Computational Chemistry
Vol.04 No.03(2016), Article ID:67963,10 pages
10.4236/cc.2016.43007
Determination of Band Structure of Gallium-Arsenide and Aluminium-Arsenide Using Density Functional Theory
J. A. Owolabi1, M. Y. Onimisi1, S. G. Abdu2, G. O. Olowomofe1
1Department of Physics, Nigerian Defence Academy, Kaduna, Nigeria
2Department of Physics, Kaduna State University, Kaduna, Nigeria

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 18 April 2016; accepted 2 July 2016; published 5 July 2016
ABSTRACT
This research paper is on Density Functional Theory (DFT) within Local Density Approximation. The calculation was performed using Fritz Haber Institute Ab-initio Molecular Simulations (FHI- AIMS) code based on numerical atomic-centered orbital basis sets. The electronic band structure, total density of state (DOS) and band gap energy were calculated for Gallium-Arsenide and Aluminium-Arsenide in diamond structures. The result of minimum total energy and computational time obtained from the experimental lattice constant 5.63 A for both Gallium Arsenide and Aluminium Arsenide is −114,915.7903 eV and 64.989 s, respectively. The electronic band structure analysis shows that Aluminium-Arsenide is an indirect band gap semiconductor while Gallium-Arsenide is a direct band gap semiconductor. The energy gap results obtained for GaAs is 0.37 eV and AlAs is 1.42 eV. The band gap in GaAs observed is very small when compared to AlAs. This indicates that GaAs can exhibit high transport property of the electron in the semiconductor which makes it suitable for optoelectronics devices while the wider band gap of AlAs indicates their potentials can be used in high temperature and strong electric fields device applications. The results reveal a good agreement within reasonable acceptable errors when com- pared with the theoretical and experimental values obtained in the work of Federico and Yin wang [1] [2] .
Keywords:
FHI-Aims, Local Density Approximation, Band Structure, Energy Band Gap, Density of State, Gallium Arsenide, and Aluminium Arsenide

1. Introduction
The understanding of the physical properties of interacting many body systems is one of the most important goals of physics after the foundation of quantum mechanics in the mid 1920’s (Rerum, 2005) [3] . However many computer simulation code has been employed in solving some challenges encountered in the calculation of the many body problem using DFT. One of the most immediate consequences of the periodic structure of crystalline solids is the arrangement of the electronic states within bands. This band has a particular importance for semiconductors as many properties of semiconductors are determined by only a small number of these bands (electronic band gap). The electronic band gap of a material may be defined as the difference between the electron affinity (the energy of adding an electron to the system) and the first ionization energy (the energy of the removing an electron from the system). The two can be calculated using conventional DFT which often leads to delta SCF method for band gap calculation. However it is only directly applicable to finite systems, for extended solids. It would be necessary to calculate the effect of addition or removal of a single electron from the infinite total.
Perdew shows that when DFT is extended to fractional occupation number, the exchange-correlation to fractional occupation number of the electron count is discontinuous at the Fermi level. This fundamental discontinuity in the exact Vxc is precisely the difference between the kohn-sham and true band gaps, but is not reproduced in LDA or GGA. Another major contribution to the band-gap error arises from the electrostatic electron-electron contribution to the Hamiltonian which is usually computed as the Hartree energy EH. Although the equation includes the correct coulomb repulsion, by using the total density, it has included a coulomb repulsion between an electron and its own charge. This spurious self-interaction is exactly cancelled by the exchange term in some non-DFT methods but is only partially cancelled by LDA (or GGA) exchange. This residual self-interaction is one of the most significant causes of the under estimation of the band gap in LDA or GGA based DFT calculations. [4] .
In Lichteinstein work, we observed the introduction of Hubbard U to correcting the self-interaction as LDA + U or DFT + U method; this phenomenon introduces a repulsion between the localized electrons on a given atom [5] . However this repulsion can cause a breaking of symmetry and lead to the operating of an insulating gap. Computationally it is a low-cost method for open-shell systems that can also be used for self-consistency in structural relaxations. The inclusion of the Hubbard-U term does not fix the self -interaction problem itself, but it can correct for the resultant under localization of particles and incorrect magnetic structure. Many works regards the strength of the Hubbard-U term as a parameter of the simulation and DFT + U has often been used incorrectly as a method for widening the band gap for general semiconductors. Empirically fixing the band gap for closed-shell system usually requires an un-physically large Hubbard U, causing over-localization and a flattening of the valence and conduction bands. Therefore DFT + U calculation are semi-empirical rather than ab-initio in nature. However cococcioni and dc curoncoli showed that the correct value of the Hubbard-U terms could in fact be computed using Density Functional Perturbation Theory. This technique removes the empiricism and ensures that the values are physically reasonable, though it is not straight forward to apply to material with many different elements [6] .
FHI-aims code solves the DFT + U issues by treating all electrons in an equivalent way. In some special cases of different element, frozen core treatment are applied where one compute the correlation energy of only the valence but not the core electron in second order Moller-Plessett (MP2) perturbation theory and for any two-elec- tron coulomb operator (hybrid functional, Hatree-forck, MP2, or RPA perturbation theory, GW correction etc.) therefore auxillary basis is used to expand the coulomb matrix (four basis functions ≡O(N4) matrix elements) into a two-center coulomb matrix, leading instead to O(N3) additional overlap matrix elements which offers an ad-hoc correction for strongly correlated systems at negligible computational cost.
FHI-aim also allows fixing the mixing factor between Around Mean Field (AMF) and Fully-localized limits (FLL). These two common schemes deal comfortably with the double counting problem in DFT + U. The AMF method assumes that the effect of the DFT + U term on the actual occupations remains small, so that the occupation can be assumed to be equal with each shell for the purpose of the double counting corrections while the FLL method assumes a maximal effect of the DFT + U term on the occupation numbers, handling double counting correctly in the case that all orbitals within the shell are either fully occupied or empty. This improves the handling of intermediate range in self-consistent mixing of both limits [7] . Therefore, Density functional theory has become the workhorse in electronic band structure calculations and FHI-aims computer code has been used in this work to address this Hartree-fork many particle problem, with more efficiency and accuracy by implementing all-electron density based on numeric atomic-centered orbital basis sets which allow the actual predictions for real materials using a computer program code. The code answers many possible general purpose electronic structure problems by varying the treatment of exchange correlation in Local density Approximation (LDA). This invokes the LDA as exchange correlation functional given by slater and the correlation energy of the electron given by Perdew and Wang [8] . Its success rate has helped in producing accurate results with reasonable computational effort.
This work therefore attempt to calculate and estimate the electronic energy band structures and density of state of Gallium-Arsenide and Aluminum-Arsenide semiconductor from ground state density using DFT computational code FHI-aims. The experimental lattice constant parameter values was used to calculate the minimum total energy and tested for different k-grids in order to determine the minimum time required for energy stability of the semiconductor in diamond structure.
2. Theoretical Background
The first principles of H-K theorem demonstrates that the ground state properties of a many-electron system are uniquely determined by an electron density that depends on only three spatial co-ordinates which reduces our problem to 3 spatial co-ordinates from 3 N spatial co-ordinates for N body problem because of the use of density functional. The N particle system of interacting particles with 3 N degrees of freedom is reduced to a significantly more tractable problem, which deals with a function (density) of only three variables. The many-body effects incorporated in the exchange-correlation potential are typically approximated within either the local density approximation or the generalized gradient approximation. The formulation applies to any system of interacting particles in an external potential 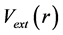 including any problem of electrons and fixed nuclei, where the Hamiltonian can be written as
including any problem of electrons and fixed nuclei, where the Hamiltonian can be written as
 (1)
(1)
The first term in this equation corresponds to the kinetic energy of the interacting electrons, the second term is the external potential acting on the electrons due to the ions, the third term is the electron Coulomb interaction, and the last term is the interaction energy of the nuclei. Since the Hamiltonian is thus fully determined (except for a constant shift of the energy), it follows that all properties of the system can be found given only the ground
state density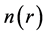 . This result allowed Hohenberg and Kohn to prove the existence of an energy functional of the density
. This result allowed Hohenberg and Kohn to prove the existence of an energy functional of the density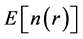 , which assumes its minimum value for the correct ground state density.
, which assumes its minimum value for the correct ground state density.
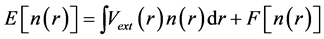 (2)
(2)
The minimization of energy functional 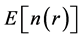 with respect to the charge density with the constraint of fixed number of electrons gives the ground state energy and the ground state charge density from which all other physical properties can be extracted. However in spite of the universality of
with respect to the charge density with the constraint of fixed number of electrons gives the ground state energy and the ground state charge density from which all other physical properties can be extracted. However in spite of the universality of 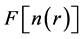 no explicit expressions for this functional are known to date. In 1965, Kohn and Sham readdressed the problem of minimizing the Hohenberg-Kohn density functional (Equation (2)) directly with an improved strategy that maps the original interacting problem into an auxiliary non-interacting one [9] . This is achieved by expressing the charge density
no explicit expressions for this functional are known to date. In 1965, Kohn and Sham readdressed the problem of minimizing the Hohenberg-Kohn density functional (Equation (2)) directly with an improved strategy that maps the original interacting problem into an auxiliary non-interacting one [9] . This is achieved by expressing the charge density  as
as
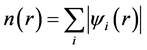 (3)
(3)
where ψi’s are the single-particle wavefunctions for the non-interacting electron gas with ground state charge density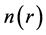 , and the sum is over all occupied singleparticle states.
, and the sum is over all occupied singleparticle states.
The 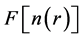 functional is now expressed as
functional is now expressed as
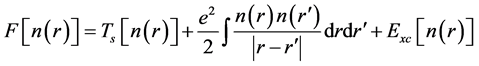 (4)
(4)
where the first term corresponds to the kinetic energy of a non-interacting electron gas at the same density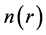 , the second term is the classical Coulomb interaction energy (the Hartree term), and the last term
, the second term is the classical Coulomb interaction energy (the Hartree term), and the last term 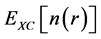 represents the quantum mechanical exchange-correlation energy. This term accounts for the differences between the non-interacting fictitious system and the real interacting one, collecting the contributions from the non-classic electrostatic interaction and the differences in their corresponding kinetic energies. The success of the Kohn-Sham approach ultimately lies in the fact that
represents the quantum mechanical exchange-correlation energy. This term accounts for the differences between the non-interacting fictitious system and the real interacting one, collecting the contributions from the non-classic electrostatic interaction and the differences in their corresponding kinetic energies. The success of the Kohn-Sham approach ultimately lies in the fact that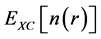 , which contains the many-body contributions, is a small fraction of the total energy, and although not known exactly, it can be approximated surprisingly well. The approximation is at present the strength and the limitation of DFT, providing efficient yet not exact reformulation of the quantum mechanical problem, respectively. If the energy functional defined in Equation (2) and Equation (4) is now varied with respect to the wave-functions ψi’s subject to the orthonormality constraint, the following set of Schrodinger equations is obtained
, which contains the many-body contributions, is a small fraction of the total energy, and although not known exactly, it can be approximated surprisingly well. The approximation is at present the strength and the limitation of DFT, providing efficient yet not exact reformulation of the quantum mechanical problem, respectively. If the energy functional defined in Equation (2) and Equation (4) is now varied with respect to the wave-functions ψi’s subject to the orthonormality constraint, the following set of Schrodinger equations is obtained
 (5)
(5)
where the effective potential  is given as
is given as
 (6)
(6)
Equations ((5) and (6)) are called the Kohn-Sham equations and have to be solved self-consistently because of the dependence of  on
on . It should be emphasized here that the Kohn-Sham procedure introduces a one-body Hamiltonian representing a single-particle electron in the mean field created by the nuclei and by all other electrons. However, it assigns no formal interpretation to the calculated orbitals and the eigenvalues. In principle, the solution of the Kohn-Sham equations would yield the exact ground state energy of the interacting electron gas problem. However, the exact exchange-correlation functional
. It should be emphasized here that the Kohn-Sham procedure introduces a one-body Hamiltonian representing a single-particle electron in the mean field created by the nuclei and by all other electrons. However, it assigns no formal interpretation to the calculated orbitals and the eigenvalues. In principle, the solution of the Kohn-Sham equations would yield the exact ground state energy of the interacting electron gas problem. However, the exact exchange-correlation functional  for an inhomogeneous interacting electron gas is not known for general
for an inhomogeneous interacting electron gas is not known for general . To proceed further, approximations to this functional are required. The most common and extensively tested approximation is the local density approximation (LDA), in which
. To proceed further, approximations to this functional are required. The most common and extensively tested approximation is the local density approximation (LDA), in which  for the inhomogeneous system is constructed from a parameterized form of the exchange-correlation energy density of the homogeneous electron gas
for the inhomogeneous system is constructed from a parameterized form of the exchange-correlation energy density of the homogeneous electron gas  [9] .
[9] .
 (7)
(7)
and
 (8)
(8)
with . These varies the treatment of exchange correlation (LDA) to Kohn-Sham DFT given by Perdew and Wang [10] .
. These varies the treatment of exchange correlation (LDA) to Kohn-Sham DFT given by Perdew and Wang [10] .
3. Methodology
FHI-aims (“Fritz Haber Institute ab-initio molecular simulations”) Code [11] is used in the analysis of this work. It is a computer program package for computational materials science based on quantum-mechanical first principles. The main production method is density functional theory (DFT) to compute the total energy and derive quantities of solid condensed matter in its electronic ground state. This allows the description of electronic single-quasi-particle excitations in molecules using different self-energy formalisms, and wave-function based on molecular total energy calculation of Hartree-Fock and many-body perturbation theory.
The focus of this work is to use DFT to estimate the local density approximations (LDA) in exchange-corre- lation potential of Ceperley and Alder work as parametrized by Perdew and Zunger [12] and an extension to Hartree-Fock theory, electron self-energies for total energies and excited state. The construction of transferable, hierarchical basis sets is demonstrated by allowing the calculation to range from qualitative tight-binding like accuracy to total energy convergence with the basis set. A scalar-relativistic treatment that has different version of approximation which can yield considerably different total energies for different systems is also included in the approximation. FHI-aims offers two reliable, effective one-component scalar relativistic treatment (by “effective one component” we mean that no coupling exist between the two spin channels of the calculation with collinear spin) atomic zero-order regular approximation (ZORA) and scalar ZORA. The basis sets provide access to all elements from light to heavy. Both low-communication parallelization of all real-space grid based algorithms and a ScaLapack-based customized handling of the linear algebra for all matrix operations becomes possible thereby guaranteeing efficient scaling (CPU time and memory) [13] .
To compute the band structure of GaAs, and AlAs, we first calculate the ground state total energies of the most stable structure of these semiconductors as a function of their lattice constants [14] [15] .
All calculations were carried out using FHI-aims code upgrade 6 (released on 17th July, 2011; version 071711_6). It works on any Linux based operating system. Computations can only be carried out after building an executable binary file since the FHI-aims package is distributed in a source code form.
A working Linux-based operating system (Ubuntu 14.04LTS is used in this case), A working FORTRAN 95 compiler, in this case we use intel’s ifort compiler (specifically Composerxe 2013_sp1.3.174) was installed and used for computation in this work. A compiled version of lapack library, and a library providing optimized linear algebra subroutines (BLAS). Standard libraries such as Intel’s mkl or IBM’s essl provide both lapack and BLAS support. Intel’s Composerxe 2013_sp1.3.174 comes with mkl. All necessary adjustment were made for building the executable binary file for running the code and the executable program was successfully built.
The FHI-aims requires two input files: the control.in which contains all runtime-specific information and the geometry.in which contains information directly related to the atomic structure for a given calculation. The two input files must be placed in the same directory from where the FHI-aims binary file is invoked at the terminal.
Our first step towards studying periodic systems with FHI-aims is to construct periodic geometries in the FHI-aims geometry input format (geometry.in). Next, we set basic parameters in control.in for periodic calculations. Finally, we compare total energies of different GaAs, and AlAs bulk geometries.
Geometry.in files for the GaAs and AlAs structures were constructed varying the lattice constants around the experimental lattice constants a of 5.63 Å for GaAs and 5.63 Å for AlAs. At each lattice constant, if the symmetry of the system allows the ions to move, a separate geometric optimization must be performed. The form of total energy as a function of lattice constant is asymmetric and is well described by Murnaghan’s equation [15] - [17] .
In setting up the geometry.in file of a periodic structure in FHI-aims, the lattice vectors of the two semiconductors as well as their atomic positions in the unit cell are specified.
The electronic band structure of the stable phases of the semiconductor were then calculated along the high symmetry lines of the Brillouin zone by computing the control.in settings to calculate the band-structures and density of state. This was done when the control.in input files for GaAs and AlAs were created with the appropriate settings.
Thus the calculation is performed as follows:
# Geometry for GaAs
lattice_vector 0.00000000 2.82500000 2.82500000
lattice_vector 2.82500000 0.00000000 2.82500000
lattice_vector 2.82500000 2.82500000 0.00000000
atom_frac 0.00000000 0.00000000 0.00000000 Ga
atom_frac 0.25000000 0.25000000 0.25000000 As
# Geometry for AlAs
lattice_vector 0.00000000 2.83000000 2.83000000
lattice_vector 2.83000000 0.00000000 2.83000000
lattice_vector 2.83000000 2.83000000 0.00000000
atom_frac 0.00000000 0.00000000 0.00000000 Al
atom_frac 0.25000000 0.25000000 0.25000000 As
while the control.in input files for the band structure of GaAs and AlAs were created with the following settings
control.in for GaAs
# Physical model
xc pw-lda
spin none
relativistic atomic_zora scalar
# SCF convergence
sc_accuracy_rho 1E-5
sc_accuracy_eev 1E−3
sc_accuracy_etot 1E−6
sc_iter_limit 100
# k-grid
k_grid 12 12 12
# Density of states
output dos -18 0 1000 0.1
dos_kgrid_factors 5 5 5
# High-symmetry k-points for diamond bandstructure output
output band 0.5 0.5 0.5 0.0 0.0 0.0 50 L Gamma
output band 0.0 0.0 0.0 0.0 0.5 0.5 50 Gamma X
output band 0.0 0.5 0.5 0.25 0.5 0.75 50 X W
output band 0.25 0.5 0.75 0.375 0.375 0.75 50 W K
control.in for AlAs
# Physical model
xc pw-lda
spin none
relativistic atomic_zora scalar
# SCF convergence
sc_accuracy_rho 1E−5
sc_accuracy_eev 1E−3
sc_accuracy_etot 1E−6
sc_iter_limit 100
# k-grid
k_grid 12 12 12
# Density of states
output dos -18 0 1000 0.1
dos_kgrid_factors 5 5 5
# High-symmetry k-points for diamond bandstructure output
output band 0.5 0.5 0.5 0.0 0.0 0.0 50 L Gamma
output band 0.0 0.0 0.0 0.0 0.5 0.5 50 Gamma X
output band 0.0 0.5 0.5 0.25 0.5 0.75 50 X W
output band 0.25 0.5 0.75 0.375 0.375 0.75 50 W K
4. Results and Discussion
The band structures of GaAs and AlAs were calculated and the aimsplot.py was used to plot the band structures. The position of the Fermi level in the band structure of these crystals is shown by the zero on the energy scale and that of symmetry points are indicated by vertical lines on the band graph in Figure 1 and Figure 2. In all the two cases, there is an important characteristic of the band structure, namely the range of energies where there are no electronic states across the entire Brillouin zone; this is the band gap. We notice that the band below zero shows that all the bands are fully occupied since there are eight valence electrons in each of these solid. The Fermi level within the band gap shows that all state below it remain occupied and all state above remain unoccupied. This is the hallmark of metallic behaviors that is, the availability of state immediately below and immediately above the Fermi level, which makes it possible to excite electrons thermally.
Figure 1 shows the GaAs band structure and the bottom valence band indicate an s-like atom character which is localized on an anion splits off from the rest of the valence band antisymmetry gap. This gap grows with increasing ionicity or charge transfer. A smaller gap arises from the antisymmetric part of the potential and is located between the first and second conduction band along the  direction. The smaller gap is important for transport properties in zinc-blende semiconductors and has been observed to have subtle effect on the reflectivity spectrum of zinc-blendes [18] . We also notice that the band gap in GaAs is narrow which shows that the semiconductor becomes more ionic. This effect arises from dehybridization accompanying the change in bonding from covalent to ionic [18] . The GaAs has three electrons in the Ga orbital and five electrons in the As orbital which makes the state near the valence band maximum have p bonding character which are associated with
direction. The smaller gap is important for transport properties in zinc-blende semiconductors and has been observed to have subtle effect on the reflectivity spectrum of zinc-blendes [18] . We also notice that the band gap in GaAs is narrow which shows that the semiconductor becomes more ionic. This effect arises from dehybridization accompanying the change in bonding from covalent to ionic [18] . The GaAs has three electrons in the Ga orbital and five electrons in the As orbital which makes the state near the valence band maximum have p bonding character which are associated with

Figure 1. Band structure of gallium-arsenide.
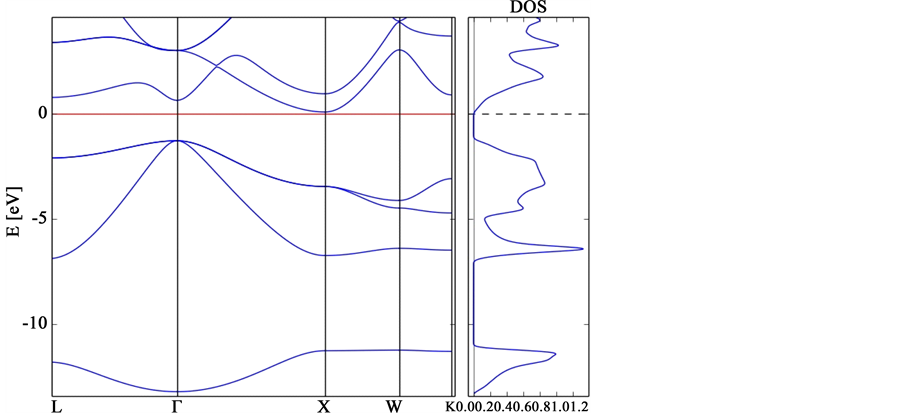
Figure 2. Band structure of aluminium-arsenide and density of state.
more electronegative element in the solid, while those near the conduction band minimum have p anti-bonding character and are associated with less electronegativity. The energy band gap shown in Table 1 is seen as the difference between the conduction band and the valence band which is calculated to be 0.38 eV.
Figure 1 also shows the Density of state of GaAs as a diamond structure semiconductor; we divide the density of state into three general regions while analyzing. The first region is the most tightly bound energy band where electron states corresponding to this band are strongly localized on the anion and are descendants of the atomic As in 4 s state.
The next region of noticed is a peak arising from the onset of the second valence band which shows there is no energy variation along the symmetry direction; in fact, it is very flat over the entire square face of the Brillouin zone. The energy band configuration results in a sharp onset of states above the antisymmetric gap. The character of state associated with the second valence band changes from predominantly cation s-like states at the bond edge to predominantly anion p-like state at the band maximum. The third region of interest in the density of state extends from the onset of the third valence band (at about 4 eV below the valence band maximum) to the valence band maximum. This region encompasses the top two valence bands and is predominantly p-like and is associated with anion state.
Showing the values of Figure 2 in Table 2, we notice that Aluminium-Arsenide (AlAs) Fermi energy is 0.093005 eV and the highest occupied state (valence band maximum) is −1.37872 eV at  (symmetry point) while the lowest unoccupied state ( conduction band minimum) is 0.04475 eV at X-point. Since the valence band maximum and the conduction bandminimum are on different symmetry point it shows that AlAs is an indirect band gap semiconductor with an energy gap value of 1.4235 eV and a large band width of 13.272 eV, the effect of the wide band width was observed to be stronger at the
(symmetry point) while the lowest unoccupied state ( conduction band minimum) is 0.04475 eV at X-point. Since the valence band maximum and the conduction bandminimum are on different symmetry point it shows that AlAs is an indirect band gap semiconductor with an energy gap value of 1.4235 eV and a large band width of 13.272 eV, the effect of the wide band width was observed to be stronger at the  and it grows into X, W and K symmetries without forming any hybridization. The three bands positioned below the Fermi level are due to 3s-like and 3p-like electrons of Al and 4p-like electrons of As. The empty conduction bands above the Fermi level are due to 3p-like states of Al and 4p-like states of As. The calculated energy gaps are in agreement with the theoretical values of AlAs. Of Federico and Yin wang [1] [2] . AlAs is more ionic and due to its increasing ionic character, have larger optical band gap and more charges transfer. These also makes it share metallic and covalent properties. The large band gap indicates the ability of AlAs for higher photon energy in reflectivity measurements and it is often utilized in applications in which higher temperature operation is required. They act as a promising candidate in semiconductor technology due to its wide and indirect bandgap [19] . This also makes it an active material in the manufacture of optoelectronic devices and also in LEDs etc. It has also gained importance in the technology applications of short wave length range of higher power and high frequency electronic devices. In particular AlAs is a basic material for light emitting diodes, lasers in the blue and ultraviolet range of the spectrum, optical pumping structures, photo detectors and hetero structure [20] .
and it grows into X, W and K symmetries without forming any hybridization. The three bands positioned below the Fermi level are due to 3s-like and 3p-like electrons of Al and 4p-like electrons of As. The empty conduction bands above the Fermi level are due to 3p-like states of Al and 4p-like states of As. The calculated energy gaps are in agreement with the theoretical values of AlAs. Of Federico and Yin wang [1] [2] . AlAs is more ionic and due to its increasing ionic character, have larger optical band gap and more charges transfer. These also makes it share metallic and covalent properties. The large band gap indicates the ability of AlAs for higher photon energy in reflectivity measurements and it is often utilized in applications in which higher temperature operation is required. They act as a promising candidate in semiconductor technology due to its wide and indirect bandgap [19] . This also makes it an active material in the manufacture of optoelectronic devices and also in LEDs etc. It has also gained importance in the technology applications of short wave length range of higher power and high frequency electronic devices. In particular AlAs is a basic material for light emitting diodes, lasers in the blue and ultraviolet range of the spectrum, optical pumping structures, photo detectors and hetero structure [20] .

Table 1. Band structure symmetry point for gallium-arsenide (GaAs).
Table 2. Band structure symmetry point for aluminium-arsenide (AlAs).
Table 3. Summary for Band gap of GaAs, and AlAs crystal structure calculated at the experimental value of the lattice constant 5.4Å for Si, 5.6Å for GaAs and 5.6Å for AlAs.
Figure 2 also shows the Density of state of AlAs which shows similar characteristics with the GaAs only that the short spikes near the Fermi energy are due to s-like and p-like electrons of As and p-like electron while the short peaks above its Fermi energy are due to the 3p, states of Al and 4p states of As. The third region is observed to encompass the top two valence bands and is predominantly p-like. The general features of the band structure and density of states are similar to that of the alkali halides [21] which is associated with anion state.
Table 3 provides the summary comparison of LDA and Experimental values for, GaAs and AlAs. It is a fact that DFT within the local density approximation (LDA) correctly predicts the existence of a band gap of semiconductors. In this table, LDA revealed a good approximation when calculating total energies and densities of bulk semiconductors. This improvements on LDA have demonstrated a very little change in the KS energy gap. The exchange correlation amount represents a significant part in the correct estimation of the LDA energy gap of the whole LDA error for the semiconductors.
However, the under estimation of the band gap from the experimental is mainly due to the fact that the exact functional in the Hohenberg-Kohn theorem is not known. Therefore the comparison of FHI-AIMS approximations for the exchange correlation predicts accurately the band gap when compared to other theoretical values [1] [2] .
5. Summary and Conclusions
This work has successfully employed Density Functional Theory method to calculate and estimate the band structure of Gallium-Arsenide (GaAs) and Aluminium-Arsenide (AlAs) using FHI-AIMS which was successfully installed and the knowledge of the input parameters which include the geometry.in and the control.in was carefully optimized for the band structure studies.
All calculations were carried out using FHI-aims code upgrade 6 (released on 17th July, 2011; version 071711_6) which works on Linux based operating system. In the calculation, Local Density Approximation (LDA) has been used to approximate the exchange correlation energy which varied the treatment of exchange correlation (LDA) to Kohn-Sham DFT leaving all other settings constant.
The experimentally lattice constant parameter value was used to calculate the minimum total energy and tested for different k-grids. The minimum total energy obtained from the experimental lattice constant of Gallium Arsenide 5.63 A and Aluminium Arsenide 5.63 A results in −114915.7903 eV for GaAs and AlAs with a computational time of 64.989 s, for the semiconductors. The result obtained shows that 12 × 12 × 12 k-grids enables the energy stability of the semiconductors in diamond structure with less computational time.
The calculated electronic band structure results shows that AlAs is an indirect band gap semiconductor with 1.42 eV while GaAs is a direct semiconductor with energy band gap of 0.37 eV. This shows significant improvement compared to other theoretical calculation obtained.
The calculated band width in this work shows large values of 13.00 eV and 13.27 eV for GaAs and AlAs and there is no interaction between the s and p orbital of GaAs and AlAs to form hybridization this gives the semiconductor an improved ionic characteristic nature.
The DOS energy level within the semiconductors shows considerable high state of electron occupation and the DOS observed around the Fermi level for the semiconductors at zero level indicate that they have conducting properties.
In general, FHI-AIMS code has shown better accuracy and prediction of band structure calculation within a reasonable computational time when compared to some other DFT theoretical programs observed in literature.
Cite this paper
J. A. Owolabi,M. Y. Onimisi,S. G. Abdu,G. O. Olowomofe, (2016) Determination of Band Structure of Gallium-Arsenide and Aluminium-Arsenide Using Density Functional Theory. Computational Chemistry,04,73-82. doi: 10.4236/cc.2016.43007
References
- 1. Iori, F. (2007) Engineering Silicon Nanostructure: Theoretical Study of the Effects of Doping with Boron and Phosphorus.
- 2. Yin, W., Ferdows, Z., Yu, Z., Lei, L., Jian, W. and Hong, G. (2013) Band Offset of GaAs/AlxGa(1?x)As Heterojunctions from Atomistic First Principles.�
- 3. Naturalium, R. (2005) Fermi Surface Calculations of Superconducting Compounds.
- 4. Perdew, J.P., Parr, R.G., Levy, M. and Balduz, J.L. (1982) Density-Functional Theory for Fractional Particle Number: Derivative Discontinuities of the Energy. Physical Review Letters, 49, 1691-1694. http://dx.doi.org/10.1103/PhysRevLett.49.1691
- 5. Lichtenstein, A.I., Anisimov, V.I. and Zaanen, J. (1995) Density-Functional Theory and Strong Interactions: Orbital Ordering in Mott-Hubbard Insulators. Physical Review B, 52, 5467. http://dx.doi.org/10.1103/physrevb.52.r5467
- 6. Cococcioni, M. and de Gironcoli, S. (2005) Linear Response Approach to the Calculation of the Effective Interaction Parameters in the LDA + U Method. Physical Review B, 71, 035105. http://dx.doi.org/10.1103/PhysRevB.71.035105
- 7. Petukhov, A.G., Mazin, I.I., Chioncel, L. and Lichtenstein, A.I. (2003) Correlated Metals and the LDA+U Method. Physical Review B, 67, 153106. http://dx.doi.org/10.1103/PhysRevB.67.153106
- 8. Perdew, J.P. and Wang, Y. (1992) Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Physical Review B, 45, 13244-13249. http://dx.doi.org/10.1103/PhysRevB.45.13244
- 9. Kohn, W. and Sham, L.J. (1965) Self-Consistent Equations including Exchange and Correlation Effects. Physical Review, 140, A1133-A1138. http://dx.doi.org/10.1103/PhysRev.140.A1133
- 10. Parr, R.G. and Yang, W. (1994) Density-Functional Theory of Atoms and Molecules. Oxford University Press, Oxford.
- 11. Blum, V., Gehrke, R., Hanke, F., Havu, P., Havu, V., Ren, X., Reuter, K. and Scheffler, M. (2009) Ab Initio Molecular Simulations with Numeric Atom-Centered Orbitals. Computer Physics Communications, 180, 2175-2196. http://dx.doi.org/10.1016/j.cpc.2009.06.022
- 12. Perdew, J.P. and Zunger, A. (1981) Self-Interaction Correction to Density-Functional Approximations for Many-Elec- tron Systems. Physical Review B, 23, 5048-5079. http://dx.doi.org/10.1103/PhysRevB.23.5048
- 13. Havu, V., Blum, V., Havu, P. and Scheffler, M. (2009) Efficient O(N) Integration for All-Electron Electronic Structure Calculation Using Numeric Basis Functions. Journal of Computational Physics, 228, 8367-8379. http://dx.doi.org/10.1016/j.jcp.2009.08.008
- 14. Hashim, F.S., Jappor, H.R. and Al-Ammar, K.H.B. (2007) Structural and Electronic Properties of Gallium Arsenide Crystal Using INDO Method. Babylon University, Babylon.
- 15. Wieferin, J., Nemec, L. and Blum, V. (2011) Tutorial II: Periodic Systems Manuscript for Exercise Problems: Presented at the Hands-on Tutorial Workshop on ab Initio Molecular Simulations at Fritz-Haber-Institut der Max-Planck- Gesellschaft Berlin.
- 16. Clark, S. (1994) Complex Structures in Tetrahedrally Bonded Semiconductors. Ph.D. Thesis, University of Edinburgh, Edinburgh.
- 17. Murnaghan, F.D. (1944) Finite Deformations of an Elastic Solid. American Journal of Mathematics, 49, 235-260.
- 18. Cohen, M.L. and Chelikowsky, J.R. (1988) Electronic Structure and Optical Properties of Semiconductors. Springer-Verlag, Berlin, 264 p. http://dx.doi.org/10.1007/978-3-642-97080-1
- 19. Hellwege, K.H. and Madelung, O.L.-B. (1982) New Series. Group III, Vol. 17a, Springer, Berlin.
- 20. Cai, J. and Chen, N. (2007) Stability of High-Pressure Phases in II-VI Semiconductors by a Density Functional Lattice Dynamics Approach D. Kirin and I. Luka?evi?. Physical Review B, 75, 172103. http://dx.doi.org/10.1103/PhysRevB.75.172103�
- 21. Nirmala, L.C. and K. Iyakutti, L. (2003) Electronic Phase Transition and Superconductivity of Vanadium under High Pressure. Physical Review B, 67, 9.