teaching polymer chemistry, we came to the conclusion that the way this discipline is taught can be improved in order to make students more interested in polymer science. A first version of our reflexions was published in French [2] and presented at the 2010 IUPAC meeting [3].
Due to very encouraging feedback we received after these publications, we decided to share our conclusions with the larger chemistry community. This article is thus intended to draw the major lines to another way of teaching polymer chemistry and highlighted some pedagogical difficulties encountered by students.
2. How to Introduce Macromolecular Chemistry?
2.1. General Considerations
After a general overview on polymers, most of the textbooks dealing with macromolecular chemistry introduce polymerization by investigating first the step-growth chemistry. Radical and ionic polymerizations and finally olefin catalytic polymerization are subsequently examined. We argue that such a pedagogical education experiences some drawbacks from both industrial and educational perspectives, as illustrated below.
Studying step-growth polymerization on a first course naturally stems from an extension of the well-known esterification reaction learned in introductory organic chemistry courses. However, applying this reaction to bifunctional monomers to build macromolecules is not straightforward. Concepts such as the progressive disappearance of initial monomers and the related concomitant extension of macromolecules end-capped with functional groups are not generally well understood by students. At any time, the system consists of a mixture of macromolecules with different chain lengths. Such phenomena are not encountered in reactions involving small molecules. Moreover, introducing non-stoichiometric conditions as well as adding monofunctional reactants to control the average molecular weight increases the difficulty to grasp concepts in polymerization.
After introducing step-growth polymerization, concept of radical polymerization is generally discussed. Understanding radical polymerization actually requires a fundamental knowledge of radicals and radical chemistry. Conceptually the very notion of a radical is not generally understood by students. For instance, the formation of the primary radical that initiates radical propagation from the decomposition of an initiator requests a fundamental understanding of the implicit kinetics. Things get more complicated when it is time to deal with the very short lifetimes of macroradicals. Difficulties still arise when copolymerization is taught, for which a composition assortment needs to be discussed.
An additional comment about traditional courses on polymer chemistry must be put forward. It concerns the use of polyethylene and polypropylene as classical examples of polymer syntheses, mainly due to their very simple structure. Unfortunately, the mechanism to get them involves olefin catalytic polymerization, which is definitely the most difficult polymerization reaction to teach. Exercises related to potential applications are not so obvious to find.
To face these aforementioned drawbacks, we thus propose an alternative way to improve student awareness regarding polymer synthesis. We argue that a course on macromolecular chemistry should begin with the anionic chain polymerization. The main reason to introduce this matter first is the fact that students who attend this course are conversant with organic chemistry, and undoubtedly with anionic reactions. Consequently, topics such as the introduction of chain generation or the determination of polymerization degree become easier to grasp and thus should be taught first. Step-growth polymerization should be relocated at the end of the introductory course on polymer chemistry.
After the description of specific features of polymers, this article introduces the concept of chain polymerizetions by first reporting anionic polymerization, followed by the other chain polymerizations (cationic, radical, coordination polymerization). Finally, step-growth polymerization (polycondensation, polyaddition) is described. All along the text, we want to emphasize the difficulties that can arise when a new concept is introduced, and we will propose ways to solve them.
Finally, whatever the approach used to deal with different sections of macromolecular chemistry, it is important to relate lessons to the basic definitions, according to IUPAC [4-7].
2.2. Some Specific Features of Polymers.
Before specifically dealing with polymerization, some general questions are firstly suggested as part of the first course on polymer chemistry. They are aimed to highlight problems or incorrectness that are generally encountered.
2.2.1. What Is a Macromolecule?
Students usually confuse the terms polymer, macromolecule and polymer chain. Although often used interchangeably in everyday life, these fundamental differences need to be emphasized. Basically, a polymer is isolated as a mixture of macromolecules, the molecular weights of which are usually higher than 1000 g/mol. Macromolecules are constituted of repeat units, whose chemical structure is directly or closely linked to the monomers.
2.2.2. What Is Polymerization?
Polymerization consists in building macromolecules through the formation of successive new chemical bonds. Such a development involves a large number of elementary steps. Due to the inherent statistical character of this process, the same chain length for all the polymers is impossible to attain, resulting in a distribution of chain lengths and hence a distribution in molecular weights. For instance, a polystyrene sample, whose measured molecular weight is 10,000 g/mol, is composed of a huge number of macromolecules that all possess the same repeating chemical structure, but differentiate from each other by their chain lengths. The measured molecular weight undoubtedly corresponds to an average. The completion of n monomers does not thus lead to a single macromolecule but to a mixture of macromolecules. Accordingly, to verify that the notion of polymerization is properly grasped by the students, they must treat a textbook case: how many monodispersed macromolecules with a polymerization degree of 1000, can be obtained, starting with one mole of monomers.
2.2.3. What Are a Monomer Unit and a Degree of Polymerization?
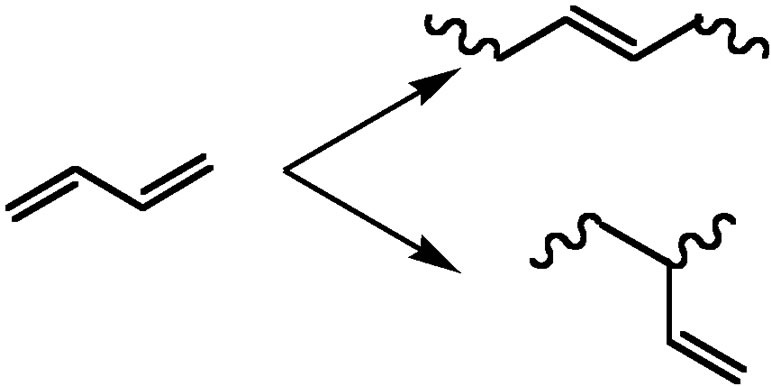
According to IUPAC, the degree of polymerization corresponds to the number of monomer units that constitute the macromolecule. It is not always matched to the number n that is actually employed in the schematic representation of a macromolecule (Figure 1), since it depends on the chemical structure of the repeat unit. Care must thus be taken in order to clearly indicate that a repeat unit is not always the synonym of a monomer unit. For instance, a repeat unit is the monomer unit when considering polystyrene or polypropylene but this is not the case with polyethyleneterephtalate (PET) or polyamide 6,6 (PA-6,6), for which the repeat unit is issued from two monomer units. Further simple examples can be given to clarify this point.
2.2.4. What Is Molecular Weight Distribution?
It is worth highlighting the general concept of molecular
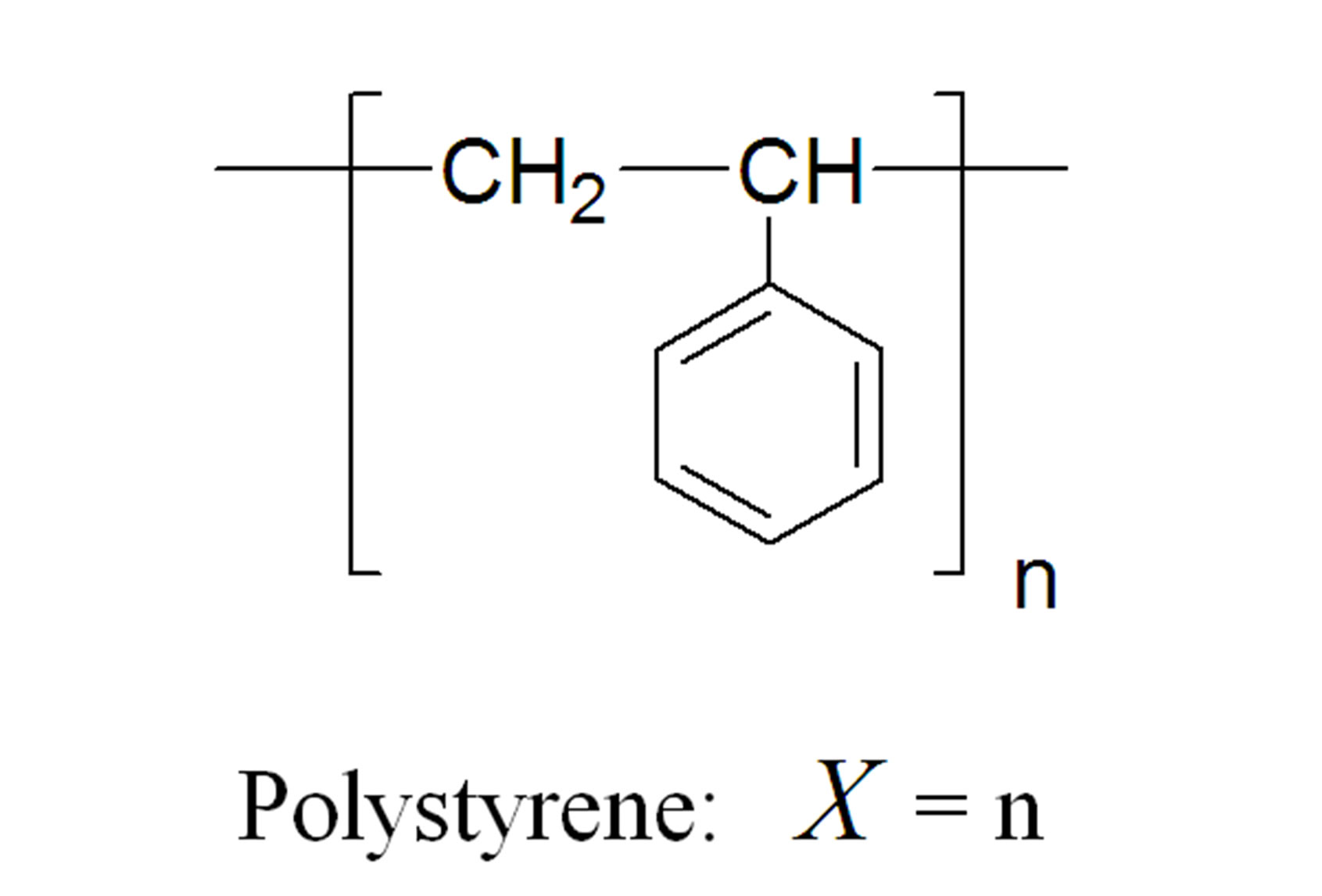

Figure 1. Two examples of the difference between the degree of polymerization and the number n which intervenes in the schematic representation of a polymer.
weight distribution since it is one of the major differences between polymers and low molecular weight molecules. To illustrate the actual molecular weight distribution in the range of relatively low molecular weights, it is suggested to display MALDI-TOF spectra, instead of SEC chromatograms, wherein each peak is attributed to a specific molecular weight (Figure 2).
The number average degree of polymerization  is defined as the ratio of the number of monomer units to the number of macromolecules:
is defined as the ratio of the number of monomer units to the number of macromolecules:
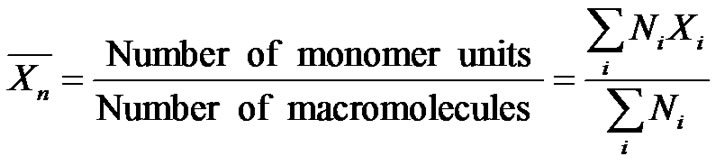 (1)
(1)
where Ni is the number of chains with a degree of polymerization Xi. This relation is of utmost importance in an elementary course. It has to be emphasized that the number of monomer units along the macromolecule, as well as the number of macromolecules, greatly depends on the reaction pathway.
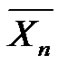 can then be written in another form by introducing molar fractions
can then be written in another form by introducing molar fractions :
:
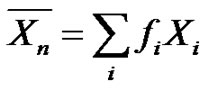 (2)
(2)
Such a notation helps to introduce the weight-average degree of polymerization 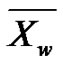 by making a parallel with the weight fraction
by making a parallel with the weight fraction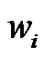 :
:
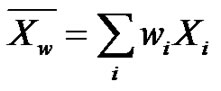 (3)
(3)
The ratio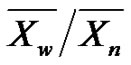 , formerly called polymolecularity index, is now referred by IUPAC as dispersity, and annotated as Đ (“D-stroke”).
, formerly called polymolecularity index, is now referred by IUPAC as dispersity, and annotated as Đ (“D-stroke”).
2.2.5. General Comment on the Different Averages
At this stage, it is useful to comment on the interest in both most used average definitions from a practical point of view. Details can easily be found in literature and lecture books However, chemistry focuses more on the number-average degree (a colligative property) while from rheological aspects, engineering is more concerned by the weight-average degree. Example from Painter and Coleman [8] is a
 (a)
(a)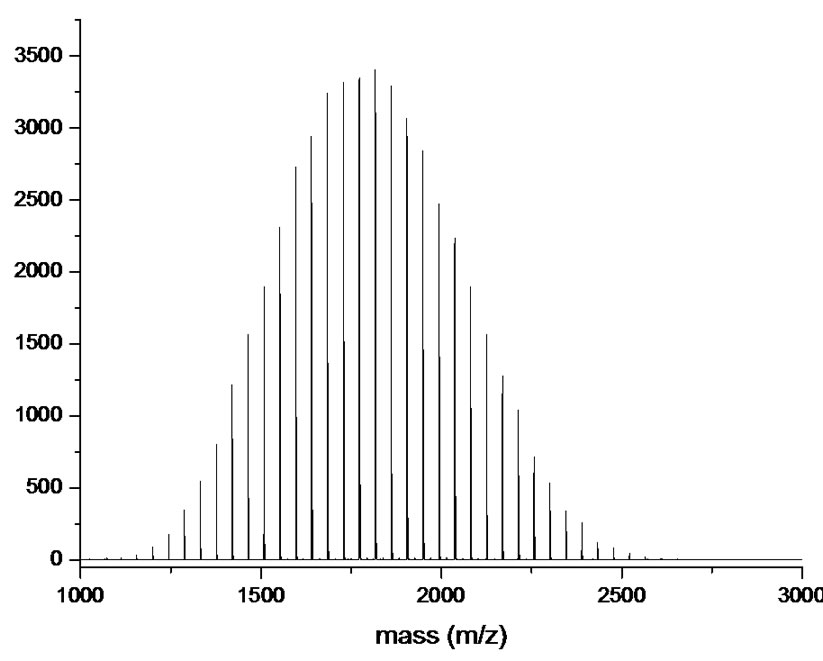 (b)
(b)
Figure 2. SEC (a) and MALDI-TOF (b) spectra of the same MPEG sample (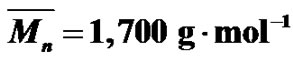 ;
;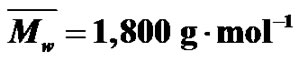 .).
.).
very nice illustration to compare the effect of both averages. The authors considered an elephant of 1,000 kg with four birds each weighting 0.5 kg in its back. Students rapidly establish a difference in the consequences of both averages.
2.2.6. Two Main Chemistries
Prior to the in depth description of the different kinds of polymerization reactions, the two main schemes, namely chain and step-growth polymerizations (polycondensation and polyaddition), should be introduced in order to highlight and comment the main differences that govern the building of macromolecular chains, according to the usual chemical schemes:
• chain polymerizations:
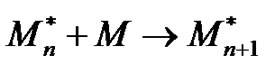
• step-growth polymerizations:

where L refers to low molecular weight molecules appearing in the case of polycondensation
3. Chain Polymerization
3.1. The Active Center and the Building of the Macromolecular Chain
The general scheme of the chain-growth polymerization is depicted in Figure 3. It is generally divided into several elementary steps. Each contribution needs to be analysed, and compared with the other ones in order to draw an overview of the involved chemistry. Whatever the elementary step, the role of the active centre is a key issue in better understanding polymerization and must be introduced at this stage.
It is worth recalling that chemistry of macromolecules is directly linked to the formation, and reactivity, of these active centres. Concepts of active centres are intimately related to the organic chemistry models (mesomeric and inductive effects, cation transpositions, coupling or disproportionation of radicals, elimination…). It is thus essential to focus the lesson on this active centre and in particular on its lifetime, which originates from its reactivity: the formation of the polymer chain greatly depends on it. Any chain formation actually begins with the generation of an active centre on a molecule, which can be an ion or a radical, than the addition of the first monomer can take place; it is the so-called initiation step. The different steps intervening during polymerization are thus introduced.
As an active centre is formed on a monomer, it enables the reaction of this activated molecule with other monomers.
3.2. Anionic Polymerization
3.2.1. The Simplest Model
Our approach is based on the fact that teaching polymerization by familiarizing the students with the concept of anionic polymerization is more educational. After introducing the very notion of active centre, polymerization with strong electronegative groups can be taught. The polymerizations of styrene from s-BuLi and ethylene oxide (EO) from EtOK are in fact immediate illustrations of both the initiation reaction and the chain growth. Interestingly, they exhibit different active centres, carbanions or alkoxydes, but they are always located at the end of the growing chain. As the occurrence of two active centres cannot give rise to bimolecular termination and the propagation steps continues until all the monomers have reacted (in an ideal case), the building of macromolecular chains becomes straightforward to describe.
The reactions stop after total use of the monomers, but it must be stressed that there are still active centers located at the end of the macromolecules which must be neutralized or which can be used for further reactions (chain end functionalization, block copolymers).
Since the students who attend this course possess basic knowledge in organic chemistry, a series of concepts of organic chemistry that help to better understand the anionic polymerization need to be reminded:
• Reactivity of vinylic monomers to foster polymerization is highly related to the electronic density of the double bond. Any effects such as withdrawing mesomer or inductive effect on the double bond, and/or stabilization of the active centre, will decrease such reactivity.
• Conjugated dienes lead to various chain structures through either 1,2-or 1,4-addition. Different chemical structures are obtained (Figure 4(a)) leading to shifts in the glass transition temperature and allowing a great variation in mechanical properties.

Figure 3. General scheme of chain-growth polymerization.
• During the propagation steps of acrylates and methacrylates (α,β-unsaturated esters), the anionic active centres may react in a Michael addition fashion at the beta carbon position (Figure 4(b)).
• Effects of the solvent and the counter-ion are important to consider: for instance, lithium alkoxides cannot be used to initiate EO polymerization. This feature is widely used to introduce hydroxyethyl groups in organic synthesis: potassium salts are therefore preferred to carry out EO polymerization (Figure 4(c)).
• The ring opening polymerization of oxygenated heterocycles is governed by both the polarity of the carbon-oxygen bond and the strain of the ring: for instance ethylene oxide can be polymerized but not tetrahydrofuran (THF) (cationic conditions have to be fulfilled to polymerize THF). Differentiation in the pathway between anionic and cationic reactions must thus be outlined
3.2.2. Kinetics
Regarding the kinetics laws, it becomes straightforward to demonstrate that, in this case, the polymerization is of first order, considering the monomer concentration.
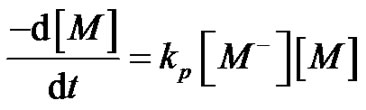 (4)
(4)
where 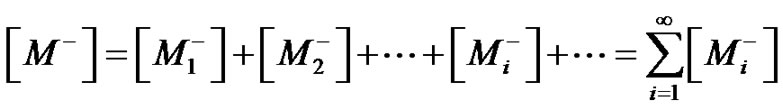
stands for the overall concentration of active species, whatever the chain length. Provided that an instantaneous initiation occurs, 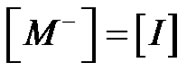 and
and 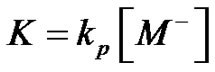 is a constant, so that we get
is a constant, so that we get . Therefore, Equation (1) can be rewritten under the following form (Equation (5)):
. Therefore, Equation (1) can be rewritten under the following form (Equation (5)):
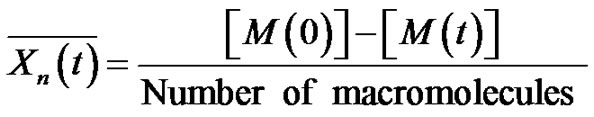 (5)
(5)
where the number of macromolecules is equal to the number of initiator. Consequently
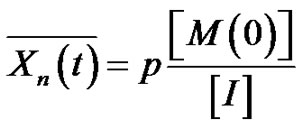 (6)
(6)
where
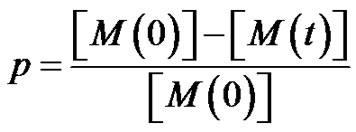
is the monomer conversion.
Although it may seem trivial, our own experiences showed that students have difficulties understanding the idea of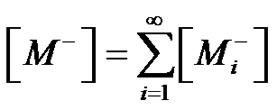 . On the other hand, they often confuse residual monomer
. On the other hand, they often confuse residual monomer  with reacted mono-
with reacted mono-
 (a)
(a)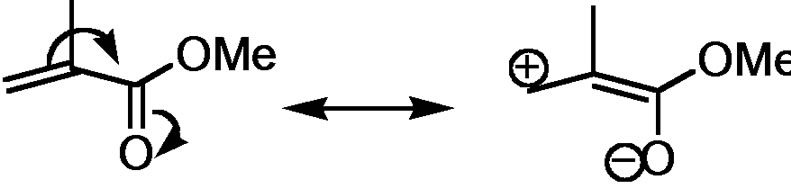 (b)
(b)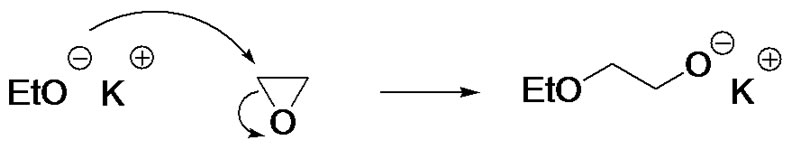 (c)
(c)
Figure 4. Some features about anionic polymerization. (a) 1,4- and 1,2 polymerization of butadiene; (b) mesomer effect with MMA; (c) ring opening polymerization of EO.
mer, that is .
.
The students are now familiar with the first concepts allowing understanding the building of macromolecules.
3.2.3. Distribution in Molecular Weights
The notion of distribution in molecular weights was already discussed. Its importance in the whole polymerization process involves recalling it. Providing an instantaneous initiation of all macromolecular chains, a narrow molecular weights distribution is expected (Poisson distribution) and can be demonstrated from kinetics. At this stage, students are familiar with the notion of binomial distribution (7) and it may be interesting to show how the assumptions made in the context of the anionic polymerization can go towards the Poisson distribution (8) [9]. On the other hand, the broadening of the molecular weight distribution (MWD) due to a non instantaneous initiation can also be specifically discussed in tutorials.
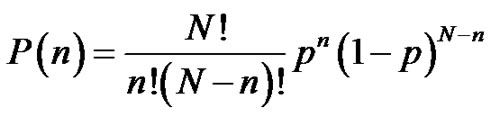 (7)
(7)
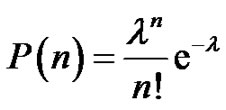 (8)
(8)
with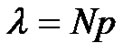 ,
, 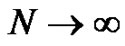 and
and 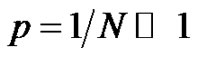
3.2.4. Concepts Learned and Possible Extensions
The students are now familiar with the first concepts allowing understanding the building of macromolecules. In the general development of the anionic polymerizationthe concept of living polymerization is introduced depending on the level of the attendees. It may be useful to stress that a narrow MWD is not a condition to prove the living character of the polymerization process since, even if the instantaneous initiation does not take place, the active centres are not destroyed. In this context, synthesis of block copolymers can also be introduced by outlining the reactivity of the active centres. Some interesting examples can be drawn from industrial cases to illustrate the synthesis of block copolymers and the structure-property relationships:
• thermoplastic elastomers based on polystyrene-b-polybutadiene copolymers and their thermomechanical properties;
• poly(ethylene oxide)-b-poly(propylene oxide) surfactants and the hydrophilic lipophilic balance dependence on chain structure.
In the next section, replacing the anion by a cation as the active centre enables to grasp cationic polymerization and to introduce another reaction, namely the transfer reaction, which frequently occurs in that case.
3.3. Cationic Polymerization
Some vinylic monomers and heterocycles can be polymerized with cationic initiators. Main differences with anionic polymerization ought to be highlighted. Vinylic polymers are thus of particular interest to deal with in the first instance.
3.3.1. Specific Features of Cationic Polymerization
Care must be taken when dealing with this polymerization process. The usual more reactive character of the cationic species can lead to a deactivation of the growing species through transfer reactions. Nevertheless, the discussion on these cationic species makes straightforward the introduction of the three kinds of reactions: namely nucleophilic substitution reactions (leading to propagation or termination), elimination reactions (leading to termination or transfer), and cationic transposition reactions. Two specific reactions are then taught:
• Initiation using Brönstedt acids can be easily captured by considering the addition reactions to the double bonds. The Markovnikov rule and the formation of the more substituted carbocation need to be reminded. The initiation step by Lewis acids and the contribution of a co-initiator can be more easily understood by referring to the Friedel-Crafts reaction.
• Rearrangement of carbocations to yield more stable active species reveals that the repeat unit in the polymer chain can be quite different from that expected from the monomer structure (Figure 5).
Finally, discussion on the influence of the counter-ion during the chain termination reveals the intimate link
 (a)
(a) (b)
(b)
Figure 5. Typical examples of a clear difference between structures of the monomer and the monomer unit: cationic polymerization of 3-methyl-1-butene and β-pinene
between chemical structure and reactivity: nucleophilicity decreases with the increasing size of the anion and/or by electronic withdrawing effects (see e.g., 
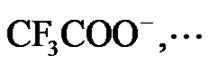 ).
).
At this point, it is worth emphasizing the fact that any transfer reaction stops a growing chain and simultaneously gives rise to another active centre. Consequently, a transfer reaction is not a polymer chain termination reaction.
3.3.2. Effect of Temperature
An important factor that was not hitherto considered, and which has great impact in the polymerization process, is the temperature. More specifically, its influence on the course of a process when facing competitive reactions is considered at this point. The principal feature that gives rise to difficulties of understanding among the students concerns the effect of the activation energy. In particular it is important to indicate that the activation energy of the transfer reaction is usually higher than that of the propagation reaction. Accordingly the ratio between propagation and transfer constants 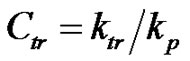 highly depends on the temperature. Moreover, discussion on the Arrhenius equation is useful: a decrease in temperature goes with a decrease in the overall polymerization rate, but also (and that has great consequence from a practical point of view) to an increase in the average molecular weight.
highly depends on the temperature. Moreover, discussion on the Arrhenius equation is useful: a decrease in temperature goes with a decrease in the overall polymerization rate, but also (and that has great consequence from a practical point of view) to an increase in the average molecular weight.
3.3.3. Cationic Ring Opening Polymerization
Apart from the fact that the reaction mechanism is of particular interest, cationic ring opening polymerization often gives rise to equilibrium between propagation and depropagation. Accordingly kinetics order of the reaction is not equal to 1. Besides THF, 1,3-dioxolane is another interesting example that can be used to discuss the carbon reactivity located in α of the oxonium ion. Students are also invited to discuss the reactivity of both carbon located in α of the oxonium ion.
3.3.4. Concepts Learned
At this stage, anionic and cationic polymerizations have been discussed. The students, now familiar with the ionic concepts of polymerization, possess sufficient knowledge to better comprehend radical polymerization and the differences induced by using radicals as the active centre.
3.4. Radical Polymerization
3.4.1. The Radical Polymerization: A Complex System
Aside from the catalytic olefin polymerization, the radical polymerization is probably the most difficult notion in polymer chemistry to conceptualize since it consists in a significant set of competitive steps that ultimately leads to the polymer. These elementary steps are so important that they dictate the final chemical structure and the chain length. Starting from the very simple instantaneous anionic scheme should clarify initiation reaction and propagation steps to the students.
Before beginning to explain the radical polymerisation, polymerization, it may be useful to recall what a radical is: conceptually the very notion of a radical is not always well understood by students, so that counting electrons and protons around a carbon or an oxygen atom in a chemical structure is not lost time.
More complex systems to grasp have to be introduced, namely non instantaneous initiation reactions, followed by propagation steps and bimolecular termination reactions. These later reactions currently occur since two radicals can react with each other (unlike ionic species). The very short lifetime of the macroradicals is probably the most important point that differentiates the radical polymerization from the ionic systems. As a consequence, the number of macromolecules at any time is quite different from the number of initiator molecules introduced in the reactor so that Equation (6) does hold no longer. In addition, some transfer reactions can occur, which complex more or less the overall reaction scheme.
It is important to clearly explain the basic steps and draw the kinetic equations relating thereto, as well as the quasi steady state hypothesis. It may be worthy to point out that the initiation reaction does not correspond to the decomposition of the initiator, but to the reaction of a primary radical coming from the initiator system with a first monomer.
3.4.2. A Too Restrictive Current Approach for the Average Degree of Polymerization
After dealing with the chemical schemes, the current approach consists in developing the kinetics protocol of radical polymerization. Equations are made easy to deal with since they are considered as first order (quasi stationary state hypothesis); consumption of the initiator is neglected. The next step is to define the kinetics of the chain formation resulting in variations of the chain length and the kinetic chain length λ defined as the number of reacting monomer molecules per active centre. Because of the very short lifetime of the growing chain, it is important to highlight the fact that the kinetics chain length refers to an instantaneous process, and thus cannot be used to determine the actual number-average degree of polymerization at the end of the polymerization. This actual degree of polymerization is in fact the conesquence of all these “instantaneous” processes occurring during the polymerization event. Although very useful in practice, it is rarely discussed and calculated.
Another educational approach to tackle the thorny problem of number instantaneous and average degrees of polymerization in radical polymerization is to start again with Equation (5), where the number of macromolecules depends closely to the initiation and the termination steps. This calculation of the number of macromolecules is then an opportunity to challenge the students about the building of the macromolecules according to a radical process.
It deserves also to note that Equation (5) is true, regardless of the reaction time and enables to get the actual number average degree of polymerization. This relation is also true in the case of a very short reaction time dt, equal to the lifetime of the macroradicals, resulting in 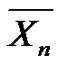 tending to the kinetic chain length
tending to the kinetic chain length . Consequently,
. Consequently, 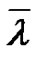 can also be seen as the limit of
can also be seen as the limit of 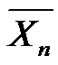 when t ® dt at any time.
when t ® dt at any time.
Through tutorials, it is therefore pedagogically very useful to compare and discuss the evolution of 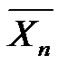 and
and 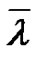 with time or conversion. Another point to consider is the rate of consumption of the initiator (do we have to take into account its actual consumption?) and the kinetic dependence on it
with time or conversion. Another point to consider is the rate of consumption of the initiator (do we have to take into account its actual consumption?) and the kinetic dependence on it
3.4.3. Extensions towards Macromolecular Engineering and Controlled Radical Polymerization Reaction
Unlike termination, a transfer reaction stops the growing chain and simultaneously generates another active centre which can be used to reinitiate another chain or take place in a termination process. The two reactions must thus be clearly distinguished. Examples are numerous to highlight their differences, such as the unwilling transfer to monomer or solvent, or the control of the molecular weight or telomerization using pertinent transfer agents. Another example of utmost importance in the field of macromolecular engineering can also be mentioned: the development of controlled radical polymerization using RAFT or ATRP. Despite the fact that controlled radical polymerization (CRP) is more difficult to comprehend in this introductory text on polymerization, it is worth introducing it by revealing analogies and differences between anionic and radical polymerization.
3.5. Chain Copolymerization
3.5.1. Copolymers and Mixtures of Homopolymers
Chain copolymerization can now be discussed. Firstly, it must be stressed to the students that copolymers are not mixtures of homopolymers. Properties of copolymers depend on both their chemical composition, and their microstructure. It becomes useful to illustrate those differences. For instance, copolymers based on butadiene and styrene are compared to mixtures of polybutadiene and polystyrene and glass transition temperatures can be discussed. It is also helpful for the students to write some sequences (for instance a tetrad or a pentad) from various comonomers. The purpose of such an exercise is for students to get acquainted with the linking of monomer units On the other hand, chain copolymerization is usually described only within the frame of radical copolymerization due to its industrial importance. It is worth mentioning that copolymerization processes can also occur in ionic systems as well, provided that active centres are able to react with each of the monomers.
3.5.2. Radical Chain Copolymerization
The actual central issue in dealing with copolymerization is the more or less important reactivity of the comonomers which can be evaluated by the radical reactivity ratios rA and rB, (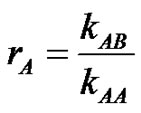 kAB being the rate constant of the macroradical A* with the monomer B).
kAB being the rate constant of the macroradical A* with the monomer B).
This results in a compositional heterogeneity due to the very short lifetime of the radicals, that causes a more or less pronounced composition drift between macromolecules Establish the well known copolymerization equation (Equation (9), 9’) can of course be an interesting exercise which incorporates the basic equations of radical polymerization, but it deserves to explain more in depth the consequences of the compositional drift on the materials properties (glass transition properties for instance).
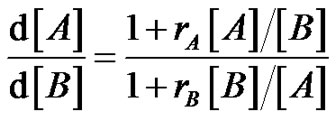 (9)
(9)
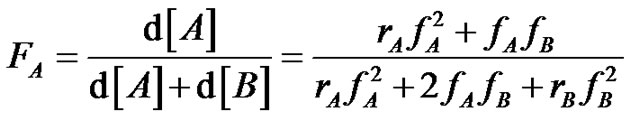 (9’)
(9’)
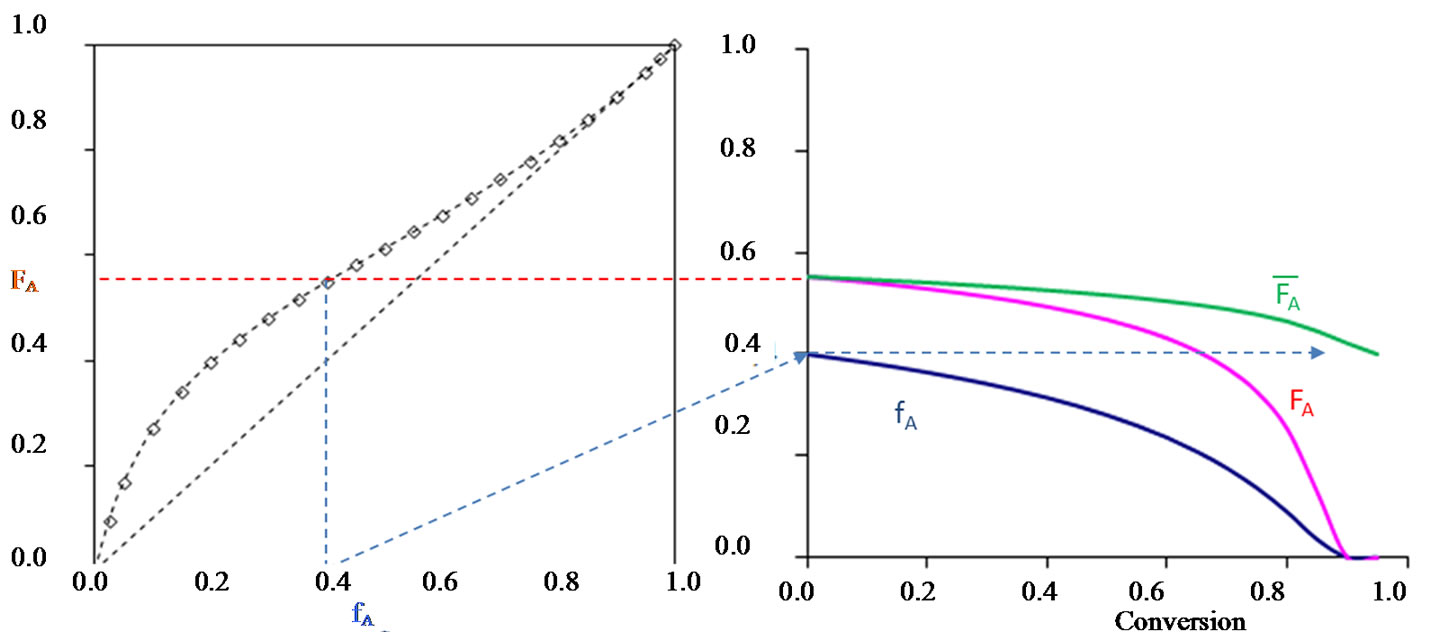
Explaining the relationship between the composition and the kinetics curves flawlessly exemplify this very significant point (Figure 6). These features can then be easily simulated in a computer, highlighting the crucial importance of the reactivity ratios.
3.5.3. Comments on Copolymerization
Some confusion amongst students can also arise between statistical and random copolymerizations; it is thus worth recalling their definitions:a statistical copolymer consists of macromolecules in which the sequential distribution of the monomer units obeys known statistical laws: they possess different reactivities, while, in a random copolymer, the probability of finding a specific monomer unit at any given site in the chain is independent of the nature of the adjacent units: they exhibit comparable reactivities.
3.5.4. Ionic Chain Copolymerization
As previously stressed, copolymerization processes can also occur in ionic systems as well, provided that active centres are able to react with each of the monomers (for instance ethylene oxide-propylene oxide or styrene-butadiene in the anionic case). In that case, provided there is no termination, the composition drift will occur all along the same macromolecular chain and not between the various macromolecules.
It may be useful to recall the differences between statistical and block copolymers. For instance, thermoplastic elastomers or polymer surfactants are quite interesting polymer materials in order to exemplify these notions.
To conclude this part, depending on the level of the class, a general overview of the various strategies to get copolymers with controlled architectures, going from block and graft copolymers to statistical structures, including gradient copolymers, can be discussed.
3.6. Coordination Polymerization
From a chemical point of view, olefin coordination polymerization is probably one of the most difficult notion to teach, and consequently to understand for students. For instance, teaching olefin catalysis polymerization involving the Ziegler-Natta catalysis starts with the CosseeArlman model, where the difficult notion of both the coordination mechanism and a heterogeneous process are introduced.
Notions stemming from the study of such a reaction help to explain the chemical process and its regiospecificity. More specifically, if we consider that the two elemental steps needed to understand this chemical process are the monomer coordination on a metal lacuna followed by its insertion into the metal-carbon bond, it becomes clear that any other homogeneous reaction can be used to exemplify this coordination-insertion mechanism
 (a) (b)
(a) (b)
Figure 6. Instantaneous copolymer composition and compositional drift during styrene (a)-butyl acrylate (b) copolymerization (rA = 0.04, rB = 0.22). Instantaneous copolymer composition and compositional drift during copolymerization. fA is the instantaneous molar fraction of styrene in feed (fA(0) = 0.4); FA and  are the instantaneous and the average molar fractions of the styrene units in the copolymer, respectively. At the end of the copolymerization, the average molar fraction
are the instantaneous and the average molar fractions of the styrene units in the copolymer, respectively. At the end of the copolymerization, the average molar fraction 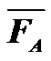 tends to fA(0) = 0.4.
tends to fA(0) = 0.4.
(Figure 7). For instance the ring opening polymerization (ROP) of ε-caprolactone from aluminium isopropoxide is an example of such reaction, even if not catalysed. These concepts can then be extended to olefin catalytic polymerization with Ziegler-Natta catalysis. The Cossee-Arlman model can be explained and its drawbacks discussed, to go to other reaction schemes models.
At this stage, our experiences showed that students have always some difficulties understanding the concept of catalysis. Comparing radical and coordination polymerisation processes of ethylene can be a versatile way to understand the interest of such catalysis.
The multi site mono-and bimetallic models can then be addressed in order to enlighten the chemical process and its regiospecificity. The metallocene catalysis will thus appear as a logical consequence and will be quite an interesting example to highlight the relationships between the catalyst structure and the polymer properties. Finally, metathesis polymerization of cyclopentene or norbornene can be presented.
4. Step Growth Polymerization
To introduce the step-growth polymerization, it is suggested that emphasis must be put on the main difference with the chain polymerization, namely the fact that no active centers are needed, only reactive groups are required: the role of a possible catalyst has to be distinguished from the initiator.
Any functional group can enter into reaction, regardless of the fact if it belongs to a residual monomer, an oligomer or a polymer. The average chain length increases continuously as the total number of species decreases. The well-known esterification reaction between diol and diacid deserves to be used as the working example. It actually characterizes the step-growth polymerizetion by revealing that it is based on the reaction of bifunctional molecules. Moreover, polymer industries provide a huge number of interesting examples. Depending on the level of the lecture, the relationships between molecular structures and physical properties can be investigated more in depth.
In addition, when dealing with equilibrated reactions such as esterification or amidification, it deserves to recall that due to the reverse hydrolysis reaction polymer chains can be cleaved anywhere. Great chain lengths cannot thus be obtained unless the equilibrium is shifted and water is removed.
Care must be taken that the conversion is defined with reference to reactive functions and not to monomers. The experience showed that students confuse both monomers and functions conversion. Monomers disappear quickly while there are still reactive functions remaining in the reaction medium.
4.1. Chemical Structures and Number Average Degree of Polymerization
Care must be taken in writing the chemical structures of repeat units (RU); some of them are not obvious and it is of great importance to specify the chain ends, as well as the relationship between the average number of repeat units and the number average degree of polymerization. Our suggestion is to introduce the molar mass of the
 (a)
(a)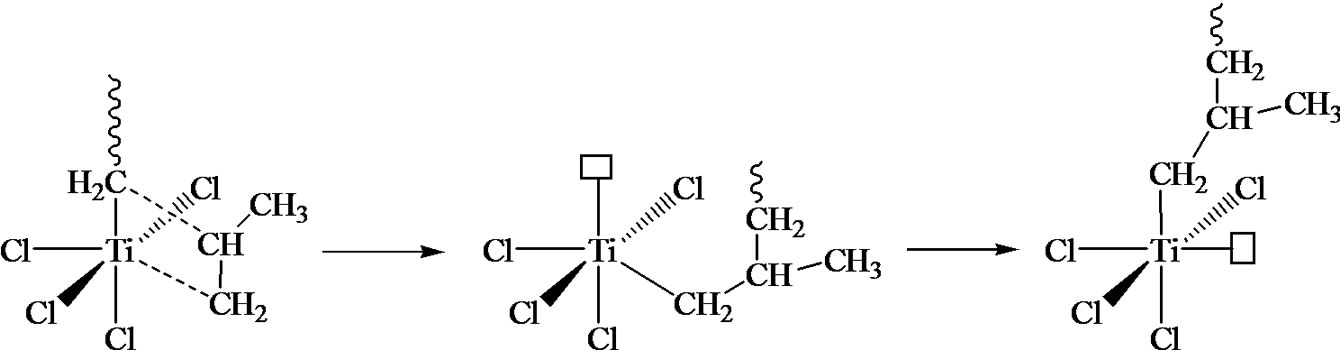 (b)
(b)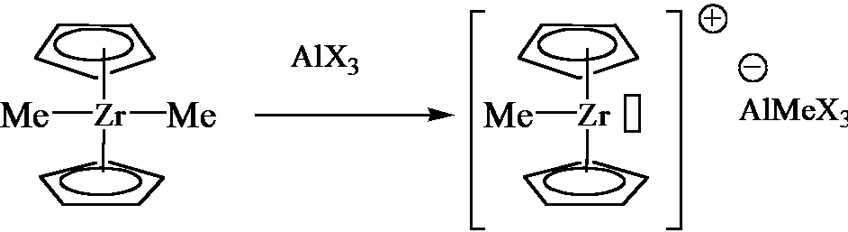 (c)
(c) (d)
(d)
Figure 7. Coordination polymerization: coordinated ring-opening polymerization of ε-caprolactone, Ziegler-Natta catalysis, and metallocene catalysis. The schemes are oversimplified and used only in order to illustrate the coordination-insertion concept.
repeating unit MRU and both extremities Mext respectively, and the average number of repeat units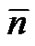 . Whatever the chain length, the number average molecular weight is given by.
. Whatever the chain length, the number average molecular weight is given by.
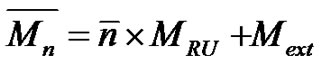 (10)
(10)
The relation between  and
and 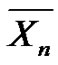 strictly depends on the chemical structure, as depicted in Table 1. For instance, in the case of PA-6,6 with two amino chain ends, we have
strictly depends on the chemical structure, as depicted in Table 1. For instance, in the case of PA-6,6 with two amino chain ends, we have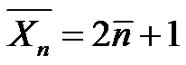 , and therefore:
, and therefore:
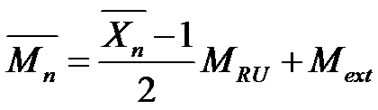 (11)
(11)
which tends to
 (12)
(12)
if the weight of the chain ends becomes negligible. The importance of these relationships must be highlighted when it is time to discuss about molar and mass balances between monomers and macromolecules, in particular when low molecular weights molecules, for instance water, are present in the system.
4.2. Determining the Average Degree of Polymerization
4.2.1. A general approach for linear macromolecules
Unlike chain polymerization, it is worth noting that all the starting monomers are considered in the counting of the average macromolecule length (the degree of polymerization of a monomer is 1).
The extent of polymerization and the conversion are defined with respect to the initial number of reactive groups and not to the number of starting monomers. Consequently, at any time t:
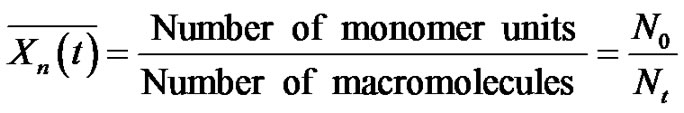 (13)
(13)
Knowing the initial number of monomers, the problem then lies in the counting of macromolecules at any time. Two cases then need to be discussed according to the functionality of the monomer unit.
In the first case, when dealing with bifunctional monomers, the number of chains will be half the number of chain ends, which corresponds to the number of non-yet reacted functions. Therefore, counting the non-yet reacted functions from the conversion enables to establish easily all the relationships, such as Equation (14) between the function conversion and the number average degree of polymerization whatever the conversion and the amount of reactants.
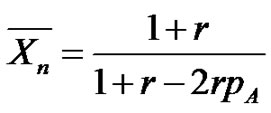 (14)
(14)
where 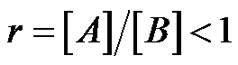 and pA is the conversion of functions A.
and pA is the conversion of functions A.
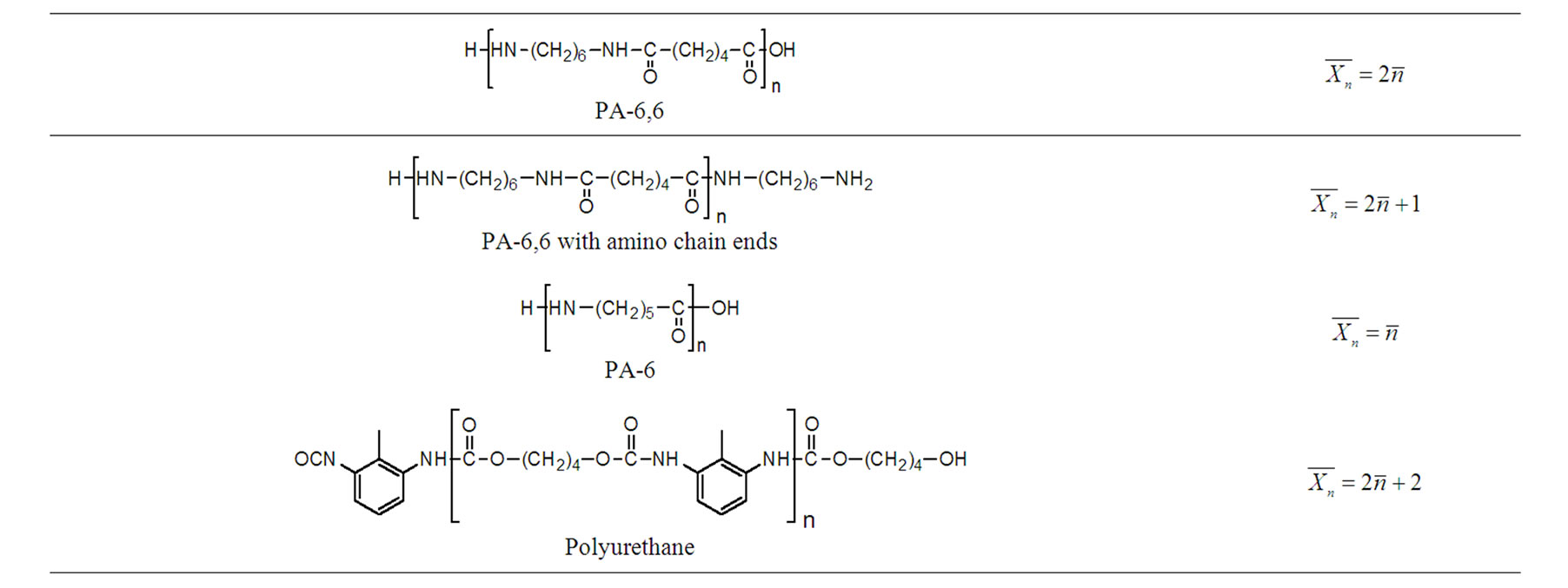
Table 1. Some relationships between the structure of the polymer and the degree of polymerization.
Monofunctional agents must be counted as a monomer unit since moieties issued from that blocking agent will be located at a chain end.
Depending on the level of the lecture, the molar and mass fractions (Equations (15) and (16)) can be introduced. Here, it is worthy of highlighting that p represents the probability that a function has reacted, i.e. the conversion, and not the probability of involvement of this function in the reaction. This latter point is highly misleading. Note that Equation (15) can also be used to get access to the amount of residual monomer when i = 1.
 (15)
(15)
 (16)
(16)
The gel point In the second case, calculation of  as described above can obviously no longer be used when implementing monomers with a functionality higher than 2 (for instance glycerol). In that case, polymerization leads first to branching and then to crosslinking, so that the number of chains will not be half the number of chain ends At this stage of the lecture, a “gel point” is usually computed according to the Carothers approach, i.e
as described above can obviously no longer be used when implementing monomers with a functionality higher than 2 (for instance glycerol). In that case, polymerization leads first to branching and then to crosslinking, so that the number of chains will not be half the number of chain ends At this stage of the lecture, a “gel point” is usually computed according to the Carothers approach, i.e , corresponding to an unique giant macromolecule and then
, corresponding to an unique giant macromolecule and then , where
, where  is the number average functionality.
is the number average functionality.
However, care must be taken that gels are weakly cross-linked systems, which are mostly liquid by weight. They behave like solids due to a three-dimensional crosslinked network within the liquid and exhibit no flow. Because of the average functionality higher than 2, giant highly ramified macromolecules are formed in the reaction medium, even if covalent gels are not yet been formed and the viscosity will become infinite. In that latter case,  then keeps a finite value, (there are many macromolecules) but, since the mass contribution of giant macromolecules become predominant,
then keeps a finite value, (there are many macromolecules) but, since the mass contribution of giant macromolecules become predominant,  (remember
(remember ) and defines the gel point [10].
) and defines the gel point [10].
5. Conclusions
Although about 50% of the polymer market is covered by standard plastics such as PE, PP, and PET that display very simple molecular structures, attractive chemical architectures are required to get specific properties.
Macromolecular chemistry covers all the fields of chemistry, starting from monomer and catalyst syntheses to the construction of polymer materials with very large domains of properties and applications. It offers a unique chance to highlight relationships between chemical structures, physicochemical properties (glass transition, melting temperature) and mechanical properties, and to link them to common objects of daily use.
This syllabus provides a new approach to teach polymer chemistry. It is based on the fact that the simplest polymerization mechanism is taught first. Despite this direct approach, complex structures are derived: complicated polymer structures combine various elementary pathways. For instance, the derivation of unsaturated polyester resins is described in several steps: the synthesis of polyester prepolymers from a diol and a mixture of maleic and phthalic anhydrides is first introduced. It actually shows how rigidity depends on the components. Finally the occurrence of crosslinking is explained through styrene radical copolymerization. On the other hand, some polycondensation or polyaddition reactions make use of prepolymers, such as polyethers (polyethylene glycol, polyTHF…), that have previously been designed by ionic polymerization. Many other examples can be drawn from technical sheets and scientific literature. Finally, sustainable development implies new constraints that are to be taken into account in order to reduce the waste and protect fragile environments, so that students must be aware of progresses of new green polymer chemistries.
6. Acknowledgements
S. Boileau, B. Charleux, H. Cramail, A. Fradet, C. Guillerme and M. Rinaudo are gratefully acknowledged for their critical reviews, as well as G. Pozza and Y. Guilbert for the SEC and Maldi Tof spectra.
REFERENCES
- GFP-Polymer French Association. http://www.gfp.asso.fr
- T. Hamaide, “Some Ideas about the Teaching of Macromolecular Chemistry,” L’Actualité Chimique, No. 319, 2008, pp. 15-24.
- H. Cramail, J. L. Six, L. Fontaine and L. T. Hamaide, “Glasgow,” IUPAC Macro, Glasgow, 2010.
- http://goldbook.iupac.org/
- S. Penczek and S. G. Moad, “Glossary of Terms Related to Kinetics, Thermodynamics, and Mechanisms of Polymerization,” Pure and Applied Chemistry, Vol. 80, No. 10, 2008, pp. 2163-2193. doi:10.1351/pac200880102163
- K. Horie, et al., “Definitions of Terms Relating to Reactions of Polymers and to Functional Polymeric Materials,” Pure and Applied Chemistry, Vol. 76, No. 4, 2004, pp. 889–906.
- “Glossary of Basic Terms in Polymer Science,” IUPAC Recommendations. http://goldbook.iupac.org/src_PAC1996682287.html
- M. M. Coleman and P. C. Painter, “Fundamentals of Polymer Science: An Introductory Text,” CRC Press LLC, Boca Raton, 1997.
- F. Reif, “Berkeley. Cours de Physique, Physique Statistique,” Armand Colin, Paris, Vol. 5, 1994, pp 350-356
- J. P. Pascault, H. Henry Sautereau, J. Verdu and R. J. J. J. Williams, “Thermosetting Polymers,” CRC Press, Boca Raton, 2002. doi:10.1201/9780203908402
NOTES
*Corresponding author.