Open Journal of Inorganic Chemistry
Vol.2 No.3(2012), Article ID:19765,9 pages DOI:10.4236/ojic.2012.23008
Synthesis, characterization and photophysical properties of quinolin-8-olato chelated osmium(II) organometallics bearing a pendant imine-phenol motif and electrogeneration of trivalent analogue
![]()
Department of Chemistry, Jangipur College, Murshidabad, India
Email: panda_bikas@rediffmail.com, bikash.panda66@yahoo.com
Received 11 March 2012; revised 13 April 2012; accepted 30 April 2012
Keywords: Osmium Organometallics; Quinolin-8-Olato Chelation; Emission Properties; Trivalent Osmium
ABSTRACT
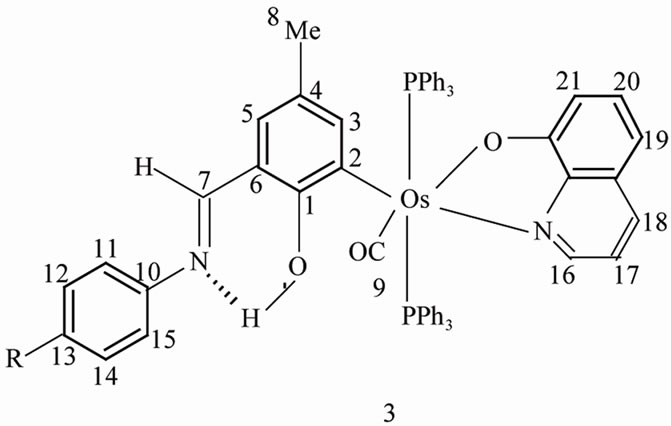
The reaction of Os(RL1)(PPh3)2(CO)Br, 1b, with quinolin-8-ol (HQ), 2, has furnished complexes of the type [Os(RL2)(PPh3)2(CO)(Q)], 3, in excellent yield (RL1 is C6H2O-2-CHNHC6H4R(p)-3-Me-5, RL2 is C6H2OH-2-CHNC6H4R(p)-3-Me-5 and R is Me, OMe, Cl). In this process, quinolin-8-olato (Q) undergoes five-membered chelation, the iminium-phenolato function tautomerizing to the imine-phenol function. The trans geometry of the Os(PPh3)2 fragment is consistent with the occurrence of a single 31P resonance near –6.0 ppm in 3. In dichloromethane solution, 3 displays a quasireversible 3+/3 couple near 0.40 V vs. SCE (3+ is the osmium (III) analogue of 3). Coulometrically generated solutions of 3+ displays a strong absorption near 340 nm, 415 nm and 500 nm and are one-electron paramagnetic (low-spin d5, S = 1/2) and show rhombic EPR spectra in 1:1 dichloromethanetoluene solution at 77 K with g values near 2.44, 2.20, 1.83. Distortion parameters using the observed g values have been computed. Solutions of 3 absorb near 420 nm and emit near 510 nm at 298 K and 580 nm at 77 K. The fluorescence is believed to originate from the 3MLCT state.
1. INTRODUCTION
The chemistry of osmium has been receiving considerable attention largely because of the variety of reactivities and due to the fascinating electron transfer properties exhibited by complexes of this metal [1-9]. Variation of the coordination environment around osmium plays a key role in modulating the redox properties of its complexes. In this article we studied the reactivity of an osmium system with 8-hydroxyquinoline because complexes incorporating quinoline moiety are important in the context of biological activity [10]. It was also reported in the literature that a series of compounds derived from 8-hydroxyquinoline showed that they could possess strong antifungal, antiasthmatic and antiplatelet activity [10-13]. In particular the chemistry of osmium compounds is of current interest in the context of synthesis, reactivity and photophysical properties [1,9,14-16].
It was demonstrated that the decarbonylative metallation of 4-methyl-2,6-diformyl phenol by M(PPh3)3X2 in the presence of primary aromatic amines (RC6H4NH2) is known to furnish four-membered ruthenium/osmium organometallics of the type M(RL1)(PPh3)2(CO)X (1) juxtaposed to a hydrogen bonded iminium-phenolato function [17-19]. We are scrutinizing the reactivity of these compounds. It has been found that different alkynes undergoes facile insertion into the Ru-C bond of the fourmembered ruthenium metallacycles leading to two carbon expansion [20-22] whereas the osmium analogue remains unreactive. Upon treatment with isonitrile ruthenium system undergoes halide substitution as well as insertion into the Ru-O bond and the resultant product then exhibits aryl migration and C-C bond formation [23, 24] while in case of osmium system isonitrile simply displaced bromide ligand furnishing the cationic complexes which are isolated as salts [25]. It is indeed found that ruthenium metallacycle is smoothly converted to the acyl system upon reaction with carbon monoxides under ambient conditions whereas the osmium analogue did not lead to insertion and afforded only an unreactive cisdicarbonyl complex [26]. We also explore the reactivity of 1 towards bidentate monoanionic donor reagents such as acetate [27], palmitate and terephthalate [28], nitrate and nitrite [29,30] xanthate [31], pyridine-2-thiolate [32] undergo four-membered chelation and we also reported earlier that 1a reacts with the electroneutral diimine ligands [33] such as bipyridine, phenanthroline and biimidazole [34] as well as 8-hydroxyquinoline [35] furnish five-membered ruthenium organometallics.
The above importance of osmium complexes and the richness of this reaction chemistry have now prompted us to explore the reactivity of 1b towards quinolin-8-ol, 2, (HQ, H stands for the dissociable phenolic hydrogen) which is suited for five-membered N, O-chelation. A facile reaction is indeed observed furnishing the novel five-membered osmium organometallics for the first time via displacement of Os-O and Os-Br bonds. Changes in the coordination mode and tautomeric state of the Schiff base ligand accompany the synthetic reaction. It may be noted that the chemistry of osmium quinolin-8-olate appears to have relatively less attention [36-39] but to the best of my knowledge organoosmium compounds incorporating 8-hydroxyquninoline are unprecedented. Another point of special interest is that such species are potentially luminescent in the visible region and are electrochemically oxidizable to the trivalent analogue.


2. EXPERIMENTAL
2.1. Materials
The compounds Os(PPh3)3Br2 [40] and Os(RL1)(PPh3)2- (CO)Br [17] were prepared by reported methods. 8-hydroxyquinoline was obtained from Merck. The purification of dichloromethane and the preparation of tetraethyl ammonium perchlorate (TEAP) for electrochemical work were done as described in previous work [41]. All other chemicals and solvents were of analytical grade and were used as received.
2.2. Physical Measurements
Electronic, IR and fluorescence spectra were recorded with a Shimadzu UV-1601 PC spectrophotometer, Nicolet Magna IR series II spectrometer and Perkin-Elmer model LS 55 luminescence spectrometer respectively. 1H NMR spectra were obtained using a Bruker 300 MHz FT NMR spectrometer. The numbering scheme used for the 1H NMR is the same as shown in drawing 3. Spin structures are abbreviated as: s, singlet; d, doublet; t, triplet; m, multiplet. 31P NMR spectra were recorded with the help of a Varian 300 MHz spectrometer using H3PO4 as standard. Microanalyses (C, H, N) were done by using a Perkin-Elmer 240C elemental analyzer. The magnetic behavior of the complexes was examined by a PAR 155 vibrating sample magnetometer. EPR spectra were recorded on a Varian E-109C X-band spectrometer fitted with a quartz dewar. Solution electrical conductivity was measured in acetone with a Phillips PR 9500 bridge using a platinized electrode (cell constant of 1.05). Electrochemical measurements were performed in a nitrogen atmosphere in dichloromethane solution using a CHI model 620A electrochemical analyzer. The supporting electrolyte was tetraethyl ammonium perchlorate and potentials are referenced to the saturated calomel electrode.
2.3. Synthesis of the Complexes [Os(RL2)(PPh3)2(CO)(Q)] (3)
[Os(RL2)(PPh3)2(CO)(Q)] (3) complexes were synthesized in excellent yield (~80%) by reacting Os(RL1)- (PPh3)2(CO)Br (1b) in MeOH-CH2Cl2 (2:1) at room temperature with excess HQ. Details of a representative case are given below. The other compounds are prepared in an analogous manner.
2.3.1. [Os(MeL2)(PPh3)2(CO)(Q)], 3(Me)
To a vigorously stirred violet solution of Os(MeL1)- (PPh3)2(CO)Br (100 mg, 0.09 mmol) in a mixture of 50 ml methanol and 25 ml dichloromethane was added 65 mg (0.45 mmol) of HQ. The mixture was refluxed for 12 h when the colour of the solution changed from violet to yellow. The solvent was then removed under reduced pressure and the yellow solid thus obtained was washed with a little amount of methanol (removal of excess HQ). The residue was recrystallized from dichloromethanehexane (1:3) mixture followed by drying in vacuo. Yield: 86 mg (81%).
Analysis: Calc. for C61H50N2O3P2Os: C, 65.93; H, 4.53; N, 2.69%. Found C, 65.88; H, 4.51; N, 2.64%. 1H NMR (300 MHz, CDCl3, ppm, 298 K): 12.56 (s, 1H, O–H, disappeared upon shaking with D2O), 7.80 (s, 1H, H7), 5.98 (s, 1H, H3), 1.92 (s, 3H, 4-Me), 2.32 (s, 3H, 13-Me), 8.01 (d, JHH = 6.2 Hz, 1H, H16), 7.19 (d, JHH = 8.5 Hz, 1H, H18), 6.11 (d, JHH = 7.6 Hz, 1H, H19), 6.45 (d, JHH = 8.2 Hz, 1H, H21), 6.33 - 6.38 (m, 2H, H17, H20). 6.65 - 7.05 (m, 35H, 2PPh3, H5, H11, H12, H14, H15). 31P NMR (300 MHz, CDCl3, ppm, 298 K): –6.196 (s, 2P). IR (KBr, cm–1): 1893 (νCO), 1602 (νC=N).
2.3.2. [Os(MeOL2)(PPh3)2(CO)(Q)], 3(OMe)
Os(MeOL1)(PPh3)2(CO)Br (100 mg, 0.089 mmol) and HQ (64 mg, 0.441 mmol) were employed. Yield: 84 mg (79%). Analysis: Calc. for C61H50N2O4P2Os: C, 65.00; H, 4.47; N, 2.48%. Found C, 65.05; H, 4.41; N, 2.50%. 1H NMR (300 MHz, CDCl3, ppm, 298 K): 12.61 (s, 1H, O-H, disappeared upon shaking with D2O), 7.82 (s, 1H, H7), 6.01 (s, 1H, H3), 1.90 (s, 3H, 4-Me), 3.74 (s, 3H, 13-OMe). 8.02 (d, JHH = 6.5 Hz, 1H, H16), 7.20 (d, JHH = 8.4 Hz, 1H, H18), 6.13 (d, JHH = 7.5 Hz, 1H, H19), 6.48 (d, JHH = 7.9 Hz, 1H, H21), 6.32 - 6.39 (m, 2H, H17, H20), 6.63 - 7.04 (m, 35H, 2PPh3, H5, H11, H12, H14, H15). 31P NMR (300 MHz, CDCl3, ppm, 298 K): –6.270 (s, 2P). IR (KBr, cm–1): 1896 (νCO), 1606 (νC=N).
2.3.3. [Os(ClL2)(PPh3)2(CO)(Q)], 3(Cl)
Os(ClL1)(PPh3)2(CO)Br (100 mg, 0.088 mmol) and HQ (64 mg, 0.441 mmol) were used. Yield: 83 mg (76%). Analysis: Calc. for C60H47N2O3P2ClOs: C, 63.68; H, 4.19; N, 2.47%. Found C, 63.65; H, 4.23; N, 2.51%. 1H NMR (300 MHz, CDCl3, ppm, 298 K): 12.51 (s, 1H, O-H, disappeared upon shaking with D2O), 7.79 (s, 1H, H7), 6.00 (s, 1H, H3), 1.92 (s, 3H, 4-Me). 7.99 (d, JHH = 6.3 Hz, 1H, H16), 7.19 (d, JHH = 8.5 Hz, 1H, H18), 6.12 (d, JHH = 7.3 Hz, 1H, H19), 6.46 (d, JHH = 7.8 Hz, 1H, H21), 6.33 - 6.40 (m, 2H, H17, H20), 6.62 - 7.02 (m, 35H, 2PPh3, H5, H11, H12, H14, H15). 31P NMR (300 MHz, CDCl3, ppm, 298 K): –6.345 (s, 2P). IR (KBr, cm–1): 1892 (νCO), 1596 (νC=N).
2.4. Electrochemical Generation of 3+
The complexes of type 3+ were generated in solution by constant potential coulometric oxidation of solutions of type 3. Details of a representative example are noted below.
A solution of 10 mg of 3(Me) in 25 ml dichloromethane solvent containing 35 mg of TEAP was oxidized coulometrically. The oxidation was performed at 0.75 V; ne = 1.108/1.086 = 1.02, ne = Q/Q1, where Q1 is the calculated coulomb count for one electron transfer and Q is the coulomb count found after exhaustive electrolysis. A part of the electrogenerated solution (5 ml) of 3(Me)+ was mixed with an equal volume of toluene and the mixture was quickly frozen at 77 K, and then used for an EPR measurement. Another part of this electrogenerated orange-red solution (10 ml) is used quickly for electronic spectral study. The oxidized complex 3(Me)+ was stable in solution at room temperature for few minutes only. Most of it (>90%) underwent reduction to produce 3, associated with some insoluble material of unknown composition. The oxidation of 3 also may be performed chemically using an aqueous solution of Ce4+. In this case the quantity of the byproducts was more than 25%. To date, we were not successful in isolating the trivalent osmium compound 3+ in its pure state. The trivalent complex 3(OMe)+ and 3(Cl)+ was generated in solution similarly as described above.
3. RESULTS AND DISCUSSION
3.1. Coordination with Quinolin-8-olato
In methanol-dichloromethane solution, Os(RL1)(PPh3)2- (CO)Br (1) reacts smoothly with five fold excess of HQ upon stirring at room temperature, according to, Equation (1), the colour of the
1 + HQ → 3 + HBr (1)
solution changes from violet to yellow, from which [Os- (RL2)(PPh3)2(CO)(Q)] (3) is obtained in excellent yield. The R groups utilized in the present work are Me, OMe and Cl. Specific compounds are identified by putting R in parentheses: thus 3(Me) stands for [Os(MeL2)(PPh3)2- (CO)(Q)]. The complex 3 has been isolated in pure form in ~ 80% yield. An isomer (because quinol-8-ol is unsymmetrical) of 3 is possible in principle but it has never been observed. Careful examination of 1H NMR spectra of crude sample (before recrystallization) did not reveal the presence of any extra signals apart from those characterizing 3. We assign structure 3 to it in analogy with the stereochemistry of other OsII(quinolin-8-olato) complexes [38,39] and also by comparing with acetate, thioxanthate, pyridine-2-thiolato chelated ruthenium/osmium organometallics [27,31,32].
The conversions 1→3 is attended with a prototropic shift within the salicyaldimine function. In 1 the metal is coordinated to phenolato oxygen and the Schiff base function occurs in the zwitterionic iminium-phenolato
tautomeric form 4. Chelation of quinolin-8-olato is attended with the cleavage of the Os-O bond and the Schiff base function becomes an imine-phenol 5. This is fully consistent with the spectroscopic data.
Thus the C=N stretching frequency in 3 is significantly lower (~1600 cm–1) than that in 1 (~1620 cm–1) as expected [42]. Also the aldimine CH signal in 1H NMR in 3 (Figure 1) occurs at lower field viz. 7.8 ppm as compared to ~7.5 ppm in 1 [17]. The O-H resonance in 3 is a relatively sharp signal near 12.5 ppm having halfheight width of ~30 Hz. In contrast, the iminium N-H resonance in 1 is broad (width, ~150 Hz) evidently due to the quadrupole moment of the nitrogen atom [17]. The prototropic transformation between 1 and 3 has certain similarities with the imine-iminium tautomerization in rhodopsins [42].
A plausible mechanism [31,32,35] for the displacement of bromide in 1 consists of cis attack by Q– is shown in 6. The anchored ligand displaces the phenolato oxygen and halide atoms achieving Q– chelation with concomitant prototropic shift and conformational reorganization as in 7.
In both 1 and 3, the carbon monoxide ligand lies cis to the metallated carbon atom. It is however, located cis and trans to the phenolato oxygen in 3 and 1, respectively which is revealed by our previous study also [32,33]. Thus the Schiff base fragment rotates by ~180˚ around the metal-carbon bond in course of the reaction of equation 1 due to steric repulsion between the phenolic and the quinolin-8-olato function as highlighted in the hypothetical anti configuration 8 for Os(CO)(Q) fragment.
3.2. Characterization
Osmium organometallics of type 3 are non-electrolytic in

solution and are diamagnetic, consistent with a metal oxidation state of +2 having idealized t2g6 electronic con-
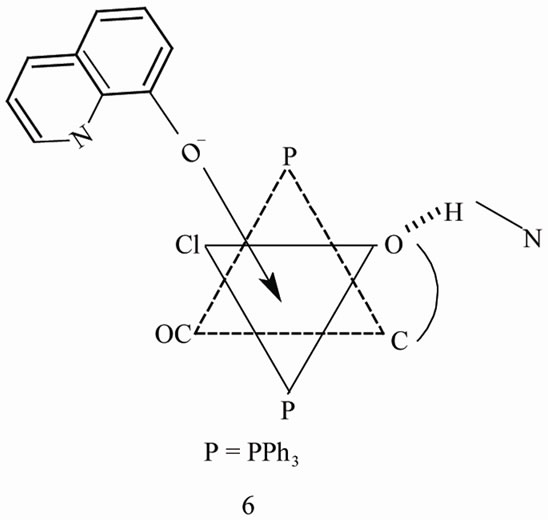
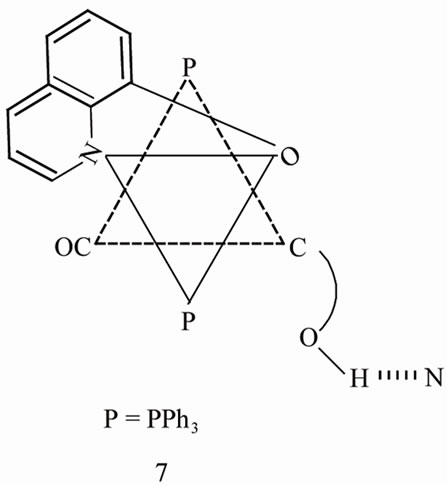
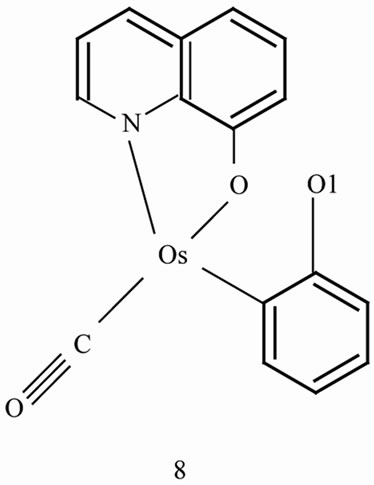
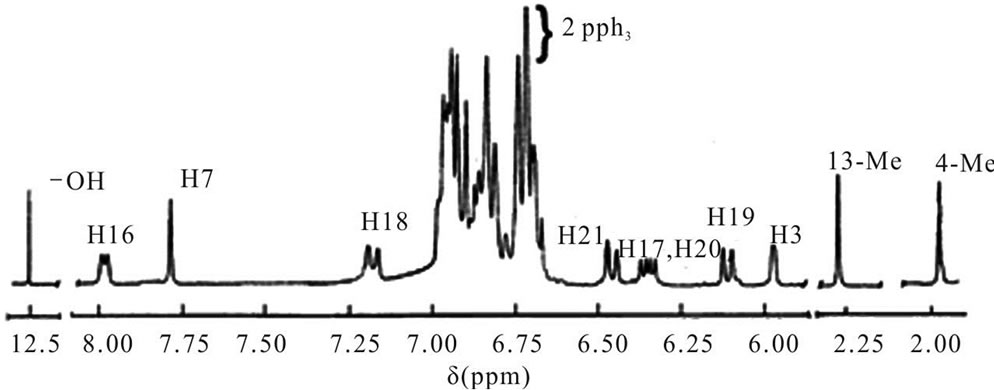
Figure 1. 1H NMR spectrum of 3(Me) in CDCl3 solution.
figuration. In IR, the CO stretch is seen as a sharp band near 1890 cm–1, the stretch is at slightly lower frequency due to the superior back bonding ability of bivalent osmium. In 1H NMR (Figure 1) the 3-H proton of the metallated ring occur as sharp singlets near 6.0 ppm, while the C(4)-Me protons resonate near 1.9 ppm. These protons are subject to shielding by phosphine phenyl rings [17,27,43]. The PPh3, Schiff base (C6H4R) aromatic protons form a complex multiplet in the region 6.60 - 7.05 ppm and the quinolin-8-olate aromatic protons appear in the 6.00 - 8.05 ppm region.
Detailed assignments of the ligand protons are given in the experimental section. In the previous section the chemical shifts of the imine-phenol protons were considered. The complexes uniformly display a single 31P signal for the two phosphine ligands implicating transOs(PPh3)2 configuration. The resonance occurs near –6.0 ppm, exact data are given in the experimental section. A representative spectrum is shown in Figure 2. The redox and photophysical properties of the complexes are examined in later sections.
3.3. Redox Properties and EPR Spectra
In dichloromethane solution 3 displays a quasireversible one-electron cyclic voltammetric response near 0.40 V (peak-to-peak separation is ~100 mV) vs. SCE corresponding to the
3+ + e → 3 (2)
couple where 3+ represents the osmium(III) analogue of 3. A representative cyclic voltammogram is shown in Figure 3. As R is varied, the E1/2 values become more positive as the electron withdrawing power of the R group increases (OMe < Me < Cl), see Table 1. The reduction potentials are systematically lower than those of the type 1 precursors [17] by ~200 mV, indicating better stabilization of the trivalent state in 3 compared with that in 1. The one-electron nature of these responses has been confirmed by comparing their current heights with the standard ferrocene/ferrocenium couple under identical experimental conditions. The exhaustive coulometric oxidation at 0.75 V affords a coulomb count corresponding to one-electron transfer (Table 1).
Low potentials for the OsII/OsIII couple in the complexes of type 3 persuaded us to try and isolate the corresponding osmium(III) complexes 3+ in their pure states. Accordingly, we tried to oxidize them both chemically and electrochemically. Unfortunately, the oxidized com-

Figure 2. 31P NMR spectrum of 3(OMe) in CDCl3.
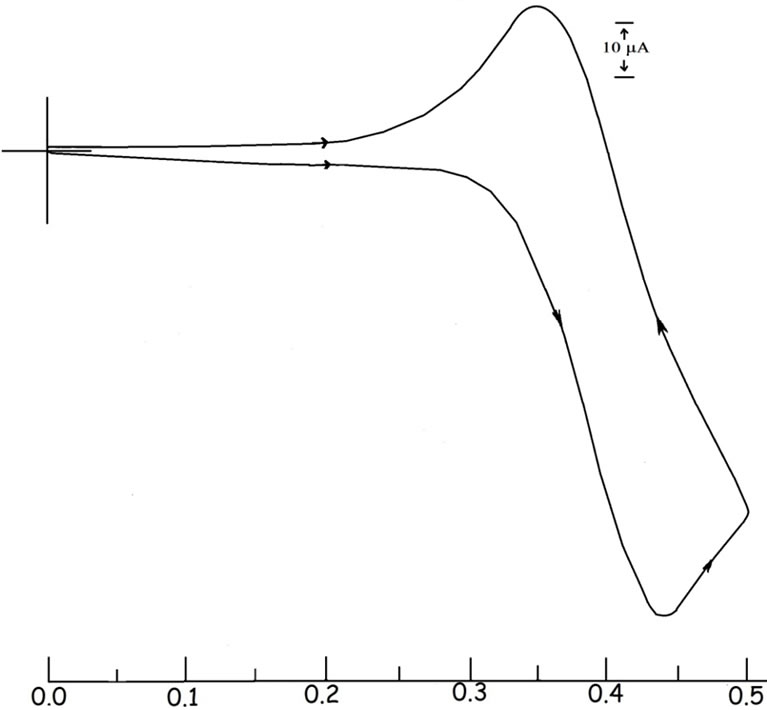
Figure 3. Cyclic voltammogram of 3(OMe) in dichloromethane solution.

Table 1. Absorption, emission spectral and electrochemical data of 3.
plexes were not stable enough for their isolation, as they revert to the parent bivalent complexes rapidly. However, we were able to generate the cationic complexes, 3+, in solution by the controlled potential bulk electrolysis of 3.
Trivalent organometallics 3+, generated coulometrically, have been examined in solution. Their cyclic voltammograms (initial scan cathodic) are virtually superimposible on those of 3 (initial scan anodic), showing that 3+ retains the gross structure of 3. Orange-red solutions of 3+ are characterized by an intense band near 340 nm, 410 nm and 500 nm. A representative spectrum is shown in Figure 4 and spectral data are given in Table 2. Electron paramagnetic resonance (EPR) spectra of the 3+ complexes, recorded in 1:1 dichloromethane-toluene solution at 77 K, show rhombic spectra with three distinct signals (g1, g2 and g3, in decreasing order of magnitude). A representative spectrum is shown in Figure 5 and the spectral data are presented in Table 2. The observed rhombicity of the EPR spectra is understandable in terms of the gross molecular symmetry of these complexes, containing the three non-equivalent P-Os-P, O-Os-C and N-Os-C axes. The rhombic distortion can be thought of as a combination of axial distortion (Δ, which splits t2 into a and e) and rhombic distortion (V, which splits e). The splitting pattern is illustrated in Figure 5. Spin-orbit coupling causes further changes in the energy gaps. Thus two electronic transitions (transition energies ΔE1 and ΔE2; ΔE1 < ΔE2) are possible in principle within these three levels. All these energy parameters have been computed (Table 2) using the observed g-values, the gtensor theory of low-spin d5 complexes and a reported method [44,45]. The axial distortion is observed to be much stronger than the rhombic. The EPR data analysis thus shows that the 3+ complexes are significantly distorted from ideal octahedral geometry which is observed previously in the crystal structure of acetate [27] and xanthate [31] derivatives. In this connection it should be pointed out that for thioxanthate chelated osmium organometallics we indicate the better stabilization of the trivalent state but isolation of osmium(III) species via electrooxidation has not been succeeded [31] but for quinolin-8-olato chelated osmium complexes we are able to generate the osmium(III) analogue in solution this is probably due to the five-membered chelation instead of four-membered chelation for the former.
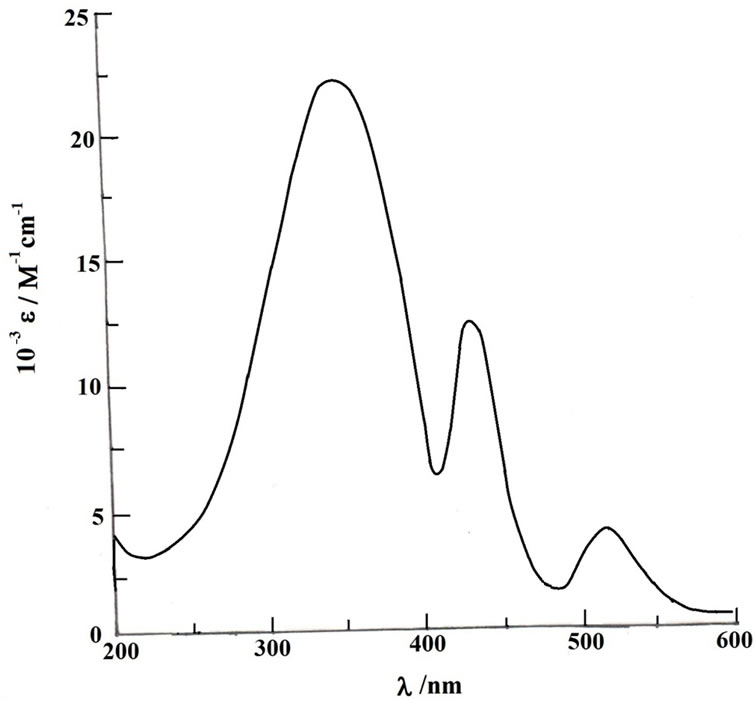
Figure 4. Electronic spectrum of 3(Cl)+ in CH2Cl2 solution.
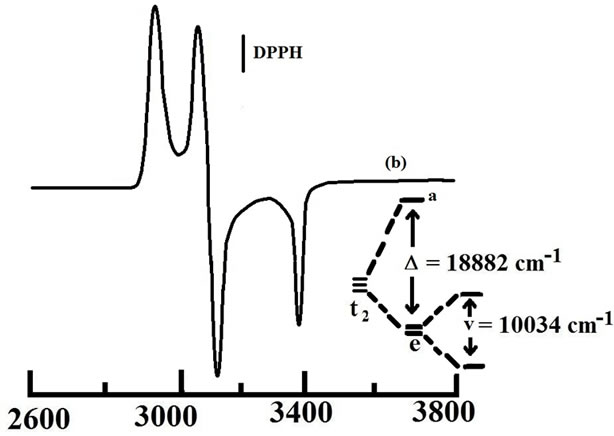
Figure 5. EPR spectra of 3(OMe)+ in frozen 1:1 dichloromethane-toluene solution at 77 K, showing the computed splitting of the t2 orbitals. DPPH = diphenyl-picrylhydrazyl.
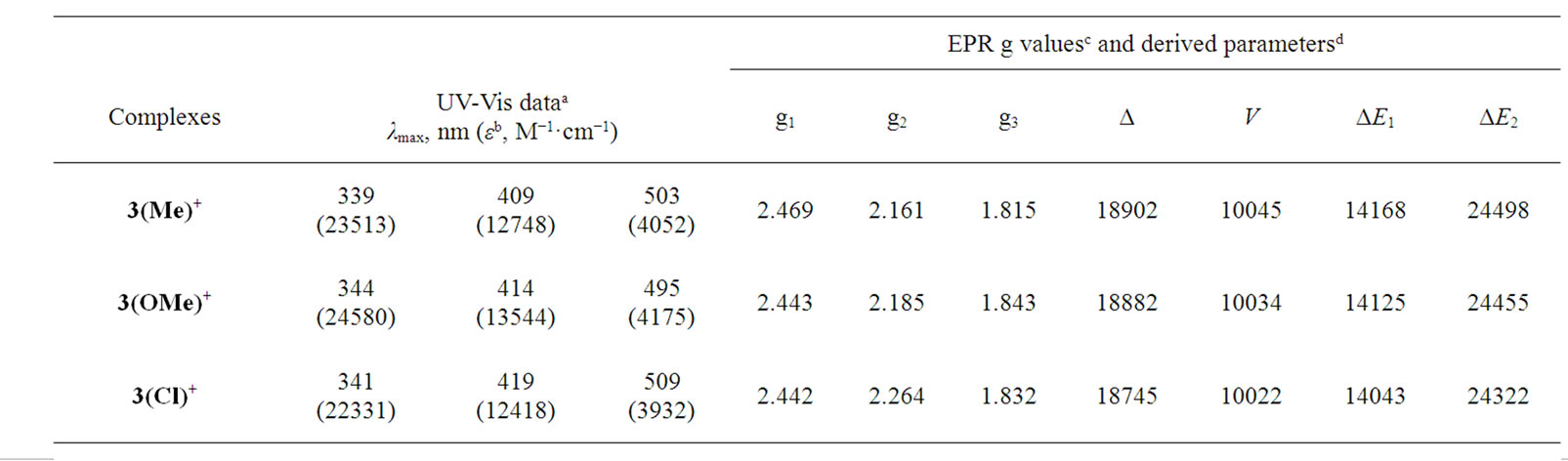
Table 2. Electronic and EPR spectral data of 3+.
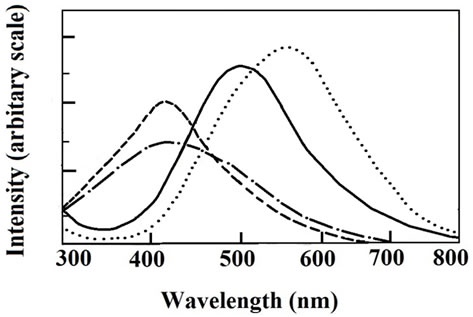
Figure 6. Absorption (− − −), excitation (─ • ─), and fluorescence spectra at 298 K (──) and at 77 K (• • •) of 3(Cl) in dichloromethane solution.
3.4. Emission Properties
In dichloromethane solution complexes of type 3 display two allowed absorption bands in the regions 345 - 355 and 410 - 425 nm. The latter band is weaker in intensity and is believed to have significant dπ(Os) – π*(Q) MLCT character. Selected UV-Vis spectral data are given in Table 1. The solutions are fluorescent at room temperature, and also at low temperature (77 K). The peaks lying in the region 505 - 515 and 570 - 590 nm at 298 K and 77 K respectively (Figure 6, Table 1) makes 3 fluorescent in the visible region. Low-temperature emission spectra are red-shifted from room temperature ones. The complexes are weak emitters, as noted from their quantum yield (Φr) values which are one order of magnitude lower than that of the [Ru(bpy)3]2+ standard [46,47] (Table 1). Quantum yields are calculated using Equation (3) as described previously [46,47] where Φstd is 0.042 at 298 K, A is the solution absorbance at the excitation wave length, I is the relative emission intensity, η are the refractive index values of the solvent for the sample (subscript r) and the standard reference (subscript std) respectively,
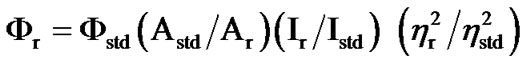 . (3)
. (3)
Excitation spectral studies show that fluorescence is associated with the absorption band near 420 nm. A representative case is shown in Figure 6. Assuming that the band has MLCT character as suggested above, the possible involvement of the 3MLCT state [15,16,33] incorporating π*(Q) contribution is implicated in the emission process. We also note that the parent organometallics of type 1 do not display any fluorescence in the visible region.
4. CONCLUSION
It is demonstrated that the metallacycles of type 1 react smoothly with HQ furnishing aryl osmium organometallics of type 3. The conversion 1→3 is attended with cleavage of Os-O and Os-Br bonds, (N, O) chelation of quinolin-8-olate, iminium-phenolato→imine-phenol tautomerization. OsIII/OsII reduction potentials are systematically lower than those of 1 and 3 is electrooxidizable to the osmium(III) analogue 3+ characterized by rhombic EPR spectra. The Os→Q MLCT absorption in the visible region makes 3 fluorescent with quantum yields one order of magnitude less than that of the [Ru(bpy)3]2+ standard.
5. ACKNOWLEDGEMENTS
The help received from Professor Animesh Chakravorty, Indian Association for the Cultivation of Science, is gratefully acknowledged. I also thank the Department of Science and Technology, and the Council of Scientific and Industrial Research, New Delhi for financial support.
REFERENCES
- Sauvage, J.P., Collin, J.P., Chambron, J.C., Guillerez, S., Coudret, C., Balzani, V., Barigelletti, F., Cola, L.D. and Flamigni, L. (1994) Ruthenium(II) and Osmium(II) bis(terpyridine)complexes in covalently-linked multicomponent systems: Synthesis, electrochemical behavior, absorption spectra, and photochemical and photophysical properties. Chemical Review, 94, 993-1019. doi:10.1021/cr00028a006
- Richmond, M.G. (1995) Annual survey of ruthenium and osmium for the year 1993. Coordination Chemistry Reviews, 141, 63-152.
- Ward, M.D. (1995) Osmium 1993. Coordination Chemistry Reviews, 146, 99-113. doi:10.1016/0010-8545(96)83891-4
- Dyson, P.J., Johnson, B.F.G. and Martin C.M. (1996) The synthesis of ruthenium and osmium carbonyl cluster with unsaturated organic rings. Cordination Chemistry Reviews, 155, 69-86. doi:10.1016/S0010-8545(96)90177-0
- Che, C.-M. and Huang, J.-S. (2002) Ruthenium and osmium porphyrin carbene complexes: Synthesis, structure and connection to the metal-mediated cyclopropanation of alkenes. Coordination Chemistry Reviews, 231, 151- 164.
- Jia, G. (2007) Recent progress in the chemistry of osmium carbine and metallabenzyne complexes. Coordination Chemistry Reviews, 251, 2167-2187. doi:10.1016/j.ccr.2006.11.018
- Che, C.-M., Ho, C.-M. and Huang, J.-S. (2007) Metalcarbon multiple bonded complexes: Carbene, vinylidene and allenylidene complexes of ruthenium and osmium supported by macrocyclic ligands. Coordination Chemistry Reviews, 251, 2145-2166.
- Esteruelas, M.A., Lopez, A.M. and Olivan, M. (2007) Osmium-carbon double bonds: Formation and reactions. Coordination Chemistry Reviews, 251, 795-840. doi:10.1016/j.ccr.2006.07.08
- Laine, P.P., Campagna, S. and Loiseau, F. (2008) Conformationally gated photoinduced processes within photosensitizer-acceptor dyads based on ruthenium(II) and osmium(II) polypyridyl complexes with an appended pyridinium group. Coordination Chemistry Reviews, 252, 2552-2571. doi:10.1016/j.ccr.2008.05.007
- Musiol, R., Jampilek, J., Nycz, J.E., Pesko, M., Carroll, J., Kralova, K., Vejsova, M., Mahony, J.O., Coffey, A., Mrozek, A. and Polanski, J. (2010) Investigating the activity spectrum for ring-substituted 8-hydroxyquinolines. Molecules, 15, 288-304. doi:10.3390/molecules15010288
- Sissi, C. and Palumbo, M. (2003) The quinolone family: From antibacterial to anticancer agents. Current Medicinal Chemistry—Anti-Cancer Agents, 3, 439-450. doi:10.2174/1568011033482279
- Musiol, R., Jampilek, J., Buchta, V., Niedbala, H., Podeszwa, B., Palka, A., Majerz-Maniecka, K., Oleksyn, B. and Polanski, J. (2006) Antifungal properties of new series of quinoline derivatives. Bioorganic & Medicinal Chemistry, 14, 3592-3598. doi:10.1016/j.bmc.2006.01.016
- Gershon, H., Gershon, M. and Clarke, D.D. (2004) Synergistic mixtures of fungitoxic monochloroand dichloro- 8-quinolinols against five fungi. Mycopathologia, 158, 131-135. doi:10.1023/B:MYCO.0000038427.42852.6a
- Bhaumik, C., Das, S., Maity, D. and Baitalik S. (2012) Luminescent bis-tridentate ruthenium(II) and osmium(II) complexes based on terpyridyl-imidazole ligand: Synthesis, structural characterization, photophysical, electrochemical and solvent dependence studies. Dalton Transaction, 41, 2427-2438. doi:10.1039/c1dt11645b
- Pennington, N.S., Richter, M.M. and Carlson, B. (2010) Efficient electrogenerated chemiluminescence from osmium(II) polypyridine systems containing tetraphenylarsine or diphenylphosphine ligands. Dalton Transaction, 39, 1586-1590. doi:10.1039/b912877h
- Hwang, K.-C., Chen, J.-L., Chi, Y., Lin, C.-W., Cheng, Y.-M., Lee, G.-H., Pi-Tai Chou, P.-T., Lin, S.-Y. and Shu, C.-F. (2008) Luminescent osmium(II) complexes with functionalized 2-phenylpyridine chelating ligands: Preparation, structural analyses, and photophysical properties. Inorganic Chemistry, 47, 3307-3317. doi:10.1021/ic7023132
- Ghosh, P., Bag, N. and Chakravorty, A. (1996) Decarbonylative metallation of diformylphenol Schiff basses: New osmium and ruthenium organometallics incorporating the iminium-phenolato zwitterionic motif. Organometallics, 15, 3042-3047. doi:10.1021/om960018b
- Bag, N., Choudhury, S.B., Pramanik, A., Lahiri, G.K. and Chakravorty, A. (1990) Ruthenium(II) phenolates. Synthesis and characterization of a novel four-membered metallacycle. Inorganic Chemistry, 29, 5013-5015. doi:10.1021/ic00350a001
- Bag, N., Choudhury, S.B., Lahiri, G.K. and Chakravorty, A. (1990) A novel zwitterionic o-metallated Ru(II)-phenolate. Journal of the Chemical Society, Chemical Communications, 22, 1626-1627.
- Panda, B.K. (2005) Metallacycle expansion via butadiyne/octadiyne insertion into a four-membered ruthenium organometallics. Transition Metal Chemistry, 30, 712- 719. doi:10.1007/s11243-005-6282-6
- Ghosh, K., Chattopadhyay, S., Pattanayak, S. and Chakravorty, A. (2001) Alkyne insertion into the Ru-C bond of a four-membered metallacycle: Insertion rate and reaction pathway. Organometallics, 20, 1419-1423. doi:10.1021/om000649c
- Ghosh, K., Pattanayak, S. and Chakravorty, A. (1998) Metallacycle expansion by alkyne insertion. Chemistry of a new family of ruthenium organometallics. Organometallics, 17, 1956-1960. doi:10.1021/om970917f
- Panda, B.K., Chattopadhyay, S., Ghosh, K. and Chakravorty, A. (2002) Isonitrile insertion into the Ru-O bond and migratory C-C bond formation. Novel organoruthenium imidic ester and acyl species. Organometallics, 21, 2773-2780. doi:10.1021/om020059
- Panda, B.K. (2005) Structurally characterized acylruthenium organometallics bearing a pendant aldehyde function. Transition Metal Chemistry, 30, 488-495. doi:10.1007/s11243-004-6970-7
- Panda, B.K. and Chakravorty, A. (2005) Chemistry of a family of osmium(II) metallacycles incorporating isonitrile coordination. Indian Journal of Chemistry, 44A, 1127-1132.
- Panda, B.K. and Chakravorty, A. (2005) Carbonylation of four-membered ruthenium and osmium metallacycles incorporating an orthometallated phenolic function: New acylruthenium and arylosmium complexes. Journal of Organometallic Chemistry, 690, 3169-3175. doi:10.1016/j.jorganchem.2005.04.012
- Ghosh, P., Pramanik, A. and Chakravorty, A. (1996) Chemistry of a new family of carboxyl chelated ruthenium and osmium aryls incorporating the imine-phenol motif. Organometallics, 15, 4147-4152. doi:10.1021/om960199h
- Pan, S. and Panda, B.K. (2005) Synthesis and properties of long chain carboxyl and dicarboxyl chelated ruthenium organometallics incorporating the imine-phenol motif. Journal of Indian Chemical Society, 82, 16-20.
- Ghosh, P. and Chakravorty, A. (1997) A family of ruthenium aryls incorporating η2-bonded nitrite or nitrate and a pendent imine-phenol function. Inorganic Chemistry, 36, 64-69. doi:10.1021/ic960358p
- Pramanik, K., Ghosh, P. and Chakravorty, A. (1997) Synthesis and structure of osmium(II) organometallics incorporating a four-membered salicylideneiminium metallacycle and Os-η1-NO2 binding. Journal of the Chemical Society, Dalton Transactions, 9, 3553-3556.
- Chattopadhyay, S., Panda, B.K., Ghosh, K. and Chakravorty, A. (2001) A family of thioxanthato ruthenium and osmium aryls. Israel Journal of Chemistry, 41, 139-144. doi:10.1560/C80W-66CR-QHHL-GYU5
- Panda, B.K., Chattopadhyay, S., Ghosh, K. and Chakravorty, A. (2002) Synthesis and structure of pyridine-2- thiolato ruthenium aryls bearing a pendant imine-phenol function. Polyhedron, 21, 899-904.
- Panda, B.K., Ghosh, K., Chattopadhyay, S. and Chakravorty, A. (2003) Chemistry of a new family of aryl ruthenium species incorporating α-diimine chelation and a pendant imine-phenol function. Journal of Organometallic Chemistry, 674, 107-115. doi:10.1016/S0022-328X(03)00211-0
- Panda, B.K., Sengupta, S. and Chakravorty, A. (2004) Synthesis, structure and properties of biimidazole-chelated arylruthenium complexes. European Journal of Inorganic Chemistry, 178-184. doi:10.1002/ejic20030029
- Panda, B.K. (2004) Synthesis, characterization and emission properties of quinolin-8-olato chelated ruthenium organometallics. Journal of Chemical Science, 116, 245- 250. doi:10.1007/BF02708274
- Peacock, A.F.A., Parsons, S. and Sadler, P.J. (2007) Tuning the hydrolytic aqueous chemistry of osmium arene complexes with N, O-chelating ligands to achieve cancer cell cytotoxicity. Journal of American Chemical Society, 129, 3348-3357. doi:10.1021/ja068335p
- Kostrhunova, H., Florian, J., Novakova, O., Peacock, A.F.A., Sadler, P.J. and Brabec V. (2008) DNA interactions of monofunctional organometallic osmium(II) antitumor complexes in cell-free media. Journal of Medicinal Chemistry, 51, 3635-3643. doi:10.1021/jm701538w
- Sinha, S., Das, P.K. and Ghosh, B.K. (1995) Synthesis and characterization of new mononitrosyl complexes of osmium-containing quinoline-8-olates. Transition Metal Chemistry, 20, 59-61. doi:10.1007/BF00135403
- Leung, C.-F., Wong, T.-W., Lau, T.-C. and Wong, W.-T. (2005) Addition of carbenes to an Os(VI) nitride complex. European Journal of Inorganic Chemistry, 773-778.
- Hoffman, P.R. and Caulton, K.G. (1975) Solution structure and dynamics of five-coordinate d6 complexes. Journal of American Chemical Society, 97, 4221-4228. doi:10.1021/ja00848a012
- Sawyer, D.T. and Roberts J.L. Jr. (1974) Experimental Electrochemistry for Chemists. Wiley, New York.
- Rhofir, C., Vocelle, D. and Sandorfy, D. (1989) Ftir study of the protonation of a retinyl schiff base in chloroform/ methanol mixtures. Research on Chemical Intermediates, 12, 131-139. doi:10.1163/156856789X00096
- Ghosh P. (1997) A four-membered C, O-Chelate: Two families of ruthenium organometallics incorporating N∙∙∙O hydrogen bonding. Polyhedron, 16, 1343-1349. doi:10.1016/S0277-5387(96)00390-7
- Griffith, J.S. (1961) The Theory of Transition Metal Ions. Cambridge University Press, Cambridge.
- Bhattacharyya, S. and Chakravorty, A. (1985) Electron spin resonance studies of distorted octahedral ruthenium(III) species. Proceedings of the Indian Academy of Sciences—Chemical Sciences, 95, 159-167.
- Houten, J.V. and Watts, R.J. (1976) Temperature dependence of the photophysical and photochemical properties of the tris(2, 2'-bipyridyl)ruthenium(II) ion in aqueous solution. Journal of the American Chemical Society, 98, 4853-4858. doi:10.1021/ja00432a028
- Rillema D.P., Taghdiri D.G., Jones D.S., Worl L.A., Meyaer T.J., Levy H.A. and Keller C.D. (1987) Structure and redox and photophysical properties of a series of ruthenium heterocycles based on the ligand 2,3-bis(2- pyridyl) quinoxaline. Inorganic Chemistry, 26, 578-585. doi:10.1021/ic00251a018