Open Journal of Animal Sciences
Vol. 2 No. 4 (2012) , Article ID: 23925 , 7 pages DOI:10.4236/ojas.2012.24030
Genetic characterization of Northeast Asian cattle based on sequence polymorphisms in the complete mitochondrial genome
![]()
1Animal Genomics and Bioinformatics Division, National Institute of Animal Science, Suwon, Korea; #Corresponding Author: thkim63@korea.kr
2Lee Gil Ya Cancer and Diabetes Institute, Gachon University of Medicine and Science, Incheon, Korea
3College of Veterinary Medicine, Seoul National University, Seoul, Korea
4Department of Animal Science, Chungbuk National University, Cheongju, Korea
Received 1 June 2012; revised 8 July 2012; accepted 22 July 2012
Keywords: Bos taurus; Complete Mitochondrial DNA Genome; Phylogeny; Polymorphism
ABSTRACT
In this study, we analyzed complete mtDNA sequences variation and genetic relationship among taurine, indicine and Bison groups. In total, 107 sequences from different breeds, 45 European (45 Italian), 16 Middle East Asian (seven Iranian and nine Iraqi), 41 Northeast Asian (34 Korean and seven Japanese), two Nellore (Bos indicus) and two American Bison bison (Ame. bison) were obtained from Gen-Bank database. One Korean Hanwoo (Bos taurus) sequence was generated using the SOLiDTM System. In total, 1370 polymorphic sites, representing 8.39% of the complete 107 mtDNA sequences (16,338 bp) were detected and of these, 1186 parsimony informative polymorphic sites were identified. Neighbor-joining tree indicated that Korean, Japanese, Iranian, Iraqi, and Italian cattle were closely related to one another, but are separated from B. bison. The B. taurus mtDNA polymorphism was greater in the D-loop than in the other regions. The ATP8, ND3, ND5, and ND6 regions were also quite parsimony informative, similar to Cyt b. In addition, this study revealed a distinct genetic difference between Korean cattle and B. indicus.
1. INTRODUCTION
Cattle are distributed worldwide and are extremely important domestic animals in Korea and other parts of the world. Due to their high economic and social values, this species has received significant attention in evolutionary and genetic diversity studies for a long time. However, there is still incomplete understanding of their association with early human civilization, and research on their origins may enlighten us on hitherto unknown aspects of prehistory.
In Northeast Asia (Mongolia, North China, Korea, and Japan) most cattle lack humps and are classified as Bos taurus [1]. Kim and Lee [2] reported that Korean cattle originated from crossbreeding between European Bos primigenius and Indian Bos indicus. These cattle subsequently moved south and east to become the ancestors of modern-day Japanese cattle, and interactions might have occurred between Japanese cattle and the cattle of other Northeast Asian countries as described by Kim et al. [3]. Moreover, over several decades introgression of several European breeds into East Asian cattle as well as interbreeding among regional breeds has occurred for upgrading native breeds in many countries of Northeast Asia [4].
To reveal their phylogenetic relationships based on DNA sequence polymorphisms, several studies have been performed on bovine mitochondrial DNA (mtDNA) [5,6]. Among them, 237 polymorphisms in two complete bovine mtDNA sequences (one B. taurus, Simmental, and one B. indicus, Dwarf Zebu) and 393 in 36 (34 B. taurus, Korean cattle/Japanese Black/Holstein/Fleckvich, and two B. indicus, Nellore/Zwergzebu) were reported by Hiendleder et al. [5] and Kim et al. [6], respectively. However, the large-scale identification of polymerphisms in complete mtDNA sequences for regional cattle breeds has not been reported. Here, we present the analysis of a large-scale data set of complete mtDNA sequences from various cattle breeds. We used complete mtDNA sequences to examine the phylogenetic relationships and sequences polymorphism among Northeast Asian cattle as well as various other regional cattle breeds.
2. MATERIALS AND METHODS
2.1. Sampling and Sequencing of Hanwoo Proven Bull mtDNA
Blood sample of Hanwoo proven bull 27,223 was obtained from the Hanwoo Experiment Station of the National Institute of Animal Science, South Korea. Genomic DNA was extracted from the blood with a QIAamp DNA Blood Maxi kit according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA). The DNA was fragmented with a Covaris S2 and HydroShear (Genomic Solutions, Chelmsford, MA, USA) using the proper settings for the targeted sizes. Libraries were prepared according to the SOLiDTM System Matepaired Library Preparation protocol for the Applied Biosystems SOLiDTM System: Library Preparation Guide (02/2009 and 10/2009 editions; Applied Biosystems, Foster City, CA, USA). Four paired-end libraries with three different size ranges (600 - 700 bp, 1 - 2 kb, and 0.6 - 2.2 kb) were constructed. Sequencing was performed according to the Applied Biosystems SOLiDTM system. Templated beads were deposited onto two fullscale slides per library, and sequencing was conducted on 50 bases using SOLiD v3.0 chemistry, with one exception: the library prepared from the 0.6 - 2.2 kb DNA fragments was used for four full-scale slides with sequencing performed on 50 bases using SOLiD v3 plus chemistry. The UMD3 mitochondrial DNA sequence (ftp://ftp.cbcb.umd.edu/pub/data/Bos_taurus/Bos_taurus_UMD_3.0/, NC 006853) was used as a reference.
2.2. Data Analysis
Complete B. taurus mtDNA sequences for 45 European (45 Italian), 16 Middle East Asian (seven Iranian and nine Iraqi), and 41 Northeast Asian (34 Korean and seven Japanese) cattle previously deposited into GenBank were used. A hundred and three B. taurus, two B. indicus (Nellore), and two Ame. bison (outgroup) complete mtDNA sequences were aligned using ClustalW version 1.8.3. [7] as implemented in MEGA 5.03 [8]. All positions containing gaps and missing data were eliminated using the complete-deletion option [8,9]. For the B. taurus mtDNA sequences, the nucleotide composition, pattern of nucleotide substitutions, and site variations were examined using MEGA5.03 [9]. A phylogenetic analysis was also performed using MEGA5.03. A neighbor-joining tree [10] was constructed using 107 sequences based on Tamura-Nei distances. The statistical confidence of each node in the neighbor-joining tree topology [10] was estimated using 1000 random bootstrap replications. The NIAS_BT_Hanwoo mtDNA that was newly sequenced in this study has been deposited into GenBank under Accession No. HQ025805.
3. RESULTS AND DISCUSSION
3.1. Nucleotide Composition and Sequence Variation
An analysis of the 103 B. taurus complete mtDNA sequences displayed 621 polymorphic sites representing 3.8% of the mtDNA genome. Similarly, four complete mtDNA sequences (two B. indicus and two Ame. bison) revealed 1370 polymorphic sites, accounted for 8.3% of the complete mtDNA sequences. From these polymerphic site comparisons, 383 (B. taurus) and 1186 (B. indicus and Ame. bison) parsimony informative polymerphic sites were identified, respectively. The average nucleotide frequencies of A, T, C, and G were 33.4%, 27.2%, 13.4% and 26.0% for most B. taurus cattle, respectively (Table 1). In Table 1, each entry shows the probability of an instantaneous substitution from one base (row) to another (column). When only entries within a row are compared, rates of different transitional substitutions are shown in bold, while those of transversional substitutions are shown in italics. The ratios of transition/transversion mutation rate were k1 = 34.14 (purines) and k2 = 34.71 (pyrimidines) for the B. taurus mtDNA sequences, and k1 = 33.66 (purines) and k2 = 30.71 (pyrimidines) for the Korean B. taurus sequences. The overall transition/transversion bias (R) was 15.99 for the B. taurus mtDNA sequences and 14.77 for the Korean B. taurus mtDNA sequences, with  .
.
These results indicate that the transition bias for B. taurus mtDNA genomic sequences was a bit higher than for the Korean B. taurus sequences. While for other breeds, the transition bias for B. taurus mtDNA genomic sequences was a little bit lower than for the each of B. taurus breed sequences as shown in Table 1. The strong transitional bias detected in this study accorded with previous studies on Mesolithic Wild Aurochs (Bos primigenius) [11] and the ancient and modern bovine populations [12]. Table 2 shows the number of polymorphic sites for different mtDNA regions and regional variation among the 103 B. taurus mtDNA sequences. The D-loop (mtDNA control region) and cytochrome b (Cyt b) gene regions contain abundant phylogenetic information and are appeared to be good markers for studying genetic differentiation and phylogenetic relationships among species within the same genus or family [3,13-16]. Of 922 (D-loop) and 1140 (Cyt b) DNA sequences in the 103 individual B.
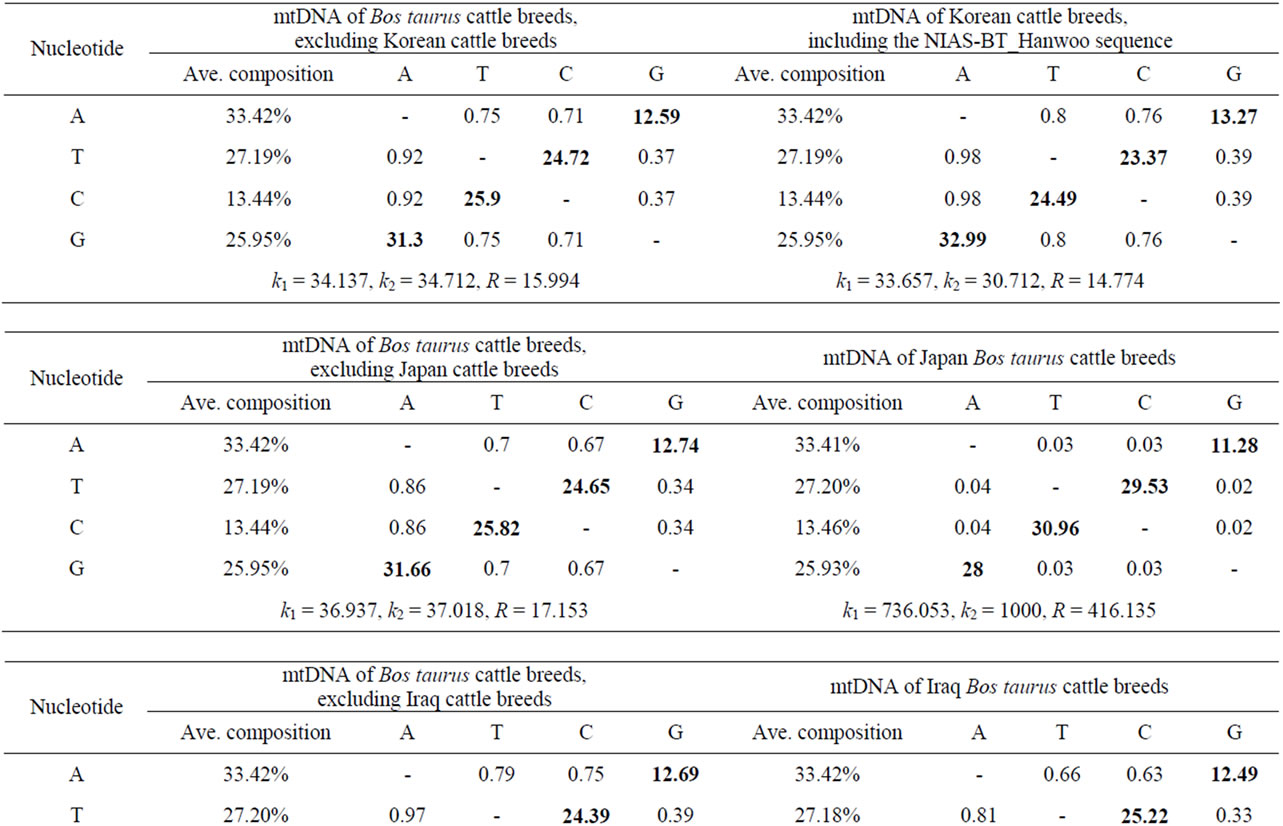
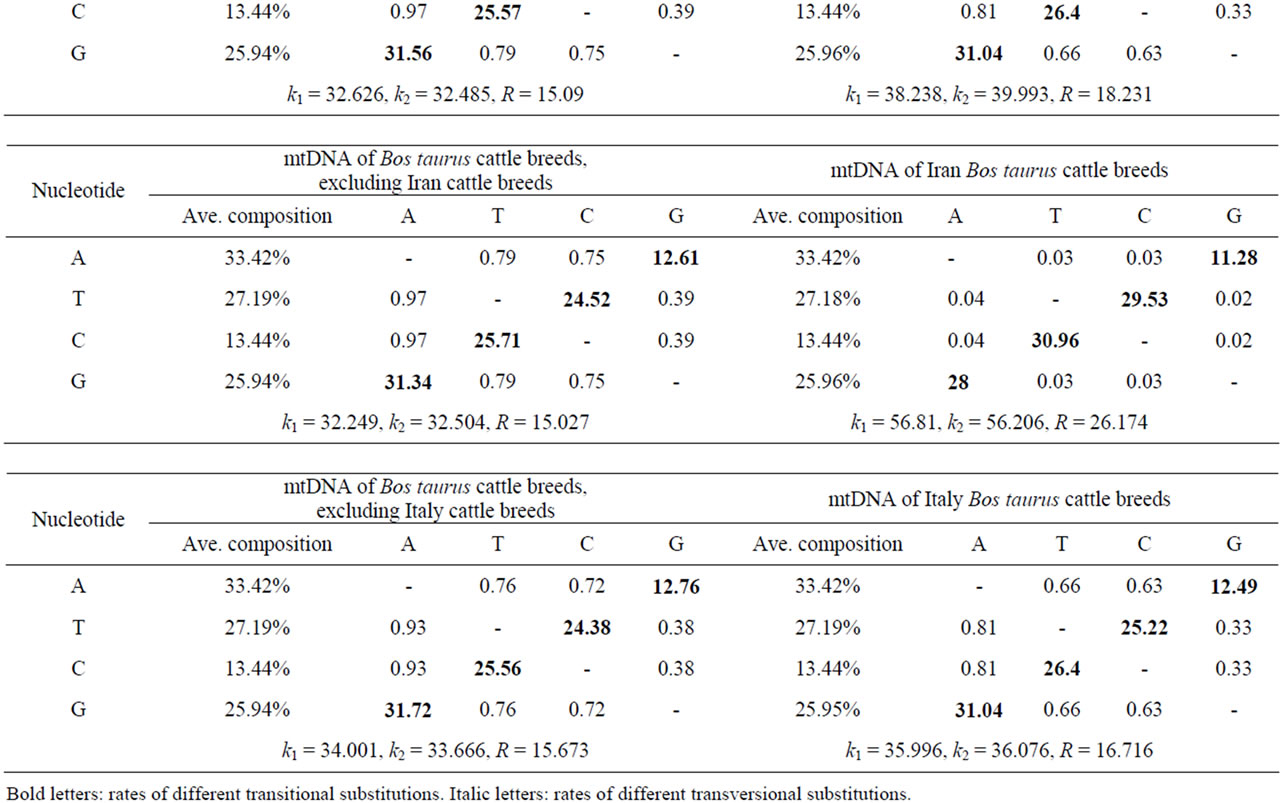
Table 1. Nucleotide composition and maximum composite likelihood estimate of the pattern of nucleotide substitution in the complete B. taurus mtDNA sequence.

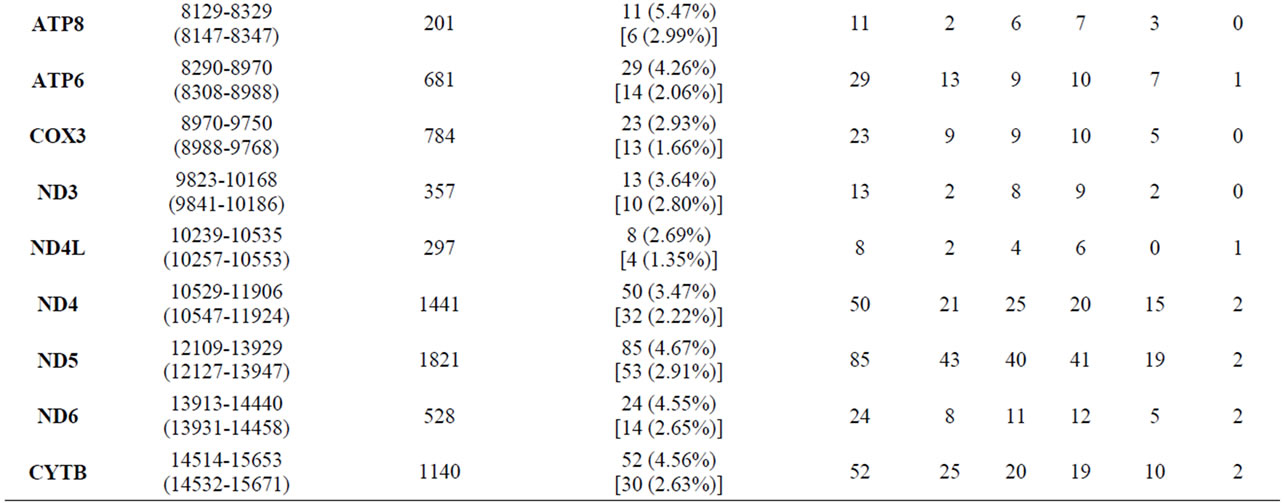
Table 2. Sequence variation of Bos taurus mtDNA complete genome sequences and variation for each product region and geographical region.
taurus mtDNA, 105 (11.39% in the D-loop) and 52 (4.56% in Cyt b) variable sites were observed, of which 77 and 30 were parsimony informative polymorphic sites, respectively (Table 2). Generally, the B. taurus mtDNA polymorphic sites were denser in the D-loop than in the other regions. Moreover, this region seemed to be more parsimony informative. The ATP8, ND3, ND5, and ND6 regions were also quite parsimony informative, similar to Cyt b. In total, 178 variable sites in the 34 Korean B. taurus mtDNA sequences and NIAS_BT_Hanwoo sequence were detected (Table 3). These SNPs reported in this study could be used as valuable markers for maternal lineage test.
3.2. Phylogenetic Analysis
The average genetic distance was calculated based on the Tamura-Nei pairwise distance (Table 4). The distance between Ame. bison and the other genus Bos populations (Korean, Japanese, Iraqi, Iranian, Italian, and B. indicus) was >0.06. Furthermore, the distance between B. indicus and the other B. taurus populations was >0.01. However, the average inter-population distance among the B. taurus population was ranged from 0.001 to 0.005, while the average intra-population distance was ranged from 0.001 to 0.006. Interestingly, the average distance for the Iraqi intra-population was slightly greater than that for the inter-population, and there was not much difference between the average distances for the intraand interpopulations with the other cattle populations. From these results, we infer that some Iraqi B. taurus cattle originated from other regional cattle populations, and that B. taurus cattle populations are closely related based on their mitochondrial genome. All B. taurus populations had similar genetic distances from B. indicus (0.012 -
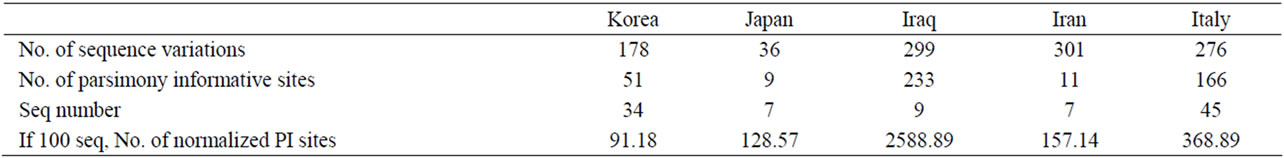
Table 3. Sequence variation between NIAS_BT_Hanwoo and each population.
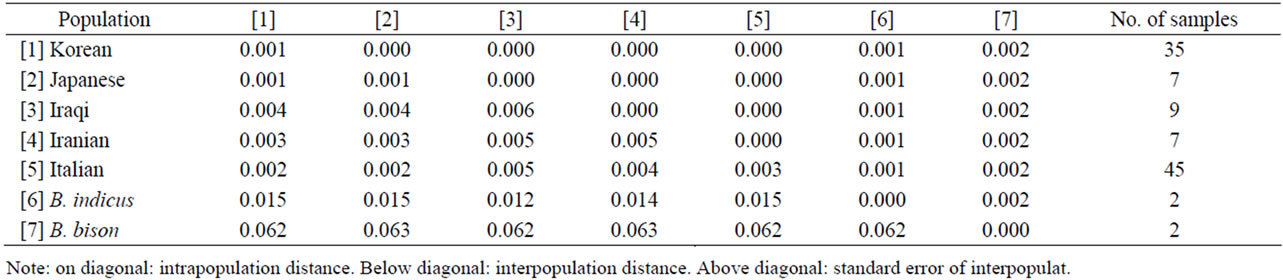
Table 4. Tamura-Nei pairwise distances within and between cattle populations, including Ame. bison.
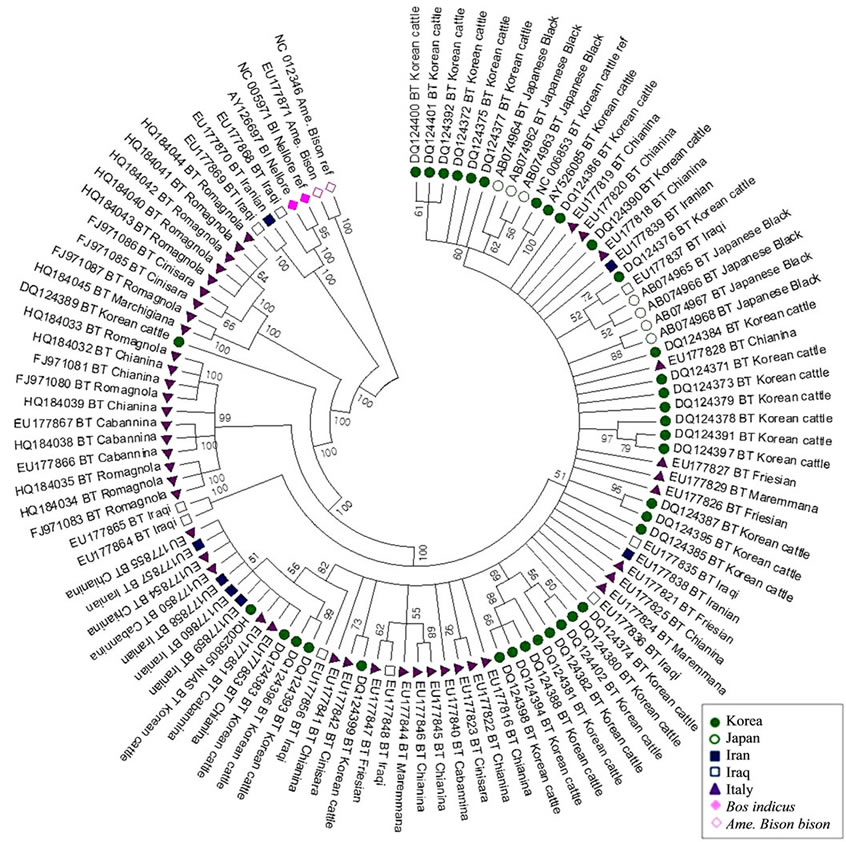
Figure 1. Neighbor-Joining tree of 107 cattle mtDNA complete genome sequences constructed from Bos taurus (Korean cattle, Japanese cattle, Iranian cattle, Iraqi cattle, and Italian cattle), Bos indicus Nellore, and Ame. bison (outgroup) on the basis of Tamura-Nei distances with 1000 bootstrapping.
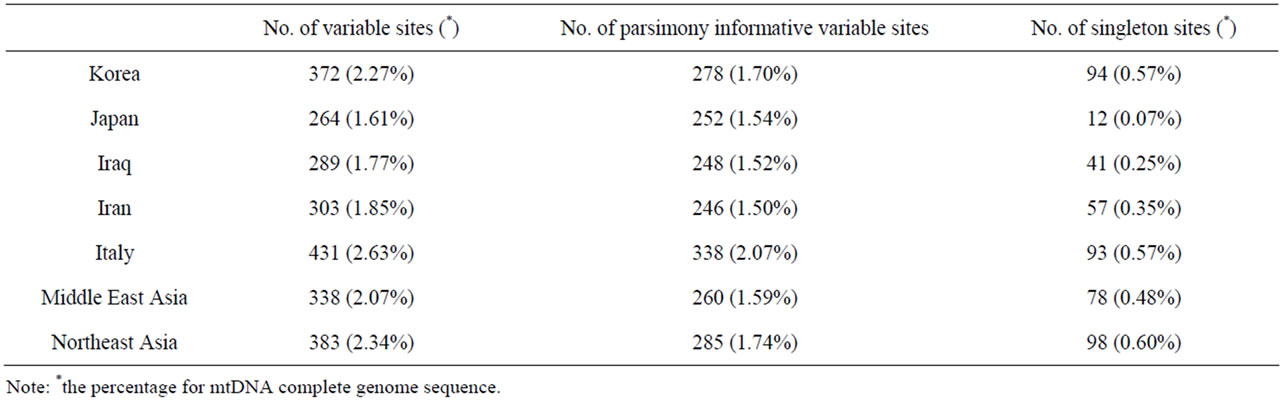
Table 5. Comparison of polymorphism between Bos indicus and each population in Bos taurus.
0.015) and Ame. bison (0.062 - 0.063).
Complete mtDNA sequences from B. taurus, B. indicus, and Ame. bison, together with a Korean cattl mtDNA sequence that was determined newly in this study were used for phylogenetic analysis. The neighbor-joining tree (Figure 1) based on Tamura-Nei distances with 1000 bootstrap values demonstrates that the B. taurus populations including Northeast Asian (Korean and Japanese), Middle East Asian (Iranian and Iraqi), and European (Italian) cattle were closely related, but that they were separated from Ame. bison. Among B. taurus populations, two of nine the Iraqi cattle appear to be more closely related to B. indicus. We also compared the polymorphism from each population of B. taurus respective to B. indicus (Table 5). If they show any trace of crossbreeding, the inter-population distance is likely close to zero or the number of singleton sites among them will be plentiful, even though there are several variations. As shown in Tables 4 and 5, B. indicus was closer to the Middle East Asian cattle (Iraqi and Iranian cattle) than to the Near East Asian cattle (Korean and Japanese cattle).
Because we used only two complete B. indicus mtDNA sequences, genetic distance and phylogenetic analyses may be insufficient. However, our results reveal a signature for distinctness between Korean cattle and B. indicus based on genetic difference (Tamura-Nei pairwise distance of 0.015).
4. ACKNOWLEDGEMENTS
This work was supported by a grant (PJ007197) from the BioGreen 21 Program, Rural Development Administration, Republic of Korea.
REFERENCES
- Phillips, W. (1961) World distribution of the major types of cattle. Journal of Heredity, 52, 207-213.
- Kim, J.B. and Lee, C. (2000) Historical look at the genetic improvement in Korean cattle. Asian-Australasian Journal of Animal Sciences, 13, 1467-1481.
- Kim, K.I, Lee, J.H., Lee, S.S. and Yang, Y.H. (2003) Phylogenetic relationships of northeast Asian cattle to other cattle populations determined using mitochondrial DNA D-Loop sequence polymorphism. Biochemical Genetics, 41, 91-98. doi:10.1023/A:1022021900205
- Kim, K.S., Yeo, J.S. and Choi, C.B. (2002) Genetic diversity of north-east Asian cattle based on microsatellite data. Animal genetics, 33, 201-204. doi:10.1046/j.1365-2052.2002.00848.x
- Hiendleder, S., Lewalski, H. and Janke, A. (2008) Complete mitochondrial genomes of Bos taurus and Bos indicus provide new insights into intra-species variation, taxonomy and domestication. Cytogenetic and Genome Research, 120, 150-156. doi:10.1159/000118756
- Kim, E., Cheong, H.S., Bae, J.S., Chun, J., Park, T.J., Lee, K., Yun, Y. and Shin, H.D. (2010) Identification of genetic polymorphisms in bovine mitochondrial deoxyribonucleic acid. Journal of animal science, 88, 2551-2555. doi:10.2527/jas.2009-2235
- Thomson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTALW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research, 22, 4673-4680. doi:10.1093/nar/22.22.4673
- Tamura, K., Nei, M. and Kumar, S. (2004) Prospects for inferring very large phylogenies by using the neighborjoining method. Proceedings of the National Academy of Sciences of the United States of America, 101, 11030-11035. doi:10.1073/pnas.0404206101
- Tamura, K., Dudley, J., Nei, M. and Kumar, S. (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution, 24, 1596-1599. doi:10.1093/molbev/msm092
- Saitou, N. and Nei, M. (1987) The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406-425.
- Edwards, C.J., Magee, D.A., Park, S.D., McGettigan, P.A., Lohan, A.J., Murphy, A., Finlay, E.K., Shapiro, B., Chamberlain, A.T., Richards, M.B., Bradley, D.G., Loftus, B.J. and MacHugh, D.E. (2010) A complete mitochondrial genome sequence from a mesolithic wild aurochs (Bos primigenius). PloS One, 5, e9255. doi:10.1371/journal.pone.0009255
- Stock, F., Edwards, C.J., Bollongino, R., Finlay, E.K., Burger, J. and Bardley, D.G. (2009) Cytochrome b sequences of ancient cattle and wild ox support phylogenetic complexity in the ancient and modern bovine populations. Animal genetics, 40, 694-700. doi:10.1111/j.1365-2052.2009.01905.x
- Loftus, R.T., Machugh, D.E., Bradley, D.G., Sharp, P.M. and Cunningham, P. (1994) Evidence for two independent domestications of cattle. Proceedings of the National Academy of Sciences, 91, 2757-2761. doi:10.1073/pnas.91.7.2757
- Mannen, H., Tsuji, S., Loftus, R.T. and Bradley, D.G. (1998) Mitochondrial DNA variation and evolution of Japanese black cattle (Bos taurus). Genetics, 150, 1169-1175.
- Cai, X., Chen, H., Lei, C., Wang, S., Xue, K. and Zhang, B. (2007) mtDNA diversity and genetic lineages of eighteen cattle breeds from Bos taurus and Bos indicus in China. Genetica, 131, 175-183. doi:10.1007/s10709-006-9129-y
- Xuan, T.P., Georgescu, S.E., Manea, M.A., Hermenean, A. O. and Costache M. (2010) Genetic diversity and phylogenetic relationships of Romanian cattle breeds inferred from cytochrome b gene partial sequences. Romanian Biotechnological Letters, 15, 5154-5158.
NOTES
*These authors contributed equally to this work.