Advances in Biological Chemistry
Vol.3 No.3(2013), Article ID:32390,7 pages DOI:10.4236/abc.2013.33032
Role of amino acid residues involved in the active cavity of proline iminopeptidase in catalytic activity
![]()
The Key Laboratory for Bio-Resources and Eco-Environment of Ministry of Education, The Sichuan Key Laboratory of Molecular Biology and Biotechnology, College of Life Sciences, Sichuan University, Chengdu, China
Email: *hfeng@scu.edu.cn
Copyright © 2013 Kangkang Xing, Hong Feng. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 7 April 2013; revised 10 May 2013; accepted 18 May 2013
Keywords: Proline Iminopeptidase; Site-Directed Mutagenesis; Oxyanion Hole; Active Cavity; Catalytic Kinetics; Phanerochaete chrysosporium
ABSTRACT
The proline iminopeptidase (PchPiPA) of the whiterot fungi Phanerochaete chrysosporium is an exopeptidase specific to catalyze hydrolysis of the N-terminal proline of peptides or proteins. Its catalytic cavity is comprised of a catalytic triad (Ser107, Asp264 and His292) and an oxyanion hole (His38, Gly39, Gly40 and Pro41). In this work, several amino acid residues involved in the catalytic cavity were selected for investigation of their influences on the catalytic activity by site-directed mutagenesis. It was shown that mutation of residues (Gly39 and Gly40) involved in oxyanion hole resulted in almost complete loss of catalytic activity largely due to changes in kcat. The other residues (Gly42 and Cys45) lined at the entrance of the active cavity also yielded a profound negative effect on the activity. Mutation of the other two residues Arg130 and Gly131 which were flanked spatially by the nucleophilic attacking active site of Ser107, caused different effects on the activity. R130A increased catalytic efficiency due to changes in both kcat and Km; while G131V decreased the value of kcat/Km mainly due to changes in kcat. And T111A also caused a negative effect on the kcat. Conclusively, these amino acid residues involved in active cavity were more susceptible to be negatively affected by mutation, suggested that the active cavity of proline iminopeptidase might evolve to be less plausible.
1. INTRODUCTION
Proline iminopeptidase (PiP, prolyl aminopeptidase; EC 3.4.11.5) is a special exopeptidase, which catalyzes cleavage of proline from the N-terminus of peptides [1]. PiP was first reported in Escherichia coli in 1959 [2]. Since then, many PiPs have been cloned and characterized from a variety of living organisms, such as bacteria [3-6], fungi [7-9], and plants [10,11]. In some bacteria, PiP activities were related to pathogenicity [12,13]. For example, disruption of the gene encoding PiP resulted in significantly attenuated virulence in Xanthomonas campestris pv. campestris [12]. In plants, PiPs may participate in the response to abiotic stress by turning over oxidatively damaged proteins or by taking part in the regulation of free amino acids pools [10,14]. In view of the industry, PiPs can be used as biocatalyst in food processing, such as for debittering of proteolysates and flavor development of Swiss-type cheeses [15,16]. In addition, PiP was also employed for synthesis of the synthesis of dipeptides, especially proline-containing peptide [17,18].
Based on the biochemical properties and sequence analysis, PiP is a serine peptidase and classified into family S33 in the MEROPS database [19]. The threedimensional structure of two PiPs from bacteria has been reported [20,21]. Overall, the structure is composed of two contiguous domains. The larger domain shows the general topology of a typical α/β hydrolase fold with a central eight-stranded β-sheet. The smaller domain is placed on top of the larger domain and essentially consists of six helices. The active cavity is consisted of a catalytic triad (Asp-Ser-His), which is located at the end of a deep pocket at the interface between these two domains. It is believed that the serine and histidine residues are directly involved in the catalytic reaction, serving as the nucleophilic attacking group and general acidbase catalytic elements, respectively. There is also an oxyanion hole involved in the active cavity, which is recognized to be important in catalysis due to acting as hydrogen bond donors to stabilize the oxyanion intermediate. However, previous studies with the PiP from Serratia marcescens have focused on the substrate recognition and identified several residues involved in substrate recognition by site-directed mutagenesis and structural analysis [22-24].
In our previous work, a PiP (PchPiPA) was cloned and characterized biochemically from the white-rot fungus Phanerochaete chrysosporium [7]. Based on its amino acid sequence, PchPiPA belongs to the subfamily S33.001, and it is somehow different from the other two fungal PiPs which have been characterized biochemically. In order to get more insights on the catalytic mechanism, site-directed mutagenesis was performed on the amino acid residues surrounding to the catalytic triad and oxyanion hole. The results showed that these amino acid residues were more susceptible to mutation, suggesting the PchPiPA may contain a relative rigid catalytic cavity.
2. MATERIALS AND METHODS
2.1. Materials and Bacterial Strains
All chemicals were reagent grade commercial products and used without further purification. L-proline-p-nitroanilide trifluoacetate (Pro-pNA) was purchased from Sigma-Aldrich. Escherichia coli strain JM109 served as the host for cloning. Escherichia coli BL21 (DE3) was used for gene expression.
2.2. Site-Directed Mutagenesis
The mutagenesis was performed following QuikChange site-directed mutagenesis method (Stratagene) according to manufacturer’s instruction with minor modification. The mutagenesis PCR was carried out using Pfu DNA polymerase in 25 μl mixture with pET28PiPA [7] as template. The primers used for site-directed mutagenesis were presented in Table 1. PCR reaction were carried out as follows: an initial denaturation at 94˚C for 3 min; followed by 25 cycles including annealing for 30 s at 94˚C and extension at 68˚C for 8 min; and a final extension for 10 min at 68˚C. The PCR products were digested with Dpn I at 37˚C for 1 h to remove the parent template and then transformed into E. coli JM109. The desired mutations were confirmed by DNA sequencing. Subsequently, the mutated plasmids of were transformed into the E. coli BL21 (DE3).
2.3. Protein Expression and Purification
Single colony of E. coli BL21 (DE3) which hosted the desired recombinant plasmid was inoculated in 3 ml LB liquid medium supplement with 50 μg/ml kanamycin. After shaking at 37˚C and 200 rpm overnight, 250 μl of the culture was transferred into 250 ml LB medium containing kanamycin (50 μg/ml). Under the same cultivat-

Table 1. Primers used for site-directed mutagenesis.*
ing condition, 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) was added to the culture after the cell density reaching 0.6 at OD600. The culture was then transferred to 18˚C and incubated for additional 16 h. The cells were harvested by centrifugation at 4000 rpm for 10 min at 4˚C. And the pellets were suspended in 25 ml TNPG buffer (20 mM Tris-HCl, pH 7.5, 250 mM NaCl, 0.5 mM PMSF, 10% glycerol). And the cells were broken by ultrasonic cell crushing apparatus (the parameter was set as: output 35%, ultrasonic pulse 8 seconds, stop pulse 5 seconds) at 4˚C for 10 min. The cell debris was removed by centrifugation at 10,000 rpm for 10 min at 4˚C.
The recombinant protein was purified by Ni2+-affinity chromatography. The supernatant was applied onto the 1.0 ml of Chelating Sepharose column (AmershamPharmacia), and washed with 5× column volume (CV) of buffer A (0.1 M Tris-HCl, pH 7.5, 250 mM NaCl, 10% glycerol). The bound proteins were eluted manually with 5 ml of the buffer B (Buffer A plus various concentration of imidazole) at flow rate of 1.0 ml/min. The fractions eluted from 200 - 300 mM imidazole were pooled and dialyzed against 1000 ml of dialysis buffer (50 mM TrisHCl, pH 7.5, 1 mM DTT, 0.1 mM EDTA-Na2) at 4˚C overnight. Finally, the recombinant protein was concentrated using PEG-2000 and kept in storage buffer [100 mM Tris-HCl, pH 7.5, 50% (v/v) glycerol] at −20˚C. Protein purity was monitored by SDS-PAGE (12%) and the concentration was quantified using AlphaImager software (Alpha Innotech Corporation, San Leandro, USA) with bovine serum albumin (BSA) as a Standard. All the mutated enzymes were diluted to 0.5 μM with adequate amount of storage buffer.
2.4. Activity Assays
The PiP activity was determined by monitoring the released nitroanilide from substrate Pro-pNA. For activety assay, 700 μl reaction mixture (100 mM Tris-HCl, pH 8.0) containing 0.5 mM or indicated amount of Pro-pNA was pre-incubated at 45˚C for 2 min, and then proper amount of each PchPiPA WT or mutant were added into the mixture to start the reaction. The absorbance at 410 nm over 10 min was recorded continuously on line in the spectrophotometer UV-2450 (Shimadzu, Japan). For determine the kinetic parameters, a range of substrate concentrations (0.5 mM to 1.2 mM) were applied. All the assays were performed at least three times.
2.5. Determination of Thermostability
The WT and mutant enzymes were diluted into 100 mM Tris-HCl buffer pH 8.0, and then incubated at 50˚C for exact 20 min. The residual activity was assayed as above. For evaluation of thermostability, percentage of the residual activity after heat treatment to the initial activity was adopted.
3. RESULTS
3.1. Construction and Protein Expression of PchPiPA Mutants
The proline iminopeptidase (PchPiPA) from Phanerochaete chrysosporium belongs to the subfamily S33.001, and their catalytic triad (Ser107-Asp264-His292) has been confirmed with site-directed mutagenesis [7]. In view of its structure (data not shown), the general topology of PchPiPA is similar with the other two PiPs from bacteria of Xanthomonos campestris pv. citri and Serratia marcescens, respectively [20,21], which was organized into two domains. The larger domain is composed of a typical α/β hydrolase fold with a central eight-stranded β-sheet. The smaller domain is placed on top of the larger domain and essentially consists of six helices. The active sites, located at the end of a deep pocket at the interface between both domains, are consisted of a catalytic triad (Ser107-Asp264-His292 in PchPiPA). A conserved oxyanion hole (His38-Gly39-Gly40-Pro41 PchPiPA) forms a turn with the amide group of Gly39 and Gly40 flanking toward the active site Ser107. The spatial arrangement of active cavity with their flanked residues was illustrated in Figure 1.
In this work, eight amino acid residues were chosen for site-directed mutagenesis. The corresponding mutants were constructed using the QuikChange mutagenesis method. After the desired mutations confirmed by DNA

Figure 1. Spatial arrangement of active sites and oxyanion hole in the active cavity of PchPiPA. The catalytic triad is consisted of Asp264, His292 and Ser107; the oxyanion hole is comprised of His38, Gly39, Gly39 and Pro41. The amide groups of Gly39 and Gly40 are flanked spatially towards the nucleophilic attacking group of Ser107, which is interacting with the Thr111 (behind the helix) and Arg130 via formation of hydrogen bonds.
sequencing, these plasmids were transformed and expressed in E. coli BL21 (DE3) cells by addition of IPTG. All the mutants as well as the wild-type were well expressed under the experimental conditions. The recombinant proteins were purified using Ni2+-affinity chromatography. The purified proteins were evaluated on SDS-PAGE (12%) with purity > 80% (Figure 2). The concentration was quantified using AlphaImager software with bovine serum albumin (BSA) as a standard.
3.2. Effect of the PchPiPA Mutants on Catalytic Activity
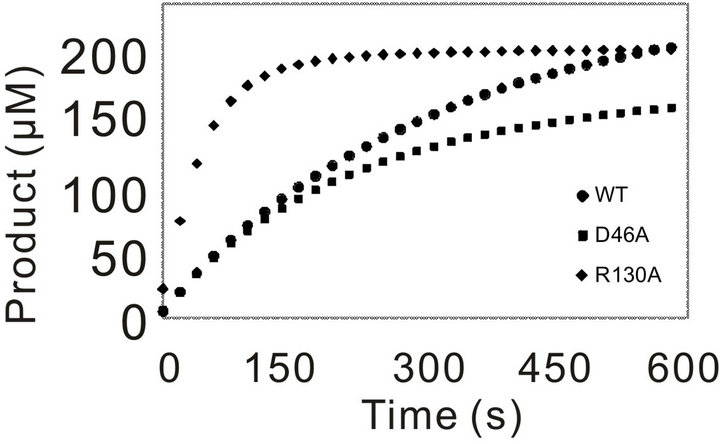
Initially, the activity of WT and the mutants was assayed using 3.57 nM of enzyme for WT and each mutants and 0.5 mM of Pro-pNA as substrate. In compareson with WT, mutation of the residues (G39) leaded to complete loss of activity towards the substrate Pro-pNA, even with the increased amount of mutation enzyme (data not shown). The progressing curves for the other mutants and WT were shown in Figure 3. R130A showed increased activity, and D46A behaved similar
Figure 2. Purification and expression of the mutation proteins of PchPiPA.
 (a)
(a)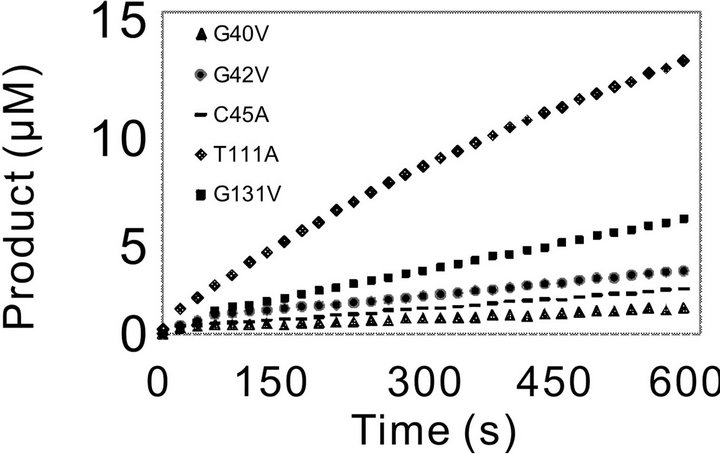 (b)
(b)
Figure 3. Catalytic progressing curves of PchPiPA and mutants towards PropNA. The catalytic reaction was assayed in 700 μl of 100 mM Tris-HCl buffer (pH 8.0) containing 3.75 nM of each mutant or WT enzyme and 0.5 mM of substrate Pro-pNA at 45˚C.
with the WT (Figure 3(a)). However, the catalytic activity of the other five mutants (Gly40, G42V, C45A, T111A and G131V) decreased much significantly (Figure 3(b)).
3.3. Effects of PchPiPA Mutants on Catalytic Kinetics
In order to get insights on effect of the mutations on catalytic mechanism, kinetic parameters were determined for each mutant except for the function-loss mutant (G39A). The parameters of Km, kcat, and kcat/Km were summarized in Table 2.
The kcat/Km values of G40V, G42V decreased by about 3084 and 1358-fold, which was largely due to change in kcat. C45A showed the similar kinetic behavior with G40V and G42V, with a decrease in kcat by about 2305- fold. For the above three mutants, Km value did not change obviously. However, replacement of Asp46 with Ala did not result in obvious change in these parameters.
Thr111 is a conserved residue in subfamily S33, which may interact with Ser107 via hydrogen bond linkage based on the structural analysis in PchPiPA. Its mutation caused a significant decrease in catalytic efficacy (kcat/ Km), largely due to change in kcat. Both of Arg130 and Gly131 are not conserved across the subfamily S33 and have been recognized as important functional residues [21,23]. The kcat/Km value of R130A increased by more than 3-fold due to change in both Km and kcat. However, mutation of Gly131 yielded greatly negative impact on the kinetic parameters, with significant decrease of kcat/ Km value to about 97-fold mainly due to change of kcat.
3.4. Effect of PchPiPA Mutants on Thermostability
Furthermore, the effect of these mutations on thermostatbility was determined. The percentage of residual activity
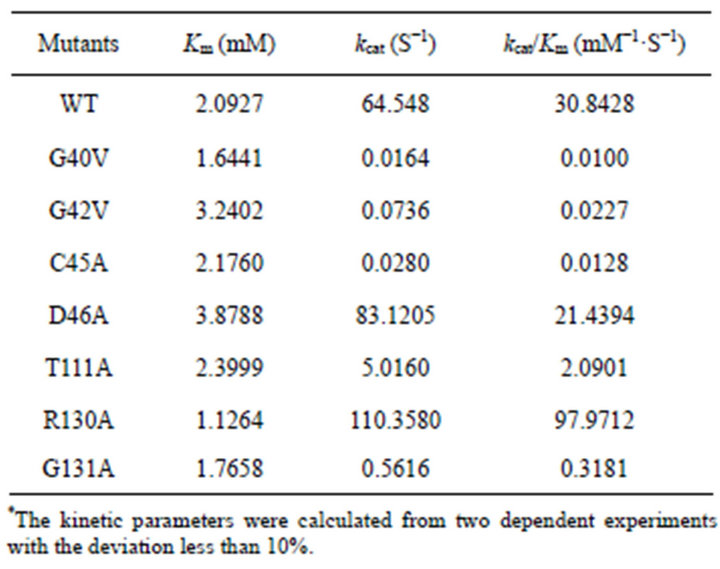
Table 2. Kinetics parameters of PchPiPA mutants and WT.*
after treat treatment in comparison to the initial activity was employed for evaluation of thermostability. The data were shown in Table 3.
After heat treatment, catalytic activity was reduced at different extent for WT and all the mutants. The residual activity of WT decreased by 33.23% in comparison to the initial activity after heat treatment at 50˚C for 10 min. Therefore, D46A, T111A, or R130A became more heatlabile than WT, with only residual activity of 10.44%, 15.63% or 10.16%, separately. It is noted that the thermostability of G131V increased significantly, which retained 92.16% of the initial activity after heat treatment. However, the other mutants like G40V, G42V, and G45V showed very low activity after heat treatment.
4. DISCUSSION
Previously, a proline iminopeptidase named as PchPiPA was biochemically characterized from the white-rot basidiomycetes Phanerochaete chrysosporium [7]. Its catalytic cavity was comprised of a conserved triad (AspSer107-His292) and an oxyanion hole (His39-G40-G41- Pro42). And all of the related residues were located on the interface between the cap domain and the α/β fold domain. In this work, sited-directed mutagenesis was performed to figure out the role of these amino acid residues on the catalytic activity.
In PiPs, the catalytic triad was confirmed by site-directed mutagenesis and the mutations caused complete loss of catalytic function [7,25,26]. These results indicated that each of the three active sites was absolute for catalytic activity. In addition, the nucleophilic Ser residue, forming a nucleophilic elbow on a sharp that turned to connect strand 5 and helix C, was embedded in a conserved motif (G-X-S-B-G-Z) [19]. Mutation of the corresponding residues of Gly105, G109, and G110 in this motif of another PiP (PepIP) of Lactobacillus delbruckii subsp. bulgaricus resulted in almost complete loss of
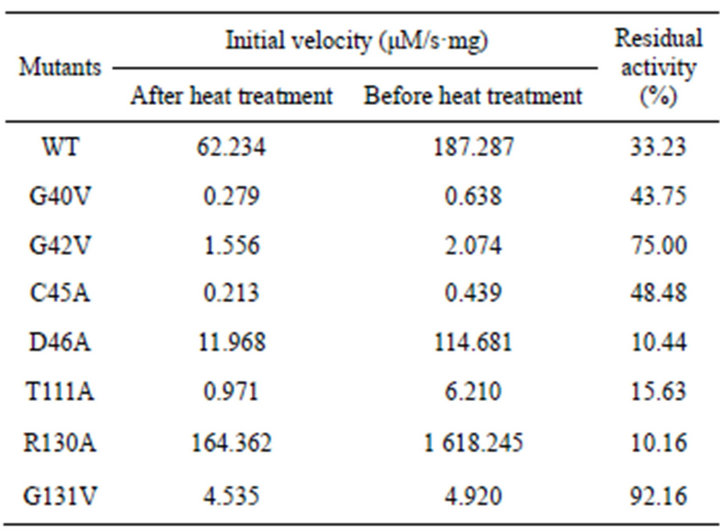
Table 3. Comparison of thermostability of PchPiPA mutants and WT by initial velocity after heat treatment at 50˚.
catalytic function [26]. Therefore, the residues with smaller side groups surrounding the nucleophilic attacking Ser residue may avoid steric hindrance and consolidate the correct position of this active site.
The oxyanion hole (His-Gly-Gly-Pro) in PiPs is another important component of active cavity, which forms a turn with the imidazole group of proline residue protruded onto the interface. And the central two Gly residues flanked spatially with Ser107 in PchPiPA, which was in general recognized to act as hydrogen bond donors to stabilize the oxyanion intermediate [19]. Gly46 in the PiP of S. marcescens in corresponding to the Gly40 in PchPiPA was indeed interacting with the imidazole ring of Pro-TBODA inhibitor in the co-structure of enzyme and inhibitor [24]. However, seldom researches have focused on this motif in PiPs. Mutation of Gly39 and Gly40 in PchPiPA leaded to almost complete loss of catalysis, which was largely due to decrease of kcat value. These results were consistent with the previous studies with another PiP (PepIP) of L. delbruckii subsp. Bulgaricus, in which mutation of the corresponding sites by chemical mutagenesis resulted in loss of catalytic activity [26]. In the other members from the α/β fold hydrolase family, such as esterase and acetylcholinesterase, mutation of the corresponding two Gly residues also had dead effect on the activity [27,28]. Furthermore, mutation of Gly42 and Cys45 also yield a profound negative influence on catalytic activity. D46A have no effect on catalytic activity, but its thermostability reduced significantly. These residues (Cys45 and Asp46), as well as Gly43 and Gly44 (corresponding to G46, G47, Cys48, and Asn49 in XCPIP) are lined at the entrance of the active cavity [19]. Besides, Gly43 are hydrogen bonded to Gly39 in the structure of PchPiPA. Taken together, the oxyanion hole as well as the flanked residues is very important for ensuring catalytic activity. And subtle changes may cause a profound negative impact on the activity.
Thr111 was located at helix C, which might interact with Ser107 via hydrogen bond in PchPiPA. Mutation of Thr111 caused a significant decrease of kcat value by about 15-fold, and almost no change in Km, indicating that Thr111 might contribute to the stability or positioning of Ser107. Arg130 and G131 were not conserved across various PiPs. And Arg130 was recognized as a putative S1’ position (because the corresponding residue Arg136 in the PiP of S. marcescens may bind the caybonyl oxygen of the P1’ residue) [21,23]. However, mutant R130A leaded to significant increase in kcat/Km, caused by changes in both kcat and Km. Since Arg130 could interact with His292 and Ser107 via formation of hydrogen bond and replacement of Arg130 with Ala leaded to loss of the interaction with His292 in the PchPiPA structural modeling, it was suggested that R130A may make more room in the cavity for increasing of flexibility of the active sites because this mutant lost thermostability obviously in comparison with the WT (Table 3). However, the same mutant in the PiP of S. marcescens showed very low activity against Pro-Ala, but retained almost the same activity as WT against Pro- βNA [23]. Mutant G131V significantly decreased kcat/Km value by about 100-fold due to changes in kcat. The larger side chain of Val protruded out might squeeze the space of active sites of His292 and Ser107, and then made the local conformation less plausible since its thermostability was increased significantly (Table 3).
In conclusion, the active sites and oxyanion hole as well as the flanked residues in PchPiPA were essential to the activity and more susceptible to be negatively affected by mutations, suggesting that the active cavity of proline iminopeptidase may evolve as a relatively compact structure.
5. ACKNOWLEDGEMENTS
This work is supported by the National Natural Science Foundation of China (31171204 to H.F.) and the High Technology Research and Development Program of China (2006AA02Z221 to H.F.).
REFERENCES
- Heikinheimo, P., Gold, A., Jeffries, C. and Ollis, D.L. (1999) Of barn owls and bankers: A lush variety of α/β hydrolases. Structure, 7, R141-R146. doi:10.1016/S0969-2126(99)80079-3
- Sarid, S., Berger, A. and Katchalski, E. (1959) Proline iminopeptidase. Journal of Biological Chemistry, 234, 1740-1746.
- Albertson, N.H. and Koomey, M. (1993) Molecular cloning and characterization of a proline iminopeptidase gene from Neisseria gonorrhoeae. Molecular Microbiology, 9, 1203-1211. doi:10.1111/j.1365-2958.1993.tb01249.x
- Gilbert, C., Atlan, D., Banc, B. and Portalier, R. (1994) Proline iminopeptidase from Lactobacillus delbrueckii subsp. bulgaricus CNRZ 397: Purification and characterization. Microbiology, 140, 537-542. doi:10.1099/00221287-140-3-537
- Kitazono, A., Kitano, A., Kabashima, T., Ito, K. and Yoshimoto, T. (1996) Prolyl aminopeptidase is also present in Enterobacteriaceae: Cloning and sequencing of the Hafnia alvei enzyme-gene and characterization of the expressed enzyme. Journal of Biochemistry, 119, 468-474. doi:10.1093/oxfordjournals.jbchem.a021265
- Kitazono, A., Kitano, A., Tsuru, D. and Yoshimoto, T. (1994) Isolation and characterization of the prolyl aminopeptidase gene (pap) from Aeromonas sobria: comparison with the Bacillus coagulans enzyme. Journal of Biochemistry, 116, 818-825.
- Li, N., Wu, J.-M., Zhang, L.-F., Zhang, Y.-Z. and Feng, H. (2010) Characterization of a unique proline iminopeptidase from white-rot basidiomycetes Phanerochaete chrysosporium. Biochimie, 92, 779-788. doi:10.1016/j.biochi.2010.02.022
- Mahon, S., O’Donoghue, A.J., Goetz, D.H., Murray, P.G., Craik, C.S. and Tuohy, M.G. (2009) Characterization of a multimeric, eukaryotic prolyl aminopeptidase: An inducible and highly specific intracellular peptidase from the non-pathogenic fungus Talaromyces emersonii. Microbiology, 155, 3673-3682. doi:10.1099/mic.0.030940-0
- Basten, D.E.J.W., Moers, A.P.H.A., van Ooyen, A.J.J. and Schaap, P.J. (2005) Characterisation of Aspergillus niger prolyl aminopeptidase. Molecular Genetics and Genomics, 272, 673-679. doi:10.1007/s00438-004-1094-5
- Szawłowska, U., Grabowska, A., Zdunek-Zastocka, E. and Bielawski, W. (2012) TsPAP1 encodes a novel plant prolyl aminopeptidase whose expression is induced in response to suboptimal growth conditions. Biochemical and Biophysical Research Communications, 419, 104-109. doi:10.1016/j.bbrc.2012.01.140
- Szaw1owska, U., Prus, W. and Bielawski, W. (2006) The molecular and biochemical characteristics of proline iminopeptidase from rye seedling (Secale cereal L.). Acta Physiologiae Plantarum, 28, 517-524.
- Zhang, L., Jia, Y., Wang, L. and Wang, R. (2007) A proline iminopeptidase gene upregulated in plant by a LuxR homologue is essential for pathogenicity of Xanthomonas campestris pv. campestris. Molecular Microbiology, 65, 121-136. doi:10.1111/j.1365-2958.2007.05775.x
- Allaker, R.P., Young, K.A. and Hardie, J.M. (1994) Rapid detection of proline iminopeptidase as an indicator of Eikenella corrodens. Letters of Applied Microbiology, 19, 325-327. doi:10.1111/j.1472-765X.1994.tb00466.x
- Delauney, A.J. and Verma, D.P.S. (1993) Proline biosynthesis and osmoregulation in plant. The Plant Journal, 4, 215-223. doi:10.1046/j.1365-313X.1993.04020215.x
- FitzGerald, R.J. and O’Cuinn, G., (2006) Enzymatic debittering of food protein hydrolysates. Biotechnology Advances. 24, 234-237. doi:10.1016/j.biotechadv.2005.11.002
- Leenhouts, K., Bolhuis, A., Boot, J., Deutz, I., Toonen, M., Venema, J., Kok, J. and Ledeboer, A. (1998) Cloning, expression, and chromosomal stabilization of the Propionibacterium shermanii proline iminopeptidase gene (pip) for food-grade application in Lactococcus lactis. Applied and Environmental Microbiology, 64, 4736- 4742.
- Yamamoto, Y., Usuki, H., Kumagai, Y., Mukaihara, T., Yamosato, A. and Hatanaka, T. (2011) Synthesis of prolyl-hydroxyproline using prolyl aminopeptidase from Streptomyces aureofaciens TH-3. Process Biochemistry, 46, 1560-1564. doi:10.1016/j.procbio.2011.04.009
- Yamomoto, Y., Usuki, H., Iwabuchi, M. and Hatanaka, T. (2010) Prolyl iminopeptidase from Streptomyces thermoluteus subsp. fuscus strain NBRC14270 and synthesis of proline-containing peptides. Applied Environmental Microbiology, 76, 6180-6185. doi:10.1128/AEM.01242-10
- Rawlings, N.D., Barrett, A.J. and Bateman, A. (2010) MEROPS: The peptidase database. Nucleic Acids Research, 38, D227-D233. doi:10.1093/nar/gkp971
- Medrano, F.J., Alonso, J., Garcıa, J.L., Romero, A., Bode, W. and Gomis-Ru, F.X. (1998) Structure of proline iminopeptidase from Xanthomonas campestris pv. citri: A prototype for the prolyl oligopeptidase family. The EMBO Journal, 17, 1-9. doi:10.1093/emboj/17.1.1
- Yoshimoto, T., Kabashim, T., Uchikawa, K., Unoue, T., Tanaka, N., Nakamura K.T., Tsuru, M. and Ito, K. (1999) Crystal structure of prolyl aminopeptidase from Serratia marcescens. Journal of Biochemistry, 126, 559-565. doi:10.1093/oxfordjournals.jbchem.a022486
- Ito, K., Inoue, T., Kabashima, T., Kanada N., Huang, H.- S., Ma, X., Azmi, N., Azab, E. and Yoshimoto, T. (2000) Substrate recognition mechanism of prolyl aminopeptidase from Serratia marcescens. Journal of Biochemistry, 128, 673-678. doi:10.1093/oxfordjournals.jbchem.a022800
- Inoue, T., Ito, K., Tozaka, T., Hatakeyama, S., Tanaka, N., Nakamura, K.T. and Yoshimoto, T. (2003) Novel inhibitor for prolyl aminopeptidase from Serratia marcescens and studies on the mechanism of substrate recognition of the enzyme using the inhibitor. Archives of Biochemistry and Biophysics, 416, 147-154. doi:10.1016/S0003-9861(03)00293-5
- Nakajima, Y., Ito, K., Sakata, M., Xu, Y., Nakashima, K., Matsubara, F., Katakeyama, S. and Yoshimoto, T. (2006) Unusual extra space at the active site and high activity for acetylated hydroxyproline of prolyl aminopeptidase from Serratia marcescens. Journal of Bacteriology, 188, 1599- 1606. doi:10.1128/JB.188.4.1599-1606.2006
- Kitazono, A., Ito, K. and Yoshimoto, T. (1994) Prolyl aminopeptidase is not a sulfhydryl enzyme: identification of the active serine residue by site-directed mutagenesis. Journal of Biochemistry, 116, 943- 945.
- Morel, F., Gilbert, C., Geourjon, C., Frot-Coutaz, J., Portalier, R. and Atlan, D. (1999) The prolyl aminopeptidase from Lactobacillus delbrueckii subsp. Bulgaricus belongs to the α/β hydrolase fold family. Biochimia et Biophysica Acta, 1429, 501-505. doi:10.1016/S0167-4838(98)00264-7
- Gall, M., Kourist, R., Schmidt, M., Bornscheuer and U.T. (2010) The role of the GGGX motif in determine the activity and enantioselectivity of pig liver esterase towards tertiary alcohols. Biocatalysis and Biotransformation, 28, 201-208. doi:10.3109/10242421003753803
- Zhang, Y., Kua, J. and McCammon, J.A. (2002) Role of the catalytic triad and oxyanion hole in acetylcholinesterase catalysis: An ab initio QM/MM study. Journal of American Chemistry Society, 124, 10572-10577. doi:10.1021/ja020243m
NOTES
*Corresponding author.