Open Journal of Genetics
Vol.07 No.02(2017), Article ID:77203,20 pages
10.4236/ojgen.2017.72008
Single Chain Fragment Variables Antibody binding to EGF Receptor in the Surface of MCF7 Breast Cancer Cell Line: Application and Production Review
Elham Omer Mahgoub
Department of Microbiology and Immunology, Faculty of Medicine, Alnelian University, Khartoum, Sudan

Copyright © 2017 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: April 24, 2017; Accepted: June 24, 2017; Published: June 27, 2017
ABSTRACT
In this review, single-chain fragment variable construction using phage-display technology as a promising anticancer immunotherapy technology is described. Cloning and the specific bio-panning selection with phage display technology, as well as the use of the epidermal growth factor receptor (EGFR) at the surface of MCF-7 cells as the antigen for the straightforward specific selection of single chain Fvs, are discussed. Moreover, phage display technologies and their application are important for vaccine production and immunotherapy against viruses and cancers. Furthermore, expression of the gene will cause the production and expression of the protein in prokaryotic and eukaryotic cells, which can be used to detect anti-cancer single chain fragment variables (scFvs). Finally, homology modelling is described to show the three-dimen- sional scFv structure that verifies the Complementary-Determining-Regions (CDRs) on the surface of the model.
Keywords:
Single Chain Fragment Variable, Epidermal Growth Factor Receptor, MCF-7, Phage Display Technology

1. Introduction
Breast cancer is the second major reason of death in women in age between 40 and 55 years after lung-cancer. Breast cancer is caused by the abnormal growth of cells in the mammary gland [1] . Moreover, the molecular conversion of the breast tissue from normal to malignant indicates that breast cancer is a genetic disease, subsequent from series of genetic lesions. Because there are restrictions to the current treatment routines for this genetic disease, novel therapeutic modalities will be beneficial for the effective treatment of breast cancer. Sum of gene therapy methods for breast cancer have been advanced. These methods can be alienated into the following six mean classes: 1) genetic assimilation therapy; 2) chemo-therapy; 3) disintegrate cell therapy; 4) gene therapy to reduce the growth of new blood vessels tumors that need to grow; 5) genetic immune-re- sistance; and 6) genetic resistance therapy. Those clinical trials indorsed the security and possibility of these different gene therapy methods for breast cancer. Gene therapy can be established by using malignant-targeted monoclonal antibodies (Mabs) alone or Mabs bind to radio-substances, drugs, which kill cancer cells while leaving normal cells unhurt.
The use of IgG monoclonal antibodies is first start antibodies produced in hybridoma technology. Those antibody targeted EGFR on the surface of MCF-7 breast cancer cells, which is function in to gene therapy and diagnosis of MCF-7 cancer cell. The growth-factor-receptors play significant parts in controlling cell propagation, that directed the attention into use them as a drug for cancer therapy. Consequently, growth-factor-receptors are best applicants for genetic therapy because they are normally over-expressed on the surface of malignant cells. Additionally, [2] described the important role growth-factors could play to diagnosis malignant cells, demonstrating that tyrosine kinase (TKGFR 1) growth- factor-receptor is produced in a wide variety of malignant cells that categorises them as unique complicated cell-surface indicators for human tumours.
Single chain fragment variables are molecular formed of IgG monoclonal antibodies. Which are constructed using phage display technology and molecular cloning and the specific bio-panning selection with the use of the epidermal growth factor receptor (EGFR) at the surface of MCF-7 cells as the antigen for the straightforward specific selection are considered as fast and accurate way of producing specific antibodies [3] . In this review, the process of constructing a single chain antibody by using phage display technologies, which are sequence of known experiments, that in the end produce high specific antibodies described in detail. The scFv-MCF-7 products are highly recommended to be used in diagnosis and gene therapy toward breast cancer cell line MCF-7.
2. Anti-EGFRvIII MCF-7 Monoclonal Antibodies
There various Mabs directed specifically towards EGFRvIII of MCF-7 with great-affinity that are produced using hybridoma technology [4] . These EGFRvIII-bound specifically with monoclonal antibodies have been produced to measure the activity and amount of the EGFRvIII protein expressed in surgically removed tissue samples that collected from human breast carcinoma. MAbs and sc-Fv manufactured specific for the mutant EGFRvIII have been developed, even though EGFRvIII does not bind EGF, these ligand-induced pathways are unrelated and are less specific to reach their aims. However, the future of an anti-EGFRvIII Mab and scFvs after binding to EGFRvIII remains uncertain. Based on immune-fluoresces-imaging and radio-EGFRvIII-labelled Mab assays, clearly, the intracellular vesicles are effected in amount of mints when, specific connection of Mabs with EGFRvIII took place [5] . Reist [5] demonstrated the expression of a human IgG2/mouse chimeric anti-EGFRvIII Mab, and the significant characteristics of the chimeric Mab are comparable to the murine parent. still, the chimeric construct showed more significant specificity in comparative tissue distribution studies of 8.2% in compared with 13.4% to the parent Mabs with P = 0.0009 towards targeting receptors in vivo. Production of MR1, and its affinity-developed generation-1 MR1 [2] , are great future therapeutics candidates.
3. General Structures of Antibodies
In human natural immunity, there are similar group of glycol-proteins that stimulate immunity response in humans [6] . Five classes (isotypes) of immune-globulins Ig (G, A, M, D, and E) have been recognised, each of them has a separated heavy-chain C region programmed by a separated C-region gene. The isotype of an immune-globulin controls the human body immune-process that it involves during antigen binding. Each domain describes the characteristic immune-globulin fold containing of two beta-sheets anti-parallel with disulphide bond. The varied choice of specificities on the surface of Mabs performed by number of amino acids consists of six hyper-variable loops or complementarity-determining-regions (CDRs), which formulate the epitopes. Those CDRs, maintained specific frame-work-region, establish the changeable regions of fragment antigen binding (Fab). The enduring V section amino acids act as a scaffold supporter of the loops and are known as framework residues as demonstrated by [7] .
The Fc fragment interacts with effector systems, such as the complement system, as shown in Figure 1. The two domains of light chain (L) is consist of 100 residues in length, called the variable-light (VL) and the constant (CL) domain as shown in Figure 2. The heavy chain (H) consist of variable-domain (VH) and the four constant C (H1, H2, H3, and H4) domains, as described [8] . Each domain contains two beta sheets filled facing each other and linked together by a well-maintained disulphide bridge and inter-strand loops as described by Lesk and Chothia [9] . Disulphide bridges are an important component of the IgG structure. The disulphide bridge stabilises domain folding, and disulphide bridge stabilise the interaction between H chain, L chain and the interaction of H chains [10] .
4. The Variable Domain Regions
Ab contacts with the antigen are helped by the variable domains V (H and L) as shown in Figure 1. Most of antibody-antigen interactions performed by the complementarity determining regions (CDRs). Regarding to the size of the antigen, different mixtures of the CDR loops are used for the contact with antigen [11] . Commonly, antibodies directed towards small antigens, such as viruses, the antibody use three complementarities determining regions. However, antibodies

Figure 1. Schematic presentation of IgG molecule and antibody fragments [35] .
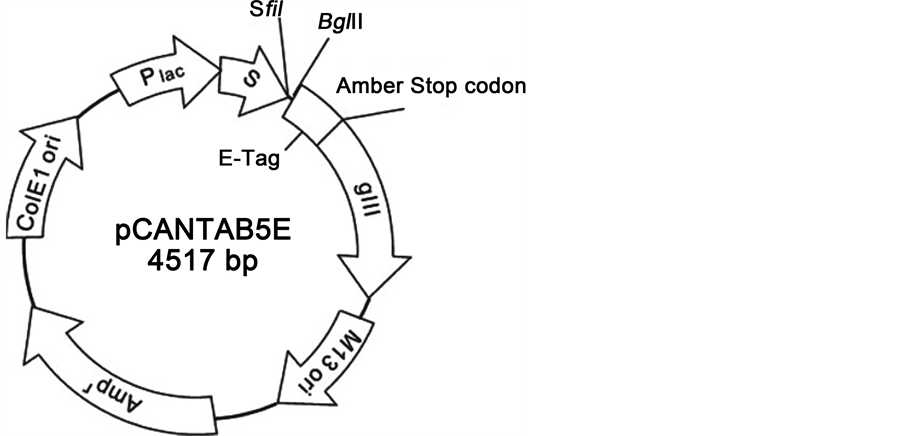
Figure 2. Phage display vector pCANTAB 5E. The restriction sites SfiI and NotI are used to insert interest gene. Arrows indicate functional direction of genes [44] .
directed towards larger proteins use more than four CDRs.
MAbs are working mostly helped by their fragment chain (Fc), collected of domains C (H2 and H3). Still, antibody-dependent cellular cytotoxicity and Ab- dependent cellular phagocytosis ((ADCC and ADCP) in humans demonstrated the finding of three major classes of Fc FcλRI, FcλRIIa/c, Fcλ RIIIa, and inhibitory FcλRIIb receptors [12] . The FcR and Fc-RI receptors of extracellular regions in humans show significant sequence similarity of 70% - 98%. The antibody binding to the components of the complement system is activated with Complement dependent cytotoxicity (CDC). In addition, the antibodies get their long half-life through active recycling of the neonatal Fc receptor [13] .
Scfv is expressed in a variety of hosts, including bacteria, yeast and plants [14] . Single-chain fragments have broad applications in medicine and have great potential in biotechnology. The unique and highly specific antigen-binding ability may be exploited to block specific enzymes or bacteria or to detect environmental pollutants present in very low concentrations (biosensors) [15] .
5. Immunoglobulin Genetics
Immunoglobulin (Ig) heavy chain variable genes are composed of variable-di- versity-joining (VH-D-JH) gene sequences. An efficient VH-D-JH compound is shaped when B-cell initiated and VH gene sequence are linked with a D and JH gene sequences [16] . In humans, there are a 100 to 200 VH gene sequences, more than 30 D sequences, and six efficient JH gene sequences. Germ-line VH sequences are divided into six groups, VH1 to 6, based on DNA sequence similarity. The same VH groups are naturally more than 80% homologous. Still, less than 70% homology between VH gene sequences and other groups sequences. Separated VH groups differ in size from 1 to more than 28 members and composed of non-functional and functional genes. The frequency with which individual VH families are used by adult peripheral blood lymphocytes (PBL) has been reported to roughly correlate with estimates of family size based on Southern blot analysis [17] .
Moreover, VH and VL sequences are amplified to build recombinant single chain F antibody fragments. The peptide linker between VH and VL sequences that manufactured single chain fragment variable. The linker contains two amino acids sequences Glycine (GlY) and serene (Ser) with repeats as GlY4 and Ser3. The linker with the flexible disulphide bonds, moves between the C-terminal of the VH chain and the N-terminal of the VL chain [18] . The stability, solubility and affinity are primarily influenced by the disulphide bond linkers in these molecules [19] . To construct the VH and VL genes, mRNA is isolate from a hybridoma, lymph cells or bone morrow. Then, the mRNA transcribed into DNA using reverse transcribe technology and antibody genes sequences template are then amplified by PCR. This technology requires oligonucleotide primers that can read most of antibodies sequences. This procedure has led to make large libraries that consist a wide range of antibody VH and VL gene sequence [20] .
Finally, the main advantage of using small fragment as antibody in experimental animals’ applications, is the easiness of the small fragment to enter the organ and fast clearance from tissues and blood stream [21] . Recombinant single chain fragments have connected with wide variety of particles, such as enzymes for pro-drug treatment, toxins for cancer treatment, viruses for gene therapy, and ligands for guiding to specific cellular targets [22] .
6. Application of scFv Antibodies
6.1. ScFv Antibodies as a Neutralising Agent
ScFv antibody is neutralising antibodies, which, they can connect with toxins and viruses and terminate them with high affinity. Additionally, scFv antibodies are smaller than parent antibodies that allows faster distribution in tissues and enhanced clearance of immuno-complexes from the blood stream. There are numbers of such cases in literature. For example, the scFv directed against F glycol-protein of respiratory-syncytial-virus, that decrease viral concentrations in the mice lungs, rabies-neutralising single chain fragment variable [23] . Moreover, wide range of neutralising single chain fragment attached to human cyto-megalo-virus glycol-proteins, isolated from a library of phage display. Furthermore, the Fab form of scFv has effective neutralising activities. The single chain fragments variable directed towards the hyper-variable loops of the Hepatitis-C-virus neutralising the C virus [24] .
6.2. ScFv Antibodies in Tumours Labelling and Bio-Therapeutics
The scFv antibodies labelled with radioisotope can be used to detect tumours in vivo. The ideal candidate tumours label that would allow faster distribution in cancer tissues and enhanced clearance from the blood stream, in the result has less contact of the healthy tissue [25] .
The advantage using of scFv antibody in tumours labelling is clear, for example when scFv antibody is fused to technetium-99 m [26] . Technetium is commonly used since it has short half-life of six-hour. This radiolabel agent is compatible with scFv antibody as carrier due to scFv antibody faster distribution in tissues and enhanced clearance of immuno-complexes from the blood stream. Moreover, scFvs or Fabs have worked successfully better, when they form complex molecule (multivalent) such as dimers, trimers, and tetramers. These complexes sequences of antibodies have advanced efficient affinity and were tried in many in vivo studies. In an earlier reported, a successful use of scFv antibody fused to chimeric-embryonic antigen (CEA) format resulted in rapid localisation to the tumour site, rapid clearance from the circulation and produced clear images.
In tumour treatment, immunotoxins are manufactured using antibodies connected to wide range of toxins, also the advantage of significant specificity of scFv antibodies in which they are connecting with cancers cells receptors with great affinity. ScFv antibodies fused to anti-cancer in various form for example cyto-toxic fragments, radio-nuclides or drugs. Gunter [27] showed that planed cancer treatment of human patient with specific scFv antibodies can handle highest drug treatment amount of 12.5 μg/kg. The tumour cells are terminated, when scFv antibody fused to cyto-toxins such as IL2 interlukin2 is attached to their receptors [28] .
6.3. ScFv Antibodies in Cancer Identification
The enormous use of scFv antibodies in identify cancer cell due to the antibodies specificity is demonstrated. The antibody can connect small fragment of the antigen or to small part of the cancer cell receptor. The scFv antibody normally constructed with tags fused to gene sequence, which they use to connect to the antibody conjugates. There are many tags in the market to be used for example: His-tags (Invitrogen) and S-tag (Merk). Still, scFv antibody face a problem of un-folding and non-soluble, when used in ELISA test, that because of its small less number of domains. To evade such problem, scFvs antibodies panned from phage display libraries can used in ELISAs, which scFv connected to the phage’s coat. This could increase activity of scFv protein since the phage can replace the heavy and light sequences [29] .
In other hand, constructed scFv fused with short tag sequences give the resulted protein extra activity and stability. There are many of these tags for example a constant light chain (CL), and alkaline phosphatase (AP) [29] . Furthermore, scFvs in two and three repeated complexes dimers for example divalent scFv or trivalent scFv, which gain better solubility and activity in protein 3D structure compared with original monovalent scFv [30] ). Finally, scFv-AP structure, won more advantage of direct connection with substrate without the need of conjugates.
7. Phage-Display Technologies
Phage display system is a technology that constructed gene and its fusion protein are connected, that gave wide range of molecular libraries selection, using antigen-specific binding and that become possible per excessive rounds of binding and washing between the target-specific and their antibodies in a procedure called bio-panning. The most popular surface was used to display is the M13KO7 bacterio-phage, described by Smith [29] .
7.1. The Phage-Mid Vector
The presentation of target-antibody on the surface of M13KO7 filamentous phage become possible with the help of phage display vectors as demonstrated by smith [29] . First, phage vectors keep all DNA information essential for the phage life-cycle [31] . The phage-mid is shorter sequence of M13KO7 used to display the antibodies in this technology [32] . The phage-mid sequence has M13KO7 middle area with its source of copying for virus strand combination, also, the hair-pin filler signal. The phage-mid also contains a source for vector copying and an antibiotics resistance.
This technology became accessible with the inventing of DNA plasmids that used selecting of target-specific antibody connected to M13KO7 phage surface in the pool of selected specific libraries. The connection of M13KO7 to the C-end of a DNA templates sequence has stop codon which, gained after special forward and reverse primers always contain translation stop codons and to avoid transcription and eventually expression of coat proteins in the beginning of connected plasmid. In M13KO7, phage-coat short amino acids sequence such as pIII and pVIII lead their forwards primers be transcript as the basic template sequence, whereas, the role of their c-end is necessary for effective phage expression as shown in Figure 2.
7.2. Bio-Panning
It is well known that bio-panning is greatly affected by the way the phage libraries coated to expose the target antigens. The coating of Target antigen is normally on immobilise magnetic surface such as ELISA magnetic wells. The specific phage receptors connected to the target antigen, and the unconnected phage is drained away. The targeted connected phage is then collected by disturbed the contacts among the phage sequence and immobilised target antigens. The improved phage is then transferred into bacteria host to grow in large scale, E. coli is one of bacterial hosts, to improve phage properties as shown in Figure 3. The bio-panning procedure could be done many times, the resulted phage- antibody fused would be accordingly gained significant specificity toward the selected target antigens. After many coating and washing process, the gene sequences from strongest phage reading should be DNA sequenced to match the result with the published gene bank libraries [33] .
The most import result from bio-panning amplification and washing procedures in phage-display is the wide range of selected numbers of target specific phage-antibody. The constructed target specific phage-antibody selected numbers can be more than 1010 [34] . The choice of selecting phage display is differ from tissue culture to sterilized mice. In tissue culture selection allows phage- antibody fusion to be coated against a various number of target antigens [35] .

Figure 3. Bio-panning is infection of bacteria with a filamentous helper phage and some incubation steps [41] .
When Yeast cell are used as target antigen, the exchanges between the fusion phage-antibody should be under specific functional environment; still, those conditions are difficult to apply in this technology, mainly in yeast cells [36] .
In other hand, phage-display selecting arrangements can be easily improved to work the screening environments and inflexibilities [37] . Both the yeast-hy- brid technologies and phage-display deliver a system capable of easy selection of specific phage-antibody in enamours scale directed toward certain targets, even though, phage-display has more quantity in favour. However, yeast hybrids could reach those enormous clones number in longer time compare to phage- display, whereas their clones would be selected in millions in not more than a week, whereas with yeast two-hybrids, millions of clones can be screened in two to four weeks [38] .
7.3. Phage-Antibody Fusion DNA Sequences
Phage-antibody fusion could be constructed with two different methods. First phage-antibody fusion constructed from spleen mRNA of immunised donors, the other type is derived from non-immunised donors. With the first library, the phage antibody repertoire is developed for antigen-specific antibodies because some have endured immune response from natural immune system [39] . The spleen mRNA of immunised donors’ yields stronger specification of phage-an- tibody than the one constructed from hybridoma [40] . For phage-antibody fusion pools, it is better to use a spleen sample with a higher serum antibody concentration of the target. Even though specific antibodies reflect higher levels of specific mRNA as shown in Figure 3.
The manufacturing of phage-antibodies pool is diversity of species of animal such as human, mice and rates has been demonstrated by Hoogenboom [41] . Moreover, the specificity of heavy and light chain fragments connected with linker directed towards target antigen is higher. However, phage-display libraries of single-library consider less specific and much low affinity. Therefore, count on the cause of the monoclonal antibodies DNA sequence is further differentiated in resulted antibody pools. The naïve pool of antibodies gives better specificity of phage-antibodies without the use for immunisation. V-sequence are constructed from B-cell cDNA template with VL and VH family primers as shown in Figure 4 [42] . The amplified heavy and light chains are linked together and amplified to produce an advanced pool of phage-scFv antibodies. This technology offers manufacturing to antibodies that did not directed towards antigen, while the number of those open “original antibodies” is dependent on B-cells source of construction. A huge pool of libraries can be needed to produce phage-antibodies fusion to many targets proteins, such as self, non-immuno- genic and quite dangerous targeted protein [43] .
The immature pool of antibody shall be enormous to contain almost the whole immune collection and permit isolation of target protein receptor with strong reaction as shown in Figure 4. The pool of immature pool of antibodies could be million or more, still most of it is hardly recover [44] . The reading of

Figure 4. Construction of recombinant scFv antibody using phage display technology [12] .
reaction of antibodies collected from the immature pool of antibody is goes per the number of the antibody pool, as such as 106-7 million for a small antibody pool, to 108-10 million for a large collection, constructed by phage display technology [45] . As mentioned earlier, the constructed pool of antibodies has been amplified using V gene sequence as template and VH a VL wide range of primers in RT-PCR are represented specially CDR3s [46] . The regions and degree of wide range of collected antibodies per points of largest species of the antibody selection. Ultimately, the value of any library is best determined by its success in yielding specific, high-affinity ligands for a variety of target molecules.
Still, antibodies with highest reaction can be constructed to connect with stronger specific antigen sequence by dimer sequences [47] ) or in vitro affinity maturation. The wide range antibody DNA sequences may be represented using different methods, such as mutator strains [48] , RT-PCR, chain scuffling; gene scuffling, and codon based mutator-genesis. The capability to change the binding reaction strength and specificity by developing monoclonal antibody source is influential development in antibody display technology. Phage-display has been mainly needed to mimic the target protein with antibodies pool, to describe epitopes for parent monoclonal antibodies, to increase antibody reaction, to isolate conjugate and to coat phage-antibody collections [49] .
Phage-display technologies have also been needed to give a clear idea of the 3Dimintional protein binding loop that mimicking the antibodies receptors and their insight limited [50] . Antibody-phage display is useful in that it permits the genuine display of scFvs, Fc sequences and diabody-sequences [51] .
8. Application of Phage Display Technology
8.1. Epitope Mapping of Antibodies
Phage display system could be utilized in various fields of study. This technology can be used to map specific antibody binding sites and is a simple and relatively low cost approach. Epitope mapping of antibodies, such as anti-HIVgp120 and HBsAg, has been successfully performed using random phage display peptide libraries. Specific epitopes of polyclonal antibodies against Nipah virus was also successfully mapped using a random peptide library of heptamers expressed on bacteriophage (M-13) [52] .
Phage display peptide libraries contain millions of foreign sequences, which represent a huge pool of antibody for a diversity of receptor ligands. Peptide selections from random peptide libraries are powerful tools for identifying receptor ligands without knowing its ligand structure. Affinity selections of random peptide libraries against receptors are a potential approach for new drug discovery. Furthermore, selected small cyclic peptides from a phage-antibody pool able to bind with activate target antigen the cytokine protein receptor is advanced. Hence, it can act as a full antagonist and may be a substitute for the larger natural hormone [53] .
8.2. Production of Vaccines and Diagnostics
To discover vaccines and diagnostics using phage display technology, antigenic and immunogenic mimicry are two important factors. Peptides selected against an antibody are antigenic mimics of the natural epitopes. If cross-reaction of the new antibodies is elicited against these antigenic mimics with the antigenic determinants of the natural epitopes, such peptides display both antigenic and immunogenic properties [52] . Sali [52] envisaged many uses for this technology such as production of malaria vaccine, which two independent libraries were panned for wide range of phage-antibody fusion expressed on the surface of bacteriophage with a mAb directed toward malarial antigen, their data showed high affinity of the antibody clones’ pool that reacted strongly with the mAb. Moreover, the DNA sequencing of the primers set combined with the templates of mAbs explained the high similarity of the selected clones.
Moreover, phage-display system was used to construct (scFv) antibodies that identification Clostridium difficile toxin B, which is a toxic type of Clostridium bacteria. Deng [54] produced a highly specific scFv antibody directed toward C- difficile toxin B that does not react with samples collected from a toxin B-negative C-difficile strain. The affinity of the soluble single-chain antibody is significantly higher than the parent monoclonal antibody as showed in ELISA test, and detect lower reading of 10 ng C-difficile B/well. Still, phage display system is a fast and actual advance technology for advance of new resulted immuno-diagnostic antibodies.
9. Expression of Recombinant scFv Protein in Prokaryotic and Eukaryotic Cells
E. coli is excessively used for the construction of antigens fragments, as well as antibody sequences, for example Fab or scFvs. Soluble scFvs are produced by expressing the target antigen DNA sequence toward the E. coli-periplasm, where the disulphide amino acids used to express the target antigen in soluble and active shape. The bacterial host manufacture scFv protein normally in small scale of 0.1 - 1 mg/L of yield, when the OD reach 600 of 1, however, the large scale of 10 mg/L can be achieved [55] .
However, scFvs active and soluble protein can be expressed in great amount in cytoplasmic space in E. coli after eliminating signal sequence from scFv DNA sequence [55] . To collect more folded and active protein from the cytoplasm of the cell, many laboratories had done alteration in the cytoplasmic oxido-redox environment by changing protein of the thiore-doxin and glutare-doxin synthesis [56] . Though, this modification results in production of folded intra-cytop- lasm-oxidised protein sequence. Also, the proper folded scFv protein produce in E. coli reducing-environment of the cytoplasm is the useful product to be manufacturing that inhibit virus and carry a toxin that lysis cancer cells. Besides, the use of intrabodies leads to many interesting possibilities for gene therapy and the in vivo study of protein function.
Accordingly, Modified E. coli cell called Origami (DE3) (Invitrogen) could produce 10 times much better soluble antibody compares with alternative hosts, even though, the production quantity is alike [56] . The Origami E. coli have designed thiore-doxin (trxB) beside gluta-thione (gor) gene sequences that activate the creation of disulphide poly peptide in the bacterial cytoplasm. Origami hosts are match with ampicillin-resistant vectors and are perfect with the pET-32 plasmid connected with the thiore-doxin fusion tag that would activate the creation of disulphide linker in the cytoplasm. The pET-32 b(+) vector is designed to produce protein that connected with the a Trx-Tag protein [55] . The multiple

Figure 5. Choosing the related structure and the step following of building correct 3D structure of protein, 3D structure of proteins from the same family is more conserved than their primary sequences [9] .
cloning site in the plasmid have His-Tag and S-Tag sequences for characterization and purification. Besides, the sequence referred to as pBR-32 resolution. For example, the T7 production site is inverted on the plasmid sequence. The functional genes areas also mapped on plasmid cycle. Besides, the DNA sequence of the plasmid copied by the T7 RNA-polymerase is described in Figure 5.
10. Protein Preparation Using Affinity-Metal-Chromatography
This system had sort of column which, purify the antibody (plasmid and insert target protein in frame) the plasmid also fused with specific tag that precisely bind to ligand manufactured in the solid part on an unsolvable backing called-matrix. The ligand holds 6x-his-tagged fragments which connects to the unsolvable backing-matrix (Ni2+) solid that can fuse to His-Bind mastics. After the non-reacted amino acids are drained, the bound amino-acids is captured by elution with imidazole. IMAC is a great system to capture specific target protein. IMAC used to precipitate and purify the scFv protein [55] .
For the capture of large amounts of target fragments, many methods have been advanced, as well as production of tagged amino-acids fragments, that capture the target specific protein to be purified by affinity chromatography [50] . However, many fusion proteins, such as glutathione-S-transferase or protein (A) are large proteins; therefore, there is a potential risk of mutation in protein sequence, because of the fusion partner. Additionally, the fusion protein is expressed in a native form for efficient isolation after affinity chromatography [50] .
Another successful approach for the purification of fusion proteins is the addition of six consecutive 6xHis-tag to the protein. The specificity of the histidine residues for solid matrix lets the protein-fused tag to hold away from unfused other proteins with high purity using metal chelate affinity chromatography. Because only six residues are fused to the protein, the risks of altering the properties of the protein of interest are minimised. Additionally, purification under denaturing conditions is possible. Several antibodies and chelator/enzyme conjugates are reported to detect histidine-tagged proteins. However, these are restricted to specific expression vectors or recognise histidine-tagged proteins with only reasonable affinity [52] .
11. Homology Protein Modelling
The use of homology modelling is to show three-dimensional protein and verify the CDRs as reported by Mahgoub [18] . Phage display can be used with molecular modelling and site-specific randomisation to advance the ways of capture the target antigens using Mabs. Prediction of three-dimension structure of target unknown protein is described. The structure would build per the highest sequence similarity to a correlated published known protein structure, which is called the template [56] . There are a few steps needed to be fallow to build a reliable model: 1) the chosen structure shall be when the proteins sequence alignment (target and templates) is correct; 2) template-target chosen with higher similarity. The structure prediction would be effected with any change in proteins (template-sequence alignment) sequences is possible since any difference in these sequences would result in totally different 3D structure as shown in Figure 5 also described by Lesk and Chothia [9] . It is also simplified the search for the best candidate 3D structure’s as the chosen model from related family sequences would be well-maintained than their own sequences (Lesk and Chothia [9] . Accordingly, the sequences alignment would show alike also the predicted 3D models will be similar. However, in some rare cases some poor aligned sequences would have a similar 3D structure. Mahgoub [15] predicted models become the reliable 3D models comparable structure with precise insight in compare to model prediction decided of wet laboratory experiment characterization of the protein affinity and specificity.
Single Chain Fragment Variables Antibody could be manufacture in divalent and trivalent complexes which are easier in folding and 4 times soluble than the mono-valet’s complexes. Also, large scale production of the antibody in yeast cells and mammalian cells hosts can be recommenced to further characterize the scFv antibody. Beside in bioinformatics direction, the EGFR model can be docked against the antibody molecule to visualize the binding sides and to produce minimized energy antibody structure that can used for stronger antigen antibody binding.
In conclusion: Single chain fragment variable is great addition to modern drug industry, which is tags to drugs and works as a diagnosis agent and a vaccine that disabled breast cancer antigens.
Cite this paper
Mahgoub, E.O. (2017) Single Chain Fragment Variables Antibody binding to EGF Receptor in the Surface of MCF7 Breast Cancer Cell Line: Application and Production Review. Open Journal of Genetics, 7, 84-103. https://doi.org/10.4236/ojgen.2017.72008
References
- 1. Howell, A., DeFriend, D. and Anderson, E. (1993) Mechanisms of Response and Resistance to Endocrine Therapy for Breast Cancer and the Development of New Treatments. Reviews on Endocrine-Related Cancer, 43, 5-21.
- 2. Kuan, C.T., Wikstrand, C.J. and Bigner, D.D. (2001) EGF Mutant Receptor vIII as a Molecular Target in Cancer Therapy. Endocrine-Related Cancer, 8, 83-96.
https://doi.org/10.1677/erc.0.0080083 - 3. Tang, C.K., Gong, X.Q., Moscatello, D.K., Wong, A.J. and Lippman, M.E. (2000) Epidermal Growth Factor Receptor vIII Enhances Tumorigenicity in Human Breast Cancer. Cancer Research, 60, 3081-308.
- 4. Wikstrand, C.J, McLendon, R.E., Friedman, A.H. and Bigner, D.D. (1997) Cell Surface Localization and Density of the Tumor-Associated Variant of the Epidermal Growth Factor Receptor, EGFRvIII. Cancer Research, 57, 4130-4140.
- 5. Reist, C.J., Batra, S.W., Pegram, C.N., Bigner, D.D. and Zalutsky, M.R. (1997) In Vitro and in Vivo Behavior of Radiolabeled Chimeric Anti-EGFRvIII Monoclonal Antibody: Comparison with Its Murine Parent. Nuclear Medicine and Biology, 24, 637-647.
https://doi.org/10.1016/S0969-8051(97)00080-2 - 6. Spiegelberg, H.L. (1974) Biological Activities of Immunoglobulins of Different Classes and Subclasses. Advances in Immunology, 19, 259-294.
- 7. Sivasubramanian, A., Sircar, A., Chaudhury, S. and Gray, J.J. (2009) Toward High-Resolution Homology Modeling of Antibody Fv Regions and Application to Antibody-Antigen Docking. Proteins, 74, 497-514.
https://doi.org/10.1002/prot.22309 - 8. Cohen-Solal, J.F.G., Cassard, L., Fridman, W. and Sautès-Fridman, C. (2004) Fc Gamma Receptors. Immunology Letters, 92, 199-205.
- 9. Lesk, A.M. and Chothia, C. (1982) Evolution of Proteins Formed by Beta-Sheets. II. The Core of the Immunoglobulin Domains. Journal of Molecular Biology, 160, 325-342.
- 10. Brambell, F.W., Hemmings, W.A. and Morris, I.G.A. (1964) Theoretical Model of Gamma-Globulin Catabolism. Nature, 203, 1352-1354.
https://doi.org/10.1038/2031352a0 - 11. Verma, R., Boleti, E. and George, A.J.T. (1998) Antibody Engineering: comparison of Bacterial, Yeast, Insect and Mammalian Expression Systems. Journal of Immunological Methods, 216, 165-181.
- 12. Pluckthun, A., et al. (1996) Producing Antibodies in Escherichia coli: From PCR to Fermentation. In: McCafferty, J., Hoogenboom, H.R. and Chiswell, D.J., Eds., Antibody Engineering: A Practical Approach, IRL Press, Oxford, 203-252.
- 13. Roopenian, D.C. and Akilesh, S. (2007) FcRn: The Neonatal Fc Receptor Comes of Age. Nature Reviews Immunology, 7, 715-725.
- 14. Shimada, N., Suzuki, Y., Nakajima, M., Conrad, U., Murofushi, N. and Yamaguchi, I. (1999) Expression of a Functional Single Chain Antibody against GA24/19 in Transgenic Tobacco. Bioscience, Biotechnology, and Biochemistry, 63, 779-783.
https://doi.org/10.1271/bbb.63.779 - 15. Elham, M.O. and Bolad, A.K. (2014) Construction and Expression of Single-Chain Variable Fragment (ScFv) against MCF7 Breast Cancer Cells by Phage Display Technology. Journal of Cancer Research and Therapeutics, 10, 265.
- 16. Reiter, Y., Brinkmann, U., Webber, K.O., Jung, S.-H., Lee, B. and Pastan, I. (1994) Engineering Interchain Disulfide Bonds into Conserved Framework Regions of Fv Fragments: Improved Biochemical Characteristics of Recombinant Immunotoxins Containing Disulfide-Stabilized Fv. Protein Engineering, 7, 667-704.
https://doi.org/10.1093/protein/7.5.697 - 17. Chua, K.H. (2003) Bioinformatics in Molecular Immunology Laboratories Emonstrated Modeling an Anti-CMV scFv Antibody. Asia-Pacific Journal of Molecular Biology and Biotechnology, 11, 93-96.
- 18. Mahgoub, E.O. and Bolad, A. (2013) Correctness and Accuracy of Template-Based Modeled Single Chain Fragment Variable (scFv) Protein Anti-Breast Cancer Cell Line (MCF-7). Open Journal of Genetics, 3, 183-194.
https://doi.org/10.4236/ojgen.2013.33021 - 19. Douglas, J.T. and Curiel, D.T. (1997) Strategies to Accomplish Targeted Gene Delivery to Muscle Cells Employing Tropism-Modified Adenoviral Vectors. Neuromuscular Disorders, 7, 284-298.
https://doi.org/10.1016/S0960-8966(97)00053-9 - 20. Nejatollahi, F., Hodgetts, S.J., Vallely, P.J. and Burnie, J.P. (2002) Neutralizing Human Recombinant Antibodies to Human Cytomegalovirus Glycoproteins gB and gH. FEMS Immunology and Medical Microbiology, 34, 237-244.
https://doi.org/10.1111/j.1574-695X.2002.tb00630.x - 21. Hudson, P.J. and Souriau, C. (2001) Recombinant Antibodies for Cancer Diagnosis and Therapy. Expert Opinion on Biological Therapy, 1, 845-855.
https://doi.org/10.1517/14712598.1.5.845 - 22. Pai, L.H. and Pastan, I. (1998) Clinical Trials with Pseudomonas Exotoxin Immunotoxins. In: Frankel, A.E., Ed., Clinical Applications of Immunotoxins, Springer, Berlin, Heidelberg, 83-96.
https://doi.org/10.1007/978-3-642-72153-3_6 - 23. Liu, S.Y., Eary, J.F., Petersdorf, S.H., Martin, P.J., Maloney, D.G., Appelbaum, F.R., Matthews, D.C., Bush, S.A., Durack, L.D., Fisher, D.R., Gooley, T.A., Bernstein, I.D. and Press, O.W. (1998) Follow-Up of Relapsed B-Cell Lymphoma Patients Treated with Iodine-131-Labeled Anti-CD20 Antibody and Autologous Stem-Cell Rescue. Journal of Clinical Oncology, 16, 3270-3278.
https://doi.org/10.1200/JCO.1998.16.10.3270 - 24. Chari, R.V. (1998) Targeted Delivery of Chemotherapeutics: Tumor-Activated Prodrug Therapy. Advanced Drug Delivery Reviews, 31, 89-104.
- 25. Gunter, M., Harder, S., Sascha, H., Jager, E., Al-Batran, S., Sibylle, L., Atmaca, A., Cimpoiasu, C., Neumann, A., Abera, A., Knuth, A., Kaufmann, M., Dirk, J., Alexander, B. and Maurer, W.S. (2005) Clinical Study of the Recombinant Antibody Toxin scFv(FRP5)-ETA Specific for the ErbB2/HER2 Receptor in Patients with Advanced Solid Malignomas. Breast Cancer Research, 7, R617-R626.
https://doi.org/10.1186/bcr1264 - 26. Mahgoub, I.O. (2012) Expression and Characterization of a Functional Single-Chain Variable Fragment (scFv) Protein Recognizing MCF7 Breast Cancer Cells in E. coli Cytoplasm. Biochemical Genetics, 50, 625-641.
https://doi.org/10.1007/s10528-012-9506-4 - 27. Griep, R.A., Prins, M., Van Twisk, C., Keller, H.J.H.G., Kerschbaumer, R.J., Kormelink, R., Goldbach, R.W. and Schots, A. (2000) Application of Phage Display in Selecting Tomato Spotted Wilt Virus-Specific Single-Chain Antibodies (scFvs) for Sensitive Diagnosis In ELISA. Phytopathology, 90, 183-190.
https://doi.org/10.1094/PHYTO.2000.90.2.183 - 28. Berek, C. and Milstein, C. (1988) The Dynamic Nature of the Antibody Repertoire. Immunological Reviews, 105, 5-26.
https://doi.org/10.1111/j.1600-065X.1988.tb00763.x - 29. Smith, G.P. (1985) Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science, 228, 1315-1317.
https://doi.org/10.1126/science.4001944 - 30. Sparks, A.B., Ade, N.B., Cwirla, S., Kay, B.K., Winter, J. and McCafferty, J. (1996) Screening Phage-Displayed Random Peptide Libraries. In: Phage Display of Peptides and Proteins, Academic Press, Cambridge, MA, 227-253.
- 31. McCafferty, J. and Johnson, K.S. (1996) Construction and Screening of Antibody Display Libraries. In: Kay, B.K., Winter, J. and McCafferty, J., Eds., Phage Display of Peptides and Proteins, Academic Press, Cambridge, MA, 79-111.
- 32. Whaley, S.R., English, D.S., Hu, E.L., Barbara, P.F. and Belcher, A.M. (2000) Selection of Peptides with Semiconductor Binding Specificity for Directed Nanocrystal Assembly. Nature, 405, 665-668.
https://doi.org/10.1038/35015043 - 33. Chowdhury, P.S., Viner, J.L., Beers, R. and Pastan, I. (1998) Isolation of a High-Affinity Stable Single-Chain Fv Specific for Mesothelin from DNA-Immunized Mice by Phage Display and Construction of a Recombinant Immunotoxin with Anti-Tumor Activity. Proceedings of the National Academy of Sciences of the United States of America, 95, 669-674.
https://doi.org/10.1073/pnas.95.2.669 - 34. Chester, K.A., Begent, R.H., Robson, L., Keep, P., Pedley, R.B., Boden, J.A., Boxer, G., Green, A., Winter, G. and Cochet, O. (1994) Phage Libraries for Generation of Clinically Useful Antibodies. The Lancet, 343, 455-456.
https://doi.org/10.1016/S0140-6736(94)92695-6 - 35. Barbas, C.F, Burton, D.R. and Silverman, G.J. (2004) Phage Display: A Laboratory Manual. Cold Spring Harbor Laboratory Press.
- 36. Vaughan, T.J., Williams, A.J., Pritchard, K., Osbourn, J.K., Pope, A.R., Earnshaw, J.C., McCafferty, J., Hodits, R.A., Wilton, J. and Johnson, K.S. (1996) Human Antibodies with Sub-Nanomolar Affinities Isolated from a Large Non-Immunized Phage Display Library. Nature Biotechnology, 14, 309-314.
https://doi.org/10.1038/nbt0396-309 - 37. Ellman, J.A. and Gallop, M.A. (1998) Combinational Chemistry. Current Opinion in Chemical Biology, 2, 317-319.
- 38. Hoogenboom, H.R. and Chames, P. (2000) Natural and Designer Binding Sites Made by Phage Display Technology. Immunology Today, 21, 371-378.
https://doi.org/10.1016/S0167-5699(00)01667-4 - 39. Hoogenboom, H.R., de Bruïne, A.P., Hufton, S.E., Hoet, R.M., Arends, J.W. and Roovers, R.C. (1998) Antibody Phage Display and Its Applications. Immunotechnology, 4, 1-20.
- 40. Low, N.M., Holliger, P. and Winter, G. (1996) Mimicking Somatic Hypermutation: Affinity Maturation of Antibodies Displayed on Bacteriophage Using Bacterial Mutator Strain. Journal of Molecular Biology, 260, 359-368.
- 41. Clackson, T., Hoogenboom, H.R., Griffiths, A.D. and Winker, G. (1991) Making Antibody Fragments Using Phage Display Libraries. Nature, 352, 624-628.
https://doi.org/10.1038/352624a0 - 42. Marks, J.D., Griffiths, A.D., Malmqvist, M., Clackson, T.P., Bye, J.M. and Winter, G. (1992) Bypassing Immunization: Building High Affinity Human Antibodies by Chain Shuffling. Biotechnology, 10, 779-783.
- 43. Stemmer, W.P. (1994) Rapid Evolution of a Protein in Vitro by DNA Shuffling. Nature, 370, 389-391.
https://doi.org/10.1038/370389a0 - 44. McCafferty, J., Griffiths, A.D., Winter, G. and Chiswell, D.J. (1990) Phage Antibodies: Filamentous Phage Displaying Antibody Variable Domains. Nature, 348, 552-554.
https://doi.org/10.1038/348552a0 - 45. Ritz, D., Lim, J., Reynolds, C.M., Poole, L.B. and Beckwith, J. (2001) Conversion of a Peroxiredoxin into a Disulfide Reductase by a Triplet Repeat Expansion. Science, 294, 158-160.
https://doi.org/10.1126/science.1063143 - 46. LaVallie, E.R., et al. (1993) A Thioredoxin Gene Fusion Expression System that Circumvents Inclusion Body Formation in the E. Coli Cytoplasm. Nature Biotechnology, 11, 187-193.
- 47. Chames, P. and Baty, D. (2000) Antibody Engineering and Its Applications in Tumor Targeting and Intracellular Immunization. FEMS Microbiology Letters, 189, 1-8. https://doi.org/10.1111/j.1574-6968.2000.tb09197.x
- 48. Blundell, T.L., Sibanda, B.L., Sternberg, M.J. and Thornton, J.M. (1987) Knowledge-Based Prediction of Protein Structures and the Design of Novel Molecules. Nature, 326, 347-352.
https://doi.org/10.1038/326347a0 - 49. Greer, J. (1981) Comparative Model-Building of the Mammalian Serine Proteases. Journal of Molecular Biology, 153, 1027-1042.
- 50. Johnson, M.S., Srinivasan, N., Sowdhamini, R. and Blundell, T.L. (1994) Knowledge-Based Protein Modelling. Critical Reviews in Biochemistry and Molecular Biology, 29, 1-68.
https://doi.org/10.3109/10409239409086797 - 51. Sali, A. and Blundell, T.L. (1993) Comparative Protein Modelling by Satisfaction of Spatial Restraints. Journal of Molecular Biology, 234, 779-815.
- 52. Sali, A. (1995) Modeling Mutations and Homologous Proteins. Current Opinion in Biotechnology, 6, 437-451.
- 53. Sanchez, R., Pieper, U., Melo, F., Eswar, N., Marti-Renom, M.A., Madhusudhan, M.S., Mirkovic, N. and Sali, A. (2000) Protein Structure Modeling for Structural Genomics. Nature Structural & Molecular Biology, 7, 986-990.
https://doi.org/10.1038/80776 - 54. Deng, X.K., Nesbit, L.A. and Morrow Jr., K.J. (2003) Recombinant Single-Chain Variable Fragment Antibodies Directed against Clostridium difficile Toxin B Produced by Use of an Optimized Phage Display System. Clinical and Diagnostic Laboratory Immunology, 10, 587-595.
- 55. Marti-Renom, M.A., Stuart, A.C., Fiser, A., Sanchez, R., Melo, F. and Sali, A. (2000) Comparative Protein Structure Modeling of Genes and Genomes. Annual Review of Biophysics and Biomolecular Structure, 29, 291-325.
https://doi.org/10.1146/annurev.biophys.29.1.291 - 56. Fiser, A., Do, R.K. and Sali, A. (2000) Modeling of Loops in Protein Structures. Protein Science, 9, 1753-1773.
https://doi.org/10.1110/ps.9.9.1753
List of Abbreviation
AP alkaline phosphatase
CL constant light domain
CH1 constant heavy domain 1
CH2 constant heavy domain 2
CH3 constant heavy domain 3
CH4 constant heavy domain 4
CDRs complementary determining regions
CEA c himeric anti?carcino-embryonic antigen
cDNA Complementary determine regions
CDC Complement dependent cytotoxicity
C3A8 mouse B-cell hybridoma cell line
CMV cucumber Mosaic Virus
dNTP Deoxyribonucleotide
EGF epidermal growth factor
EGFR epidermal growth factor receptor
ELISA Enzyme-linked immunosorbent assay
ERRAT novel structure evaluation programme
Fab Fragment antigen binding
Fc F ragment crystalline
GlY Glycine
6x his-tag 6x histidine-tagged
IgG Immunoglobulin G
IgA Immunoglobulin A
IgM Immunoglobulin M
IgE Immunoglobulin E
IgG Immunoglobulin G
IL2 Interleukin2
IMAC Immobilized Metal Affinity Chromatography
gB Glycol proteinB
gH Glycol protein
HIV Human immunodeficiency virus
HBsAg Hepatitis B virus surface antigen
Kg Kilo gram
MCF-7 Michigan cancer foundation-7
M13KO7 helper phage
mM Milimolar
ml microlitre
mg microgram
Mab Monoclonal antibody
MR1 affinity-matured derivative-1
mRNA messenger ribonucleic acid
PCR Polymerase chain reaction
RT-PCR Reverse Transcriptase-Polymerase chain reaction
protein 3D Protein three dimensional
RE Restriction endonuclease
scFv Single Chain Fragment Variable
Ser serene
trxB thioredoxin reductase part B
OD Optical Density
VL Hyper variable light chain
VH Hyper variable heavy chain

Submit or recommend next manuscript to SCIRP and we will provide best service for you:
Accepting pre-submission inquiries through Email, Facebook, LinkedIn, Twitter, etc.
A wide selection of journals (inclusive of 9 subjects, more than 200 journals)
Providing 24-hour high-quality service
User-friendly online submission system
Fair and swift peer-review system
Efficient typesetting and proofreading procedure
Display of the result of downloads and visits, as well as the number of cited articles
Maximum dissemination of your research work
Submit your manuscript at: http://papersubmission.scirp.org/
Or contact ojgen@scirp.org