Open Journal of Pediatrics
Vol.07 No.02(2017), Article ID:76838,5 pages
10.4236/ojped.2017.72010
First Case of Liver Glycogen Documented in the Pediatric Ward of the University Hospital of Treichville (Côte d’Ivoire)
Jean-Jacques Yao Atteby, Lassina Cissé, Jacob Enoh, Kouadio Richard Azagoh, Germaine Niamké, Daouda Koné, Joseph Ouattara, Evelyne Kouadio, Beugre Martine Niangué, Soumahoro Mathurin Oulai, Joseph Andoh
Pediatric Ward, Teaching Hospital Center of Treichville, Abidjan, Côte d’Ivoire

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: January 26, 2017; Accepted: June 10, 2017; Published: June 13, 2017
ABSTRACT
The authors report a case of hepatic glycogen storage diseases type III (Cori- Forbes disease) of a Métis 3-year-old girl born and resident in Côte d’Ivoire. The discovery of this disease was made at the age of 2 years in the diagnosis of abdominal mass in a child subject to frequent hypoglycemia. The management currently consists of a diet enriched with glucose and maltose dextrin. We note in our case, a failure to thrive despite the persistent support.
Keywords:
Hepatic, Glycogenosis, Metabolic Disease, Child

1. Introduction
The glycogen storage diseases are long term autosomal recessive disease, which reach the liver glucose metabolism. It is rare conditions [1] . In this study, we report the case of a 2-year-old girl. This case was accidentally discovered during abdominal exam.
2. Observation
2.1. Clinical Feature
B Maeva, 2-year-old female gender, has taken to the hospital for large abdominal mass.
She has two half-brothers, David and Morgan, 17 and 11 years old from the paternal side who are healthy. The father is mixed (Franco-Ivorian) and the mother is originally from Burkina Faso in West Africa. They have no particular antecedent. They are not inbred.
B Maeva has been hospitalized in the neonatal period (first day of life) for respiratory distress and for repeated seizures without febrile context.
The monitoring of the disease showed a gradual installation of this abdominal mass without altering the general condition.
The liver clinical examination noticed a large hepatomegaly. Its lower edge was smooth. Its length was 9.5 cm long below the costal margin. Figure 1 showed the association with splenomegalia type II (HACKET Classification). From Figure 2, we could find a lack of growth with size 80 cm (−1 SD), weighing 10 kg is the 10th percentile according to Sempé curve, head’s perimeter is 44 cm (−2 SD) and arm circumference was 15 cm (−1 SD). In addition, it has been noted in Figure 3, a facial obesity and trunk with distended abdomen and chubby face contrasting with flat but muscular buttocks and limbs.

Figure 1.Distended abdomen by a bulky hepatomegaly.
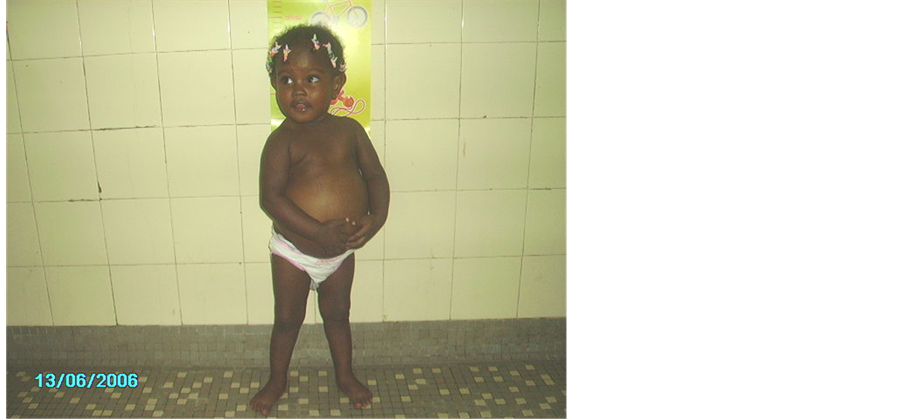
Figure 1. We notice the height-weight delay.

Figure 3. Chubby face.
2.2. Complementary Exams
The laboratory tests carried out showed a constant hypoglycemia of 0.22 g/l, hyperlactacidemia of 468 UI/l, CPK 406 IU/L, transaminase (AST) six times higher than normal, a lipid and normal kidney. The hemostasis tests were normal but a normochromic normocytic anemia moderate to 11g/dl with constant thrombocytosis to 510,000 cells/mm3 was noticed
Abdominal ultrasound exam highlighted hepatic steatosis. The abdominal scanner found homogeneous hepatomegaly overload (hemochromatosis? This fact evoked a Glycogen storage disease).
The biopsy of the liver confirmed the existence of hepatic glycogen. The enzymatic study (in Hôpital Antoine Beclere, Paris, Pr service Labrune) concluded a Glycogen storage disease type III.
2.3. Support and Outcome
It consisted of a diet enriched in glucose and dextrin-maltose in several fractional meals (every 3 hours).
After a year of medical monitoring, we see the disappearance of hypoglycaemia, a susceptibility to infections (skin and lung) and a slow change in weight (weight = 11 kg, 82 cm size = PC = 45.5 cm, PB = 15.5 cm).
3. Discussion
The glycogen storage diseases are rare and very diverse genetic diseases. We notice about 450 cases in France which comprise of 22.1% of type III [2] .
The genetic basis of these diseases has now well been established [2] . The autosomal recessive mode affecting chromosome 17 suggests consanguinity which was not found in our case. There is no similar case in the family. The paternal grandparents are Caucasian race and maternal parents are black.
Type I was the first form described in 1929 by Von Gierke [3] . Since then several forms were discovered, the type III in 1992 [1] . It is related to a challenge of the debranching enzyme (amyloid-glucosidase 1-6) in muscle and/or liver. Because of this failure, the body is no longer able to use the liver glycogen and/or muscle. This accumulates and compromises variably the operation or viability of muscle cells and/or liver with consequent permanent hypoglycemia. This will cause hypoglycemia subsequently a reaction hypersecretion of glucocorticoids responsible for muscle wasting (gluconeogenesis from protein catabolism) and a facial-central obesity [3] [4] .
The clinical manifestations of type III glycogen storage disease are highly variable from one person to another. The liver damage is always detected at some time in childhood or may be caused by another disease. Hypoglycemic seizures occur due to the inability of the body to convert liver glycogen into usable energy immediately. Liver pain can also result in an increase of liver enzyme blood levels (transaminases) and certain fats (hypertriglyceridemia) [5] . Growth retardation (delay) and dysmorphic features are often observed [5] . All these clinical and biological signs were found in our case.
The differential diagnosis is mainly with the type I or von Gierke disease; the most common form of glycogen storage diseases and related glucose 6 phosphatase deficiency [1] [6] .
The treatment is essentially diet and rests. The diet is rich in glucose, protein and low in fat and it is administered several meals spaced three hours.
The aim of this treatment is to obtain good blood glucose control, hypoglycaemia and prevent their own metabolic disorders and complications in order that they do not induce (Hyperlactacidemia, Hyperuricemia, Hyperlipemia) and the resulting pathologies (slow growth, bone mineralization defect ...) [5] . Evolution is studded with hypoglycemic events and their own consequences. The fear is the degeneration in adulthood (hepatocellular carcinoma) [3] .
4. Conclusion
The glycogen storage diseases remain poorly understood. Although they are infrequent conditions, they remain serious and deserve to be recognized. The types I and III are the most common. The diagnosis should be considered in a large smooth hepatomegaly and hypoglycemia constant height and weight delay. Diet plays a major role in treatment. However, despite treatment, growth may remain weak, requiring a long-term monitoring because it is an incurable disease.
Acknowledgements
Thanks to Professor Labrune (Hôpital Antoine Beclere Paris) and his team.
Cite this paper
Yao Atteby, J.-J., Cissé, L., Enoh, J., Azagoh, K.R., Niamke, G., Kone, D., Ouattara, J., Kouadio, E., Niangue, B.M., Oulai, S.M., Andoh, J. (2017) First Case of Liver Glycogen Documented in the Pediatric Ward of the University Hospital of Treichville (Côte d’Ivoire). Open Journal of Pediatrics, 7, 72-76. https://doi.org/10.4236/ojped.2017.72010
References
- 1. Manaranche, R. (1998) Neuromuscular Diseases. Scientific Informations of the French Association against Myopathies, 33, 3-13.
- 2. Perelman, R. and J Desbois, Cl. (1979) Pathologies of Metabolism and Nutrition. In: Perelman, R., Ed, Pediatric Practice, Maloine, Paris, 1489-1503.
- 3. Poenaru, L. and Baussau, C. (1991) Glycogenoses, Pediatrics. Technical Publishing—Medical and Surgical Encyclopedia, Paris, 11 p.
- 4. Chen, M.A. and Weinstein, D.A. (2016) Glycogen Storage Diseases: Diagnosis, Treatment and Outcome. Translational Science of Rare Diseases, 1, 45-72.
https://doi.org/10.3233/TRD-160006 - 5. Vidailhet, M. and Favre, E. (2006) The Food Balance in Glycogen Storage Diseases. The Glycogen Storage Magazine, 26. 23-25.
- 6. Wapper, R.S. (1999) Disorders of Metabolism in Carbonate. Oski’s Pediatrics: Principles and Practice, 3rd Edition, Williams and Lippincolt Wilkins, Philadelphia, 1853-1862.