Open Journal of Immunology
Vol.05 No.04(2015), Article ID:59973,10 pages
10.4236/oji.2015.54018
Deleterious Nonsynonymous SNP Found within HLA-DRB1 Gene Involved in Allograft Rejection in Sudanese Family: Using DNA Sequencing and Bioinformatics Methods
Mohamed M. Hassan1,2*, Sofia B. Mohamed3, Mohamed A. Hussain4, Amar A. Dowd1
1Faculty of Medical Laboratory Sciences, University of Medical Science and Technology, Khartoum, Sudan
2Faculty of Medical Laboratory Sciences, Al Zaiem Al Azhari University, Khartoum, Sudan
3Tropical Medicine Research Institute, Khartoum, Sudan
4Department of Pharmaceutical Microbiology, International University of Africa, Khartoum, Sudan
Email: *mhassan0210@gmail.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution-NonCommercial International License (CC BY-NC).
http://creativecommons.org/licenses/by-nc/4.0/



Received 14 August 2015; accepted 25 September 2015; published 28 September 2015
ABSTRACT
Renal transplantation provides the best long-term treatment for chronic renal failure. Single- nucleotide polymorphisms (SNPs) play a major role in the understanding of the genetic basis of many complex human diseases. Also, the genetics of human phenotype variation could be understood by knowing the functions of these SNPs. It is still a major challenge to identify the functional SNPs in a disease-related gene. This work explored how SNPs mutations in HLA-DRB1 gene could affect renal transplantation rejection. This study was carried out in Ahmed Gasim Hospital, Renal Dialysis Center during the period, from September 2012 to November 2013. Blood samples from five Sudanese patients (different families) with known renal transplantation rejection were collected before hemodialysis, furthermore one blood sample for control. DNA sequences results and detected SNPs were analyzed using bioinformatics tools (BLAST, SIFT, nsSNP Analyzer, PolyPhen, I-mutant, BioEdit, CPH, Chimera, Box shade and Project Hope). In addition, international databases were used for datasets [NCBI, Uniprot]. Results showed that, three SNPs were detected; two of three SNPs were predicted as tolerant or benign (rs1059575, novel) and one was deleterious (rs17885437). This study concluded that the identification of pathological SNPs could be an answer to unknown causes for a lot of organ transplantation rejection cases.
Keywords:
Renal Transplantation Rejection, Single Nucleotide Polymorphisms (SNPs), Nonsynonymous Variant, HLA-DRB1 Gene, Sudanese Families

1. Introduction
Around the world in 2002 there were over 1.1 million patients estimated to have end stage renal disease (ESRD) with addition of 7% annually [1] . In the same token and according to world health organization in 2011 Sudan was ranking number nine in kidney disease mortality (2.3%) [2] . In USA the incidence and prevalence of ESRD are expected to increase by 44% - 85% gradually between 2000 and 2015 [3] ; in other side the incidence rate in Sudan is expected to be around 70 - 140/million inhabitants/year [4] . In conclusion we can say that chronic renal disease (CRD) is becoming a real major public health problem worldwide [5] .
Human leukocyte antigen (HLA) system is the name of human major histocompatibilty complex (MHC). A group of cell-surface antigen-presenting proteins are encoded by a region on the short arm of chromosome: 6 in the distal portion of the 21.3 band; several different types of gene are arranged in the form of three regions: class I, II and III. Most of these genes are polymorphic, arranged close together and are generally inherited as a haplotype [6] . Class II MHC antigens are encoded by genes HLA (DP, DM, DOA, DOB, DQ, and DR) loci, and involved in list of the immunoglobulin supergene family [7] .
HLA-DRB1 belongs to the HLA class II beta chain paralogs. The class II molecule is a heterodimer consisting of an alpha (DRA) and a beta chain (DRB), both anchored in the membrane. It plays a central role in the immune system by presenting peptides derived from extracellular proteins. Class II molecules are expressed in antigen presenting cells (APC: B lymphocytes, dendritic cells, macrophages). The beta chain is approximately 26 - 28 kilo Dalton (kDa). It is encoded by 6 exons. Exon one encodes the commander peptide; exons two and three encode the two extracellular domains; exon four encodes the transmembrane domain; and exon five encodes the cytoplasmic tail. Within the DR molecule the beta chain contains all the polymorphisms specifying the peptide binding specificities. DRB1 is expressed at a level five times higher than its paralogs DRB3, DRB4 and DRB5; also DRB1 is present in all individuals and the Allelic variants of DRB1 are linked with either none or one of the genes DRB3, DRB4 and DRB5. In addition, there are five related pseudogenes: DRB2, DRB6, DRB7, DRB8 and DRB9 [8] .
Single nucleotide polymorphisms (SNPs) are variations of a single base, either between two homologous chromosomes within a single individual, or between two individuals [9] . Genetic polymorphisms are well- recognized sources of individual differences in disease risk and treatment response [10] . While the majority of SNPs have no biological consequences, a fraction of gene substitutions have functional significance and provide the basis for the diversity found among humans [10] . However, during the last decade, single nucleotide polymorphisms (SNPs) have become increasingly used because of their abundance in the genome, ease of replication in different laboratories and simplicity of analysis [11] . Recent studies have shown that single-nucleotide polymorphisms (SNPs) are associated with allograft rejection in kidney transplantation recipients [12] .
2. Material & Methods
2.1. Patient Recruitment
This study was a hospital-based case control study, in Ahmed Gasim hospital in the period from September 2012 to November 2013. Five individuals from different families, had undergone renal transplantation whether they develop (acute/hyper acute/chronic) rejection. Sample size include five patients and one control, convenience sampling technique was choose which is based on elements selected from a population on the basis of what elements are easy to obtain. Sometimes a convenience sample is called a grab sample as we essentially grab members from the population for our sample. This is a type of sampling technique that does not rely upon a random process, such as we see in a simple random sample, to generate a sample. Committee of ministry of health, Khartoum state approved the protocol and a written informed consent was obtained from all participants prior to study participation.
2.2. Mutational Detection Using Automated DNA Sequencer
Genomic DNA (gDNA) were extracted from the patient’s peripheral blood leukocytes using CinnaPure DNA extractions kits. Sequencing done for the area within HLA-DRB1 gene (301 base pairs), region location of chromosome 6: 32584100, 32584400. Using Roche/454 Genome Sequencer FLX + Titanium, Deep sequencing up to 1000 bp read length and up to 1.1 Gb/run.
2.3. Data Prediction and Analysis Using Bioinformatics Tools
Firstly; DNA sequences were compared with the NCBI human reference genome (www.ncbi.nlm.nih.gov) to check DNA sequencing quality and specificity by using BLAST (basic local alignment search tool), it’s a fast sequence similarity searching (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (Figure 1). Secondly; sequences were submitted―in sequence alignment form―to BoxShade (online tool) and BioEdit (software package) for multiple sequence alignment results, Figure 2 and Figure 3. BoxShade available at:
http://www.ch.embnet.org/software/BOX_form.html. Thirdly; three identified SNPs were submitted to NCBI human reference genome for the second time to check if the identified SNPs are known or novel, then protein sequence of SNPs were get from UniProt database (Table 1) (http://www.uniprot.org/) and submitted respectively to SIFT-Sorting Intolerant From Tolerant―(An online server was used to rate intolerant from tolerant nonsynonymous single-base nucleotide polymorphism (nsSNP), and alignments between an order sequence with a large number of homologous sequences to predict if an amino acid substitution will have a phenotypic effect, score result of each new residual ranges from zero to one, when score is below or equal to 0.05 the amino acid substitution is predicted to be intolerant, and tolerated if the score is greater than 0.05 [13] . Poly Phen (Polymorphism Phenotyping) is a server which predicts possible impact of an amino acid substitution on the structure and function of a human protein by analysis of multiple sequence alignment and protein 3D structure, using eight sequence and three structural based which selected automatically by masterful interactive algorithm, in addition that calculates position-specific independent count scores (PSIC) for each of two variants, and then computes the PSIC scores difference between two variants, where the higher a PSIC score difference, the higher functional impact a particular amino acid substitution is likely to have. Prediction outcome can be one of probably/possibly damaging or benign [14] . nsSNP Analyzer is a tool used to predict whether a nsSNP has a phenotypic effect. Its predict results contains information at functional and structural level, provides additional information about the
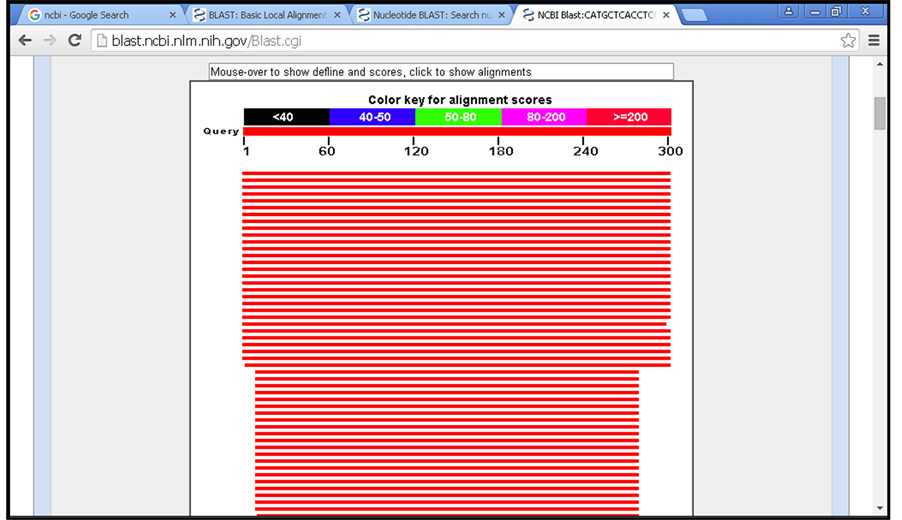
Figure 1. BLAST results. The score of each alignment is indicated by one of five different colors, which divides the range of scores into five groups, where red color shows the highly similar (≥200).

Figure 2. Multiple sequence alignment using BoxShade software. Black colour shows conserved regions, gray and white colour shows the mutants regions.
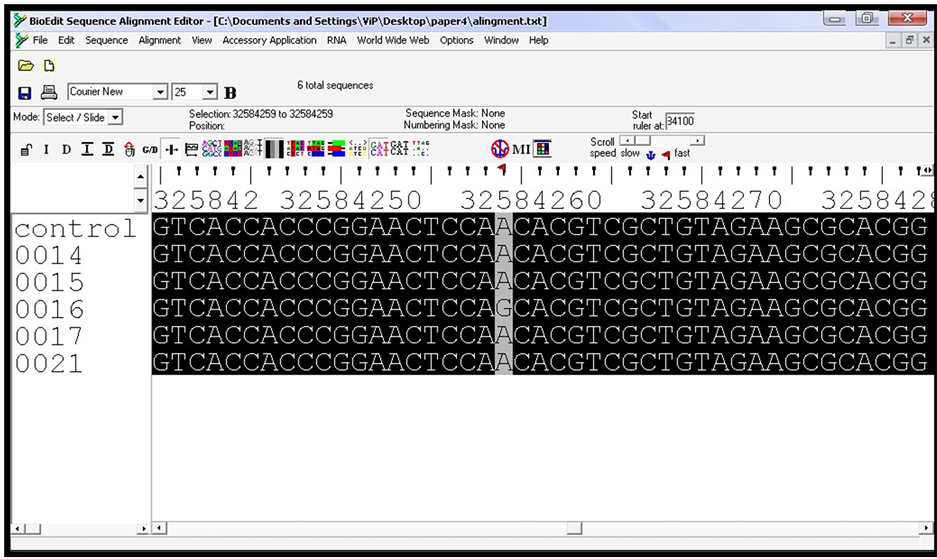
Figure 3. Multiple sequence alignment using BioEdit (software package), the upper rule numbers show the chromosome location, gray colour shows the pathological SNP (rs17885437) location (chr:6, 32584259).

Table 1. Gene and protein information of identified SNPs obtained from (NCBI & UniProt) databases. (www.ncbi.nlm.nih.gov, http://www.uniprot.org/)
SNP to facilitate the interpretation of results, e.g., structural environment and multiple sequence alignment [15] . Results summarized in Table 2 and Table 3. Previous servers available at: SIFT (http://sift.bii.a-star.edu.sg/), nsSNP Analyzer (http://snpanalyzer.utmem.edu/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/). After that protein sequence of pathological or damaging predicted SNP was submitted to I-mutant and Project Hope servers to predict the stability index and some physiochemical properties change due to SNP variant [16] [17] , Table 4 and Table 5. I-mutant (version 3.0) available at:
(http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi), Project Hope
(http://www.cmbi.ru.nl/hope/home). Fourthly; protein sequence of deleterious SNP placed to get protein secondary structure using GOR IV tool (secondary structure prediction tool) and result shown in Figure 4, (https://npsa-prabi.ibcp.fr/cgibin/npsa_automat.pl?page=npsa_gor4.html), then for the same sequence, homology modeling (3D structure) was predicted using CPH-models 3.2 server, after that for visualization of protein 3D structure and identification of native/new residues types, chimera software was used Figure 5 and Figure 6. CPH server available at: http://www.cbs.dtu.dk/services/CPHmodels/. Lastly; protein sequence of deleterious SNP placed in ProtParam tool (a tool which allows the computation of various physical and chemical parameters for a user entered sequence. The computed parameters include the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index and grand average of hydropathicity-GRAVY-) [18] (Table 6). http://web.expasy.org/protparam/.
Table 2. Predicted results of SIFT and nsSNP analyzer servers.
Environment: The structural environment of the SNP calculated by the environment program, Area buried: Solvent accessibility score, Frac polar: Environmental polarity score, SIFT and Score: Ranges from 0 to 1, the amino acid substitution is predicted damaging if the score is ≤0.05, and tolerated if the score is >0.05.
Table 3. Output for PolyPhen-2 server.
PolyPhen-2 score (range from 0 to 1): Probably damaging (~ >0.80), possibly damaging (~ >0.60 to 0.80) and benign (~ ≤0.60).
Table 4. Protein stability based on standard free energy change using I-mutant 3.0.
WT: Wild type amino acid, MT Mutant type amino acid, DDG: DG (New Protein)-DG (Wild Type) in Kcal/mol (DDG < 0: Decrease stability, DDG > 0: Increase stability), RI: Reliability index.
3. Results
3.1. Check the Sequencing Results
BLAST results showed high similarity to Homo sapiens HLA-DRB1 gene for all sequences (samples and control), with identical percentage between 92% - 93%, and these results showed great sequencing results (Figure 1).
3.2. Multiple Sequence Alignment
Alignment showed the highly conserved target sequencing region for all sequences. Three SNPs were detected, first SNP was in sample (0016) and other two SNPs were within sample (0014) (Figure 2 and Figure 3).
Table 5. Physicochemical properties of deleterious nsSNP (G74R) using Project Hope server.
Table 6. Computation of some physical and chemical parameters of protein (ID: NP_002115) using Prot Param tool.
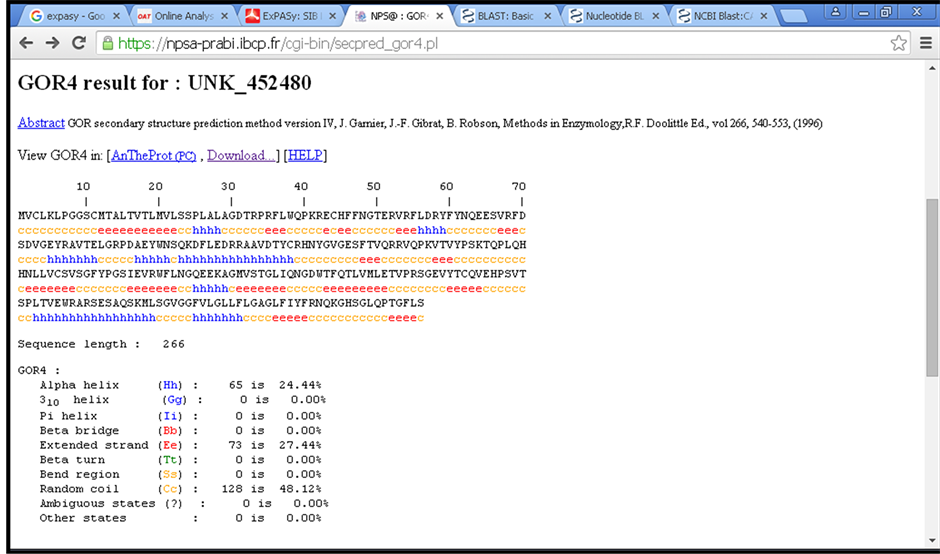
Figure 4. Shows primary and secondary structure of deleterious nsSNP related protein (NP_002115) using GOR IV tool, small box within the figure refers to mutant amino acid position of (rs17885437).
 (a)
(a)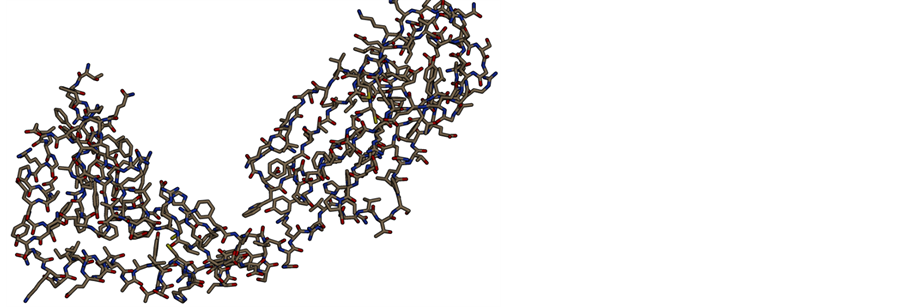 (b)
(b)
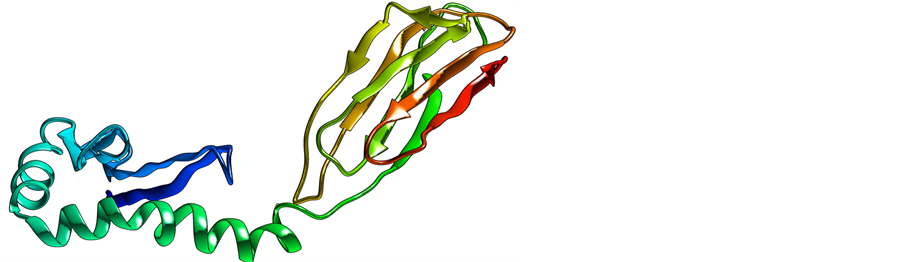
Figure 5. Shows protein tertiary structure (homology modeling) of deleterious nsSNP related protein (NP_002115). (a) Overview of the protein in ribbon-presentation; (b) Overview of all protein atoms.
 (a) (b)
(a) (b)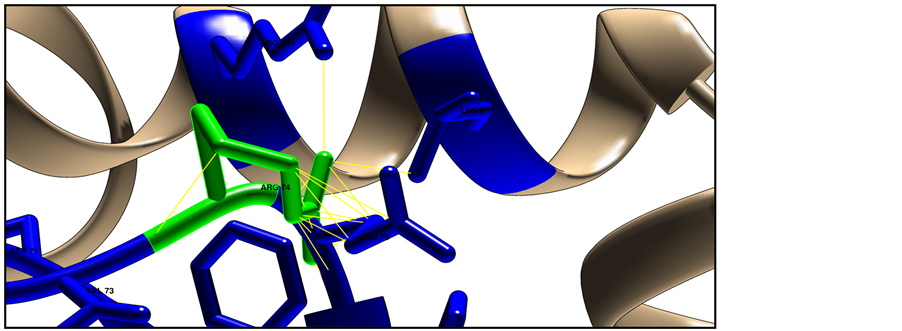 (c)
(c)
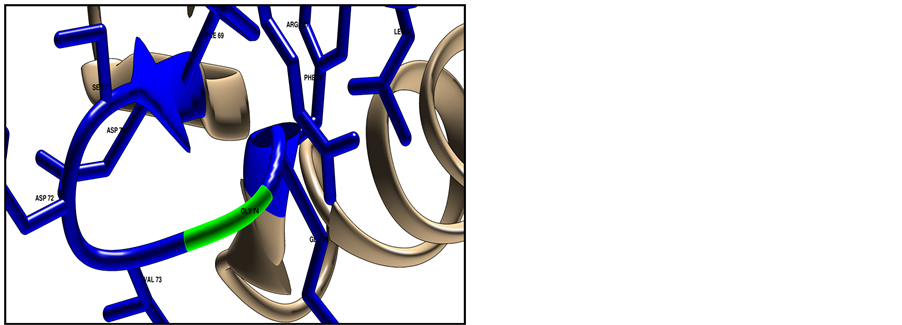
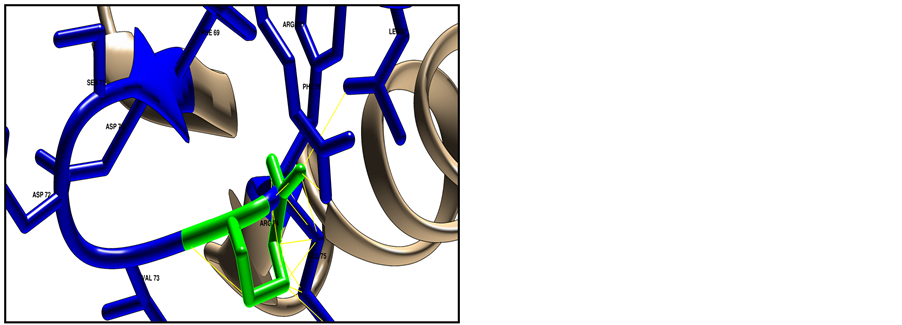
Figure 6. Shows HLA-DRB1 protein structure (ID: NP_002115) using Chimera software (v 1.8). The target residue colored green and the surrounding closest residues colored blue, where (a) represent the native structure with Glycine in position 74; (b) represent the mutated structure with Arginine in position 74, mutant amino acid shows clashes (26 points contacts in fine yellow lines) with other neighbor residues; (c) Shows clashes from different side angle.
3.3. Differentiates between Deleterious and Tolerant (Non Damaging) SNPs
Three tools were used to predict the functionality of three detected SNPs, results showed that two of three SNPs were predicted as tolerated and third one as damaging (Table 2 and Table 3).
Prediction of Change in Stability Due to SNPs Used I-Mutant 3.0 Server
Protein sequence of target SNPs was submitted, results show stability change (decrease) for two SNPs (rs1059575, rs17885437) with deferments DDG values (Table 4).
3.4. Predicted Physical and Chemical Properties Change Due to SNP
Protein sequence of just a predicted pathological SNP was submitted to Project Hope server, results show that there were wide physiochemical changes due to single variant from wide type (Glycine) to new type (Arginine) (Table 5).
3.5. Visualized the Protein Homology Modeling
Homology modeling of target pathological SNP did used CPH model 3.2 server, and then model was visualized used chimera software, results shown the structural difference between wide and new amino acid in position 74 with 26 point contact (clash contact) of the mutant residue with neighbor atom, this contact point will increase the severity of damaging (Figure 6).
4. Discussion
The study involved a group of five renal transplant known rejection patients on hemodialysis (plus one control), patients were dialyzing two to three times per week, and they were at different duration of renal transplant rejection from two to ten years when samples had been collected. Our main objective was to detect presence of SNPs through Sudanese genome of 23 pathological nsSNPs which identified located worldwide in this chromosome location through in silico study done by Hassan (2014).
Three SNPs were detected in this study using DNA sequencing technology, two of the three were known SNPs in dbSNPs (SNPs database) (rs1059575, rs17885437), and the third SNP was a novel SNP (not found in dbSNP). Then bioinformatics tools were used to determine the effect of three detected SNPs on the protein structure and function, we found that two were tolerant and one is damaging. The damaging SNP in HLA complex genes may affect renal transplantation rejection. From the above, the results are similar to Hassan’s results [8] . Novel SNP located in chromosome 6: 32584307, where nucleotide changed from thiamine to cytosine (T/C) and the codon had been changed from TCT to TCC, both codon encoded by the same amino acid Serine (degeneracy of genetic code), which means that SNP has no effect on protein level (tolerant SNP).
5. Conclusion
HLA typing using SNPs analysis is a suitable, accurate and cheap way to cover all types of HLA genes and could be used whole over the world. Damaging SNPs detections also could be an answer for unknown causes of many organ transplantation rejection cases. Results showed the power and impact of in silico tools on biomedical research and their ability to uncover the cause of genetic variations in different genetic diseases.
Disclosure Statement
The authors declare that they have no conflicts of interest related to this work.
Cite this paper
Mohamed M.Hassan,11,Sofia B.Mohamed,Mohamed A.Hussain,Amar A.Dowd, (2015) Deleterious Nonsynonymous SNP Found within HLA-DRB1 Gene Involved in Allograft Rejection in Sudanese Family: Using DNA Sequencing and Bioinformatics Methods. Open Journal of Immunology,05,222-232. doi: 10.4236/oji.2015.54018
References
- 1. Lysaght, M.J. (2002) Maintenance Dialysis Population Dynamics: Current Trends and Long-Term
Implications. Journal of the American Society of Nephrology, 13, 37-40.
http://jasn.asnjournals.org/content/13/suppl_1/S37.long - 2. (WHR) World Health Rankings. http://www.worldlifeexpectancy.com/world-health-rankings
- 3. Gilbertson, D.T., Liu, J., Xue, J.L., Louis, T.A., Solid, C.A., Ebben, J.P. and Collins, A.J. (2005) Projecting
the Number of Patients with End-Stage Renal Disease in the United States to the Year 2015.
Journal of the American Society of Nephrology, 16, 3736-3741.
http://jasn.asnjournals.org/content/16/12/3736.long
http://dx.doi.org/10.1681/ASN.2005010112 - 4. Elsharif, M.E. and Elsharif, E.G. (2011) Causes of End-Stage Renal Disease in Sudan: A Single-Center
Experience. Saudi Journal of Kidney Diseases and Transplantation, 22, 373-376.
http://www.sjkdt.org/article.asp?issn=13192442;year=2011;volume=22;issue=2;spage=373;epage=376;aulast=Elsharif - 5. Barsoum, R.S.
(2006) Chronic Kidney Disease in the Developing World. New England Journal
of Medicine, 354, 997- 999.
http://www.nejm.org/doi/full/10.1056/NEJMp058318
http://dx.doi.org/10.1056/nejmp058318 - 6. Mehra, N.K. (2001) Histocompatibility Antigens. Encyclopedia of Life Sciences.
Nature Publishing Group.
http://web.udl.es/usuaris/e4650869/docencia/segoncicle/genclin98/recursos_classe _%28pdf%29/revisionsPDF/HLA.pdf - 7. Bodmer, W.F. (1987) The HLA System: Structure and Function. Journal of Clinical Pathology,
40, 948-958. http://jcp.bmj.com/content/40/9/948.full.pdf
http://dx.doi.org/10.1136/jcp.40.9.948 - 8. Hassan, M.M., Dowd, A.A., Mohamed, A.H., Mahalah, S.M.O.S., et al. (2014) Computational Analysis
of Deleterious nsSNPs within HLA-DRB1 and HLA-DQB1 Genes Responsible for Allograft Rejection.
International Journal of Computational Bioinformatics and In-Silico Modeling, 3, 562-577.
http://bioinfo.aizeonpublishers.net/content/2014/6/562-577.html - 9.
Uricaru, R., Rizk, G., Lacroix, V., Quillery, E., et al. (2015) Reference-Free
Detection of Isolated SNPs.
Nucleic Acids Research, 43, e11.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4333369/pdf/gku1187.pdf
http://dx.doi.org/10.1093/nar/gku1187 - 10.
Berno, G., Zaccarelli, M., Goril, C., Tempestilli, M., et al. (2014)
Analysis of Single-Nucleotide
Polymorphisms (SNPs) in Human CYP3A4 and CYP3A5 Genes: Potential Implications for the Metabolism of HIV Drugs. BMC Medical Genetics, 15, 76.
http://www.biomedcentral.com/content/pdf/1471-2350-15-76.pdf
http://dx.doi.org/10.1186/1471-2350-15-76 - 11. Zhan, X., Dixon, A., Batbayar, N., Bragin, E., et al. (2015) Exonic versus Intronic SNPs: Contrasting
Roles in Revealing the Population Genetic Differentiation of a Widespread Bird Species.
Heredity (Edinb), 114, 1-9.
http://www.nature.com/hdy/journal/v114/n1/pdf/hdy201459a.pdf
http://dx.doi.org/10.1038/hdy.2014.59 - 12.
Kim, S.K., Park, H.J., Seok, H., Jeon, H.S., et al. (2014) Association Studies
of Cytochrome P450, Family 2, Subfamily E, Polypeptide 1 (CYP2E1) Gene
Polymorphisms with Acute Rejection in Kidney
Transplantation Recipients. Clinical Transplantation, 28, 707-712.
http://onlinelibrary.wiley.com/doi/10.1111/ctr.12369/pdf
http://dx.doi.org/10.1111/ctr.12369 - 13. Ng, P.C. and Henikoff, S. (2001) Predicting Deleterious Amino Acid Substitutions. Genome Research,
11, 863-874. http://genome.cshlp.org/content/11/5/863
http://dx.doi.org/10.1101/gr.176601 - 14.
Ramensky, V., Bork, P. and Sunyaev, S. (2002) Human Nonsynonymous SNPs: Server
and Survey.
Nucleic Acids Research, 30, 3894-3900.
http://nar.oxfordjournals.org/content/30/17/3894.long
http://dx.doi.org/10.1093/nar/gkf493 - 15. Bao, L., Zhou, M. and Cui, Y. (2005) nsSNP Analyzer: Identifying Disease-Associated Nonsynonymous
Single Nucleotide Polymorphisms. Nucleic Acids Research, 33, W480-W482.
http://nar.oxfordjournals.org/content/33/suppl_2/W480.abstract
http://dx.doi.org/10.1093/nar/gki372 - 16. Capriotti, E., Fariselli, P., Calabrese and Casadio, R. (2005) Predicting Protein Stability Changes from
Sequences Using Support Vector Machines. Bioinformatics, 2, 54-58.
http://bioinformatics.oxfordjournals.org/content/21/suppl_2/ii54.abstract
http://dx.doi.org/10.1093/bioinformatics/bti1109 - 17.
Venselaar, H., Beek, T., Kuipers, R.K., Hekkelma, M.L. and Vriend, G.
(2010) Protein Structure Analysis
of Mutations Causing Inheritable Diseases. An e-Science Approach with Life Scientist Friendly Interfaces. BMC Bioinformatics, 11, 548.
http://www.biomedcentral.com/1471-2105/11/548
http://dx.doi.org/10.1186/1471-2105-11-548 - 18. Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., et al. (2005) Protein Identification and Analysis
Tools on the ExPASy Server. In: Walker, J.M., Ed., The Proteomics Protocols Handbook, Humana Press, 571-607.
http://link.springer.com/protocol/10.1385%2F1-59259-890-0%3A571
http://dx.doi.org/10.1385/1-59259-890-0:571
NOTES

*Corresponding author.