Journal of Quantum Information Science
Vol.05 No.04(2015), Article ID:62462,16 pages
10.4236/jqis.2015.54016
High Capacity Hydrogen Storage in Ni Decorated Carbon Nanocone: A First-Principles Study
S. Abdel Aal*, A. S. Shalabi, K. A. Soliman
Department of Chemistry, Faculty of Science, Benha University, Benha, Egypt

Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 21 July 2015; accepted 24 December 2015; published 30 December 2015
ABSTRACT
Hydrogen adsorption and storage on Ni-decorated CNC has been investigated by using DFT. A single Ni atom decorated CNC adsorbs up to six H2 with a binding energy of 0.316 eV/H2. The interaction of 3H2 with Ni-CNC is irreversible at 603 K. In contrast, the interaction of 4H2 with Ni-CNC is reversible at 456 K. Further characterizations of the two reactions are considered in terms of the projected densities of states, electrophilicity, and statistical thermodynamic stability. The free energy of the reaction between 4H2 and Ni-CNC, surface coverage and rate constants ratio meet the ultimate targets of DOE at 11.843 atm, 0.925 and 1.041 respectively. The Ni-CNC complexes can serve as high-capacity hydrogen storage materials with capacities of up to 11.323 wt.%. It is illustrated that unless the access of oxygen to the surface is restricted, its strong bond to the decorated systems will preclude any practical use for hydrogen storage.
Keywords:
DFT, Hydrogen Storage, Nickel Complexes, CNC, Competitive Adsorption

1. Introduction
Hydrogen is considered to be an ideal fuel for many energy converters because of its low mass density and nonpolluting nature. Hydrogen can also be directly used in fuel cells in transportation applications. However, hydrogen storage, which is safe, effective and stable, remains a notable challenge to be overcome before hydrogen’s use in any automotive applications [1] [2] . Since the U.S. Department of Energy (DOE) has revised the targets for an on-board hydrogen storage medium (HSM), carbon-based materials have been considered candidates for a hydrogen storage media [3] . Accordingly, the medium should have a storage capacity of 4.50 wt.% by 2010 and 5.50 wt.% by 2015 [4] [5] .
Carbonaceous nanostructures have attracted considerable interest due to the search for new materials with specific applications. Zero-dimensional C60, one-dimensional nanotubes, and two-dimensional graphene sheets have been in focus due to their mechanical and electric transport properties [6] -[8] . Modification of the carbon nanostructure surface can be achieved by doping with B and Al [9] [10] or decorating with alkali metal [11] , alkaline earth metal [12] , transition metals (Ti, Ni, Pd and Pt) [13] -[17] , and metal hydrides [18] . These modifications can be used to increase the interaction binding energy and therefore the hydrogen storage capacity. Lee et al. [19] have reported that functionalized SWCNT with Ni atoms can yield a storage capacity of 10 wt.%.
Various types of non-planar graphitic structures, such as carbon nanocones (CNCs), have been generated by using carbon arc and other related techniques [20] . CNCs are defined as hollow structures that are composed of carbon with a conical shape. The CNCs with different disclination angles have been observed in the pyrolysis of hydrocarbons [21] . Mechanical stability and sharp tip structures at the interface of CNCs usually have a lower density than carbon nanotubes, which makes them appropriate for field emissions due to the screening effect [22] .
Carbon nanohorns (CNHs) are a subclass of the carbon nanocones CNCs family. CNHs are the fifth allotropic form of carbon. CNHs have been selected and investigated for the use in hydrogen storage capacity because a significant amount of hydrogen is evolved at ambient temperatures [23] . Although the theoretical hydrogen storage on CNCs has not been heavily researched, nickel-doped CNCs are not reported. Ming-Liang Liao [24] investigated hydrogen adsorption behaviors of single walled carbon nanocones (SWCNCs) by molecular dynamics simulations. A. Gotzias et al. [25] examined hydrogen adsorption on CNHs and CNCs by using the grand canonical Monte Carlo method. Q. Wang et al. [26] tested the ability to store hydrogen by using the gradient corrected density functional theory. A. S. Shalabi et al. [27] investigated the reactions for hydrogen storage on Ti decorated carbon nanocones (CNC) by using density functional theory (DFT) calculations. Finally, J. Yang, et al. [28] presented an overview of experimental and computational techniques employed in the field of hydrogen storage materials research.
Theoretical studies have predicted that carbon-based materials decorated with transition metal (TM) atoms, such as Ti, Ni, Sc, and V, should be capable of binding up to five hydrogen molecules per metal atom with a binding energy between 7 and 12 kcal∙mol−1 and a gravimetric density higher than 7 wt.% [11] [29] -[31] . Lee et al. [19] suggested that a single Ni atom deposited on a carbon nanotube could store up to five hydrogen molecules with a binding energy of 0.26 eV/H2 at a desorption temperature of 328 K [19] . Nickel decoration on carbonaceous nanostructures can be used to improve the capacity of these compounds toward hydrogen storage [19] [28] [32] [33] . To date, the focus of previous studies has been on hydrogen adsorption on single metal atom [14] [30] . It was suspected that metal clustering might have reduced the area of hydrogen holding and thus reduced the hydrogen uptake [34] . It has been observed that the low diffusion barrier of the Ni atom on the SWCNC leads to high mobility of metal atoms and therefore leads to large formation energy of the Ni clusters that drives atomic aggregation. For the aggregation of metals on the SWCNCs, the major driving force is metal-to-metal attraction.
The aim of this work is to examine hydrogen storage capacity and the possibility of hydride formation upon hydrogen storage operation and to determine hydrogen storage capacity in the presence of oxygen molecules at the Ni decorated CNC. Finally, aggregation of the metal atoms on the adsorption media may occur (e.g., at ambient and elevated operational temperature) and should be carefully considered before one can assess the potential of the material for hydrogen storage.
2. Computational Methodology
Electronic calculations to determine structural optimization and total energy were performed using DFT. The DFT calculations were performed by simultaneously using Becke’s three parameter exchange function (B3) and the Lee Young Parr (LYP) correlation function [35] -[38] . B3LYP correctly reproduced the thermochemistry of many compounds, including transition metal atoms [39] -[42] . The analysis of electronic and thermochemical properties of CNC molecules was performed using the Gaussian 09 program [43] . The optimal geometries were visualized using the corresponding Gauss View software.
Full geometry optimizations, without symmetry constraints, were performed for CNC with a disclination angle of 120˚, a height of 7 Å and with 72 carbon atoms. The optimal geometries of CNC, Ni-CNC, nH2-Ni-CNC, nH2-O2-Ni-CNC and nH2-Ni2-CNC (n = 1 - 6) were determined at the B3LYP level of theory using 6 - 31 G (d, p) as the basis. This set uses Gaussian type functions (GTOs), adds d-type polarization functions to carbon, f-type polarization functions to nickel, and p-type polarization functions to hydrogen. The adsorption mechanisms were determined with natural bond orbital (NBO) analysis and partial density of states (PDOS) plots, which are capable of providing a definitive description for charge redistribution. The projected densities of states (PDOS) and Fermi levels were performed using the Gauss Sum 2.2.5 program [44] .
3. Results and Discussion
3.1. Structure of Pure and Ni Doped CNC
It is well-known that the decoration of carbonaceous nanostructures with TM atoms may be an attractive alternative to improve hydrogen storage capacity. Hence, the first SWCNC, decorated with single Ni atom, has been investigated.
The adsorption energy (Eads.) of Ni atom on the surface was calculated as
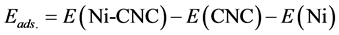 (1)
(1)
where E(Ni-CNC) and E(CNC) are the total energy of the fully relaxed Ni-CNC and CNC, respectively. E(Ni) is the energy of the isolated Ni atom. The negative binding energy corresponds to an exothermic process. The adsorption energy of Ni atom is −4.369 eV, and the average distance between Ni atom and the nearest C atoms is 1.843 Å. This strong binding interaction may originate from the hybridization between the nickel atom and CNC as the atomic projected density of states (PDOS). The corresponding interactions to the pure Ni atom and doped Ni atom have been shown in Figure 1 and Figure 2. Due to low ionization potentials of the Ni atom, the s electrons are easily donated to the CNC. These donated electrons partially fill the unoccupied states of CNC as is indicated by the PDOS near the Fermi level (shown in Figure 1). The occupied s orbital of Ni, just below the Fermi energy at −6 eV Figure 2(a), reduced in Figure 2(b) due to the charge transfer from the Ni atom to the CNC. Conversely, there is a finite probability for the CNC to donate back part of its received electrons to the low-lying d orbitals of the Ni atoms, resulting in strong hybridizations between the Ni and CNC.
The NBO analysis shows that a charge of 0.590 e is transferred from Ni to the CNC, as indicated by the decreased peak of Ni if it is supported on the CNC (Figure 2(b)). Consequently, the increased dipole moment of Ni-CNC (19.097 D), compared to the case of pristine CNC (at 12.774 D), is sufficient to enhance the van der Waal’s interaction between H2 molecules and the Ni-CNC. A fraction of the charge, between −0.223 and −0.225, is transferred from Ni and distributed to the nearest neighbor carbon atoms. This finding confirmed that Ni donates electrons to the neighboring C atoms of CNC, thereby resulting in the d-orbitals of the Ni atom overlapping with the sp2 orbitals of the C atoms. Such strong hybridizations can be observed from the resonated peaks in PDOS near −7 eV, as shown in Figure 2(b).
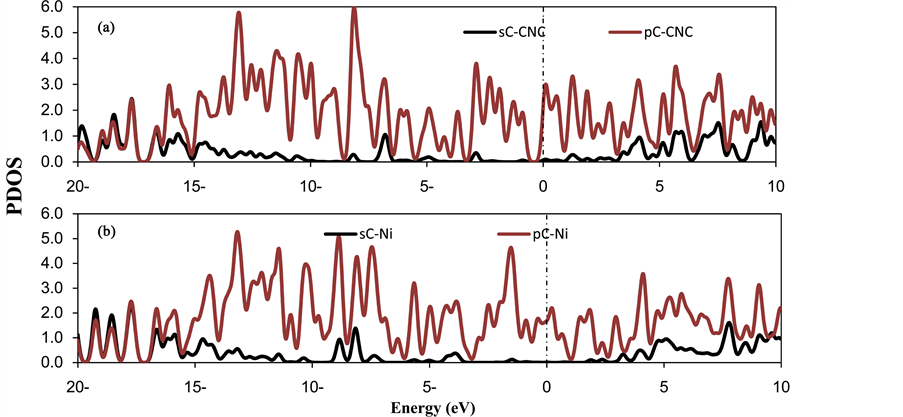
Figure 1. The partial density of states (PDOS) of (a) CNC (b) Ni doped CNC. The Fermi level is set to zero and indicated by a dotted line.
3.2. H2 Adsorption in Ni Doped CNC
Next, the interaction between the Ni/CNC complexes with H2 molecules has been investigated. The average adsorption energy of a H2 molecule at a Ni decorated CNC surface is obtained from the expression
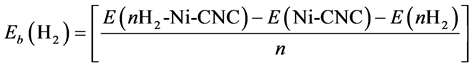 (2)
(2)
In this equation, “E” is the total energy of the optimized structure, and “n” represents the number of adsorbed H2 molecules. Negative energy indicates a stable exothermic process.
Figure 3 shows optimized geometries corresponding to a supported Ni atom that is surrounded by one to six H2 molecules. The average adsorption binding energies and their corresponding distance per hydrogen molecule are presented in Table 1. From the viewpoint of adsorption geometry, it was noticed that the first 3 H2 molecules were adsorbed on Ni by chemisorption. Further adsorption occurred as physisorption. This finding suggests that the first three hydrogen molecules interacted with the supported Ni-CNC via chemical bonds instead
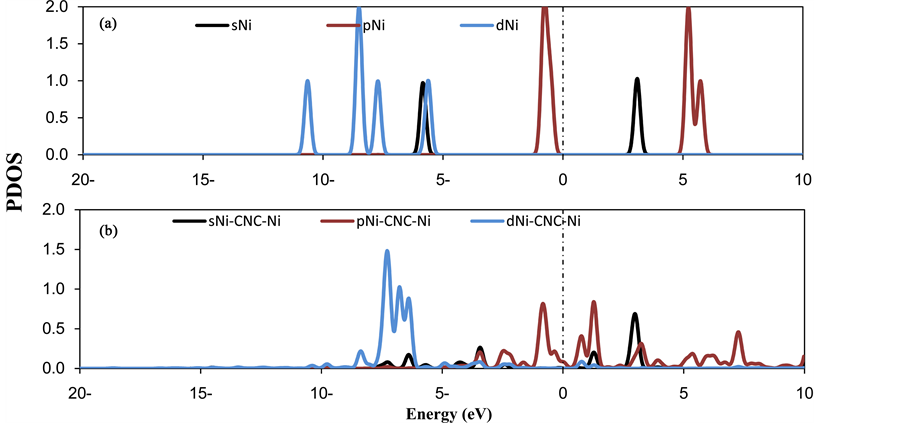
Figure 2. PDOS of (a) pure nickel atom (b) nickel doped CNC. The Fermi level is set to zero and indicated by a dotted line.

Figure 3. Optimized structure of one to six H2 adsorbed at nickel doped CNC (nH2-Ni-CNC) where n (1 - 6). Circles in grey, blue and white denote carbon, nickel and hydrogen atoms, respectively. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.

Table 1. Structural and energy properties of the optimized systems. Average adsorption energies of H2 on Ni/CNC complex Eb(H2) in eV, average distances between Ni and H2 (dNi-H), average H2 bond length (dH-H) in (Å), charges (Q) in a.u.
of the Kubas interaction. Under this condition, the H-H bonds were slightly elongated from 0.743 to 0.826 Å compared to the bond distance of 0.74 Å of an isolated H2 molecule. This result confirmed that the bonds between H2 and the supported Ni atom had both physical and chemical bond characteristics. The s electrons of hydrogen were slightly hybridized with the d orbital of Ni, which weakened the interaction between Ni and C atoms of CNC. Therefore, the average adsorption distances between the CNC and the decorated Ni atom were enhanced due to the subsequent addition of H2 molecules. The corresponding Ni-C distance also elongated to between 1.896 and 1.926 Å. The average H2-Ni adsorption distances were also found to have increased when more H2 molecules were adsorbed. These results are reported in Table 1.
It is observed that there are three different mechanisms for hydrogen adsorption on metal decorated Carbonaceous compounds: 1) polarization under the effect of the electrical field induced by the supported metal atoms; 2) the hybridization between H2 molecules and metal atoms that is modulated by the electrostatic potential induced by metal atoms; and 3) the formation of hydrogen super-molecules. The substantial charge redistribution (due to the Ni atoms donating their s electrons to the C-sp2 in the CNC) can lead to a high electric field near the Ni atoms. Consequently, the electric field causes the polarization of H2 molecules. In the interactions of H2 molecules with a decorated Ni atom, the positive charge on the Ni atom and the Coulomb’s repulsive energy also reduces. Theoretically, the interaction of H2 with TM basically arises due to the hybridization of the d levels of TM with H2 levels. To elucidate the hybridization between the Ni atoms and H2 molecules, the PDOS of three and four H2 per Ni atom on CNC have been plotted in Figure 4 and Figure 5. In Figure 4, the peaks are centered at approximately −14 - 16 eV, which corresponds to the hybridization of the Ni 3d with the H2 σ orbitals. The peaks that are centered at approximately 1 - 3 eV correspond to the hybridization of the Ni 3d with the H2σ* orbitals. Conversely, the hydrogen PDOS are shown in the range between −17 eV and −12 eV and are split into several peaks. This finding confirms the formation of bonding and antibonding states between the H2 molecules, which results in the formation of the hydrogen super-molecule.
3.3. Interactions of 3H2 and 4H2 with Ni-CNC
Two types of interactions between nH2 and Ni-CNC can be identified from Table 1: 1) irreversible interactions between nH2 (n = 1 - 3) and Ni-CNC 2) reversible interactions between nH2 (n = 4 - 6) and Ni-CNC. The irreversible interactions are outside the range of the desirable energy (−0.2 to −0.6 eV), as recommended by the DOE for practical applications. In contrast, the reversible interactions fall within this range. To characterize the nature of these two types of interactions, we considered the following theoretical descriptors.
3.3.1. Electronic Properties
The electronic descriptors, formation energy (ε), ionization potential (IP), electron affinity (EA), chemical potential (l), electronegativity (χ), chemical hardness(η) and electrophilicity index (ω) of the complexes nH2-Ni- CNC (where n = 3, 4) were considered in Table 2.
The IP and EA can be calculated from the highest occupied (HOMO) and the lowest unoccupied (LUMO)
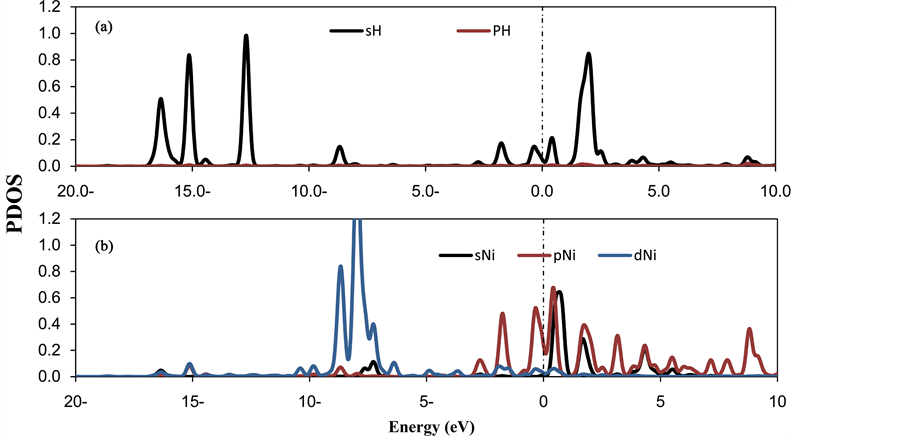
Figure 4. PDOS of (a) three H2 molecules, (b) decorated Niatomsin the (3H2-Ni-CNC) system. The Fermi level is set to zero and indicated by a dotted line.

Figure 5. PDOS of (a) four H2 molecules (b) decorated Ni atoms in the (4H2-Ni-CNC) system. The Fermi level is set to zero and indicated by a dotted line.
Table 2. The total energy (Etotal/Eh), formation energy (ε/eV), energy gap (Eg), ionization potential (I/eV), electron affinity (A/eV), chemical hardness (η/eV), electronegativity (χ/eV), and electrophilicity (ω/eV) of the complexes nH2-Ni-CNC (for n = 3, 4).
molecular orbital energies using Koopmans’ approximation [45] where, IP―HOMO and EA―LUMO. The chemical potential μ and χ are defined as [45]
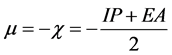 .
.
Pearson [46] introduced two parameters ‘chemical hardness (η)’ and ‘chemical softness (S)’ to account for the stability of a molecule. Hardness (η) can also be expressed in terms of HOMO and LUMO, implying a finite
difference approach [45] , as follows: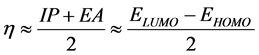 . The softness can be defined as
. The softness can be defined as
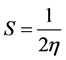 [47] . Parr [48] defined electrophilicity index (ω) as:
[47] . Parr [48] defined electrophilicity index (ω) as:
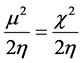
which measures the energy stabilization when the molecule accepts an additional electrical charge from the environment. It is noted that a lower energy gap (Eg) between the LUMO and HOMO of a compound implies a greater and easier possibility of the electron transition between these energy levels. Additionally, a small value for Eg for the compound is an indicator of lower chemical stability. In other words, the respective chemical hardness (η) should be low and electrophilicity (ω), which is a parameter indicating reactivity, should be high.
The results presented in Table 2 show that the 4H2-Ni-CNC shows a notably lower value of Eg and η and a maximum value compared to the 3H2-Ni-CNC. The η value for a compound essentially represents how chemically hard a compound is. Because 4H2-Ni-CNC is chemically softer, it implies that it is a better candidate in electronic transport. The electrophilicity index () is one of the DFT based parameters which quantifies how reactive a molecule or compound is.It is clear from the present analysis that the lower Eg and η values and higher value for the 4H2-Ni-CNC (compared to the 3H2-Ni-CNC compound) implies its favorable nature for the possible electronic transport and conductivity. Polarizability is closely associated with the softness (S) of a system, where the more polarizable a chemical system is, the softer the compound, and vice versa.
3.3.2. Thermodynamic Properties
The thermodynamics of the hydrogen storage reaction is one of the most fundamental properties of the hydrogen storage material. Thermodynamic properties indicate that the pressure of desorbed hydrogen and operating temperature are required for a fuel cell. The heat requirements for desorption and the potential for on-board recharge (or “reversibility”) are also associated with the thermodynamic properties of the storage reaction.
The thermodynamic properties of the 3H2-Ni-CNC and 4H2-Ni-CNC complexes can be calculated from standard statistical mechanical equations to include the finite-temperature translational, rotational and vibrational energies. The enthalpy (Hr) can be calculated as follows:
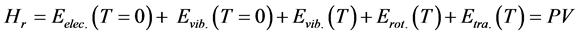 (4)
(4)
where Eelec.(T = 0 K) is the total electronic energy, Evib.(T = 0 K) is the zero point vibrational energy (ZPVE), which is a linear sum of the fundamental harmonic frequencies, and Evib.(T), Erot.(T) and Etra.(T) are vibrational, rotational, and translational contributions, respectively.
Similarly, the total entropy (S) can be expressed as
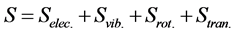 (5)
(5)
where Selec., Svib., Srot., and Stran. are the electronic, vibrational, rotational, and translational terms, respectively. The change in the standard Gibbs free energy is given by
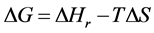 (6)
(6)
where
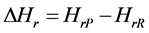 and
and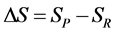 , or simply
, or simply
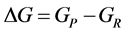 (7)
(7)
where P = product, and R = reactants.
The results of thermochemical properties of entropy, enthalpy, Gibbs free energy changes, thermal energies (Et), and heat capacities at constant volume (Cv), for the reactions 8 and 9 processed from 100 K to 700 K are presented in Table 3.
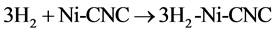 (8)
(8)
and
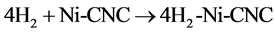 (9)
(9)
It can be clearly deduced from this table that as temperature (T) increases, the values of enthalpy (H) and entropy (S) increase, while the Gibbs free energy (G) decreases. The negative Gibbs free energy (G) also indicates an exothermic process, where the system releases energy to its surroundings during the adsorption process. The system then gradually reaches a stable condition of equilibrium. Thus, the reaction with G = −31.276 kcal/mol at 100 K has a higher probability of occurring than that of G = 6.036 kcal/mol at 700 K, (shown in Figure 6).
The polynomial regression
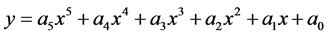 (10)
(10)
was subsequently applied to reactions (8) and (9) from 100 to 700 K after replacing (y) by (T) and (x) by (ΔG). The residual sum of squares (rss = 0) value at ΔG = 0 occurs at T = 603 K for reaction (8), and at T = 454 for reaction (9). Therefore, reactions (8) and (9) reverse above 603 K and 454 K, respectively. This implies that the two complexes, 3H2-Ni-CNC and 4H2-Ni-CNC, tend to release all hydrogen molecules above 603 and 454 K, respectively. In other words, the higher the amount of hydrogen molecules at the Ni-CNC interface, the lower temperature of hydrogenation. This can be explained by the relatively lower stability of the higher hydrogenated Ni-CNC. The other statistical thermodynamic parameters also characterized the two types of interactions, where ΔS, Et, and Cv values of the reversible interaction were always greater than those of irrreversible interaction at the same temperature range.
3.3.3. Optimal Reaction Enthalpy
The temperature and pressure range at which a hydrogen storage system should operate is dictated by the environment and the requirements of the fuel cell. This approach translates vehicle operating constraints into thermodynamic constraints, which can be used to guide material development. Enthalpy is considered as the quantity of heat that must be added to (or subtracted from) the system during hydrogen release (or uptake). It is demonstrated that materials that have large enthalpies of desorption are undesirable because they require high temperatures for hydrogen release. In principle, a system with a small desorption enthalpy is capable of liberating
Table 3. Temperature (T/K), Gibbs free energy ΔG (kcal/mol), enthalpy change ΔH (kcal/mol), entropy change ΔS (cal/mol.K), thermal energy Et (K.cal/mol), and heat capacity at constant volume Cv (cal/mol.K) of the complexes nH2-Ni-CNC (for n = 3, 4).
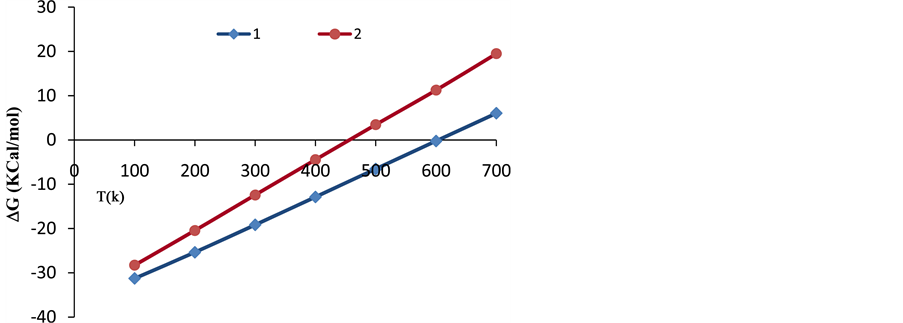
Figure 6. Variations of Gibbs free energy (ΔG) with temperature (T) of the complexes 1: 3H2-Ni-CNC and 2: 4H2-Ni-CNC.
hydrogen at low temperatures but will require notably high pressures to recharge. Consequently, the enthalpy is an important engineering design parameter [28] . It is possible to identify a range of reaction enthalpies that satisfy the ultimate DOE targets of temperatures and pressures by using the van’t Hoff equation. These targeted temperatures and pressures are presented in Table 4. The van’t Hoff equation can describe the equilibrium between gas phase hydrogen and one or more condensed phases [49] :
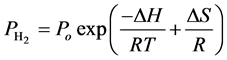 (11)
(11)
where
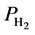 and Po are referred to the equilibrium pressure and a reference pressure (typically atmospheric pressure), respectively, T is the absolute temperature, R is the gas constant, S and H respectively represent the change in entropy and enthalpy, which accompanies the hydrogen storage reaction to describe H2 uptake in sorbent materials [28] .
and Po are referred to the equilibrium pressure and a reference pressure (typically atmospheric pressure), respectively, T is the absolute temperature, R is the gas constant, S and H respectively represent the change in entropy and enthalpy, which accompanies the hydrogen storage reaction to describe H2 uptake in sorbent materials [28] .
The thermodynamics of 4H2-Ni-CNC complex have been calibrated with the ultimate targets of DOE at (−0.2 to −0.6 eV) for physisorption, (−40˚C to 105˚C) for (min./max.) temperature, (0.3/1.2MPa) for (min./max.) pressure, and (7.5%). The results shown in Table 4 illustrate that the equilibrium pressure of 1.2 MPa is equal to the ultimate target for maximum H2 pressure that is required in the fuel cell. The desorption reaction requires a desorption enthalpy of −25.45 kJ/mol H2, which is in agreement with the previous theoretical calculations of Yang et al. [28] , who reported efficiency considerations that target the lower third of this range (H = 20 - 30 kJ/mol H2), are optimal.
In addition, the Langmuir isotherm is based on the monolayer adsorption on the active sites of the adsorbent. Langmuir suggested that the mechanism of the adsorption process is defined as Ag + S = AS, where A is a gas molecule and S is an adsorption site. The direct and inverse rate constants are k and k−1. Surface coverage, which is defined as the fraction of the number of adsorption sites occupied in the equilibrium, is shown in Equation (12).
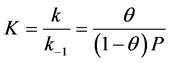 (12)
(12)
where P is the partial pressure of the gas. Substituting the values of (0.925) and P (11.843 atm.) of the reversible reaction from Equation (9) gives the value (k = 1.041).
3.4. Effect of Clustering Ni on CNC
For the next step, we examined the interactions and clustering effects of Ni ((e.g. Ni dimer at CNC) on the nature of hydrogen uptake. Full geometry optimizations at the B3LYP/6-31g (d, p) level of theory were carried out for the complexes nH2-Ni2-CNC (n = 2, 4, 6, 8, 10, 12). The corresponding geometry is shown in Figure 7. The average adsorption energy, of n = 2, 4, 6, 8, 10, 12 hydrogen molecules at Ni dimer decorated CNC, is obtained from the expression,
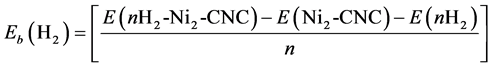
The average adsorption energies of with their geometric parameters are listed in Table 5. The results indicate that the bond length of Ni-Ni is 2.43 Å and it is elongated compared to that of the free Ni dimer at 2.01 Å. The bond lengths of Ni-C are higher than those in Figure 3, due to the internal interactions between the two Ni atoms that weaken the Ni?C interaction. Conversely, the interaction between CNC and Ni dimer weakens the Ni-Ni bond. The average adsorption energies of nH2-Ni-CNC (n = 1 - 6) complexes are approximately −1.068, −0.926, −0.622, −0.468, −0.387 and −0.316 eV per H2, which is stronger than that on Ni dimer at −1.057, −0.820, −0.546, −0.416, −0.333 and −0.278 eV, respectively. These values indicate that the formation of the Ni dimer also weakens the interaction between the H2 and the doped CNC. Figure 7 shows the saturated hydrogen uptake on a Ni2 cluster. Note that 2 of the 6 adsorbed hydrogen molecules are completely dissociated and bonded in the atomic form (irreversible reaction), while the other four H2 remain in molecular form (reversible reaction). This suggests that clustering of Ni not only alters the binding energy of the H2 molecules but also the nature of H bonding. This adsorption behavior may be explained based on the charge-transfer mechanism discussed earlier. In fact, the average charge transfer from the Ni dimer of nH2-Ni2-CNC (n = 2 - 12) complexes to the CNC is only 0.48 and 0.39 eV per Ni atom, which is less than that of a single Ni atom of nH2-Ni-CNC (n = 1 - 6)
Table 4. The (min./max.) temperature (T), (min./max.) pressure (P), free energy change (ΔG), enthalpy change (ΔH), and surface coverage (θ) of the highest hydrogen storage capacity reaction 4H2 + Ni-CNC = 4H2-Ni-CNC.
Table 5. Average adsorption energies of H2 Eb(H2) on Ni dimer at CNC complex, average distance between Ni and CNC d(Ni-CNC), average distances between Ni and H2 (dNi-H), average H2 bond length (dH-H), charges (Q) in a.u. and the expected hydrogen storage capacity (wt.%) of the complexes nH2-Ni2-CNC (n = 2, 4, 6, 8, 10, 12). Complete surface coverage affords up to 24 Ni and 144 H2. Energies are given in eV and lengths in Å.
Optimized Ni―Ni free without CNC = 2.0085 Å.
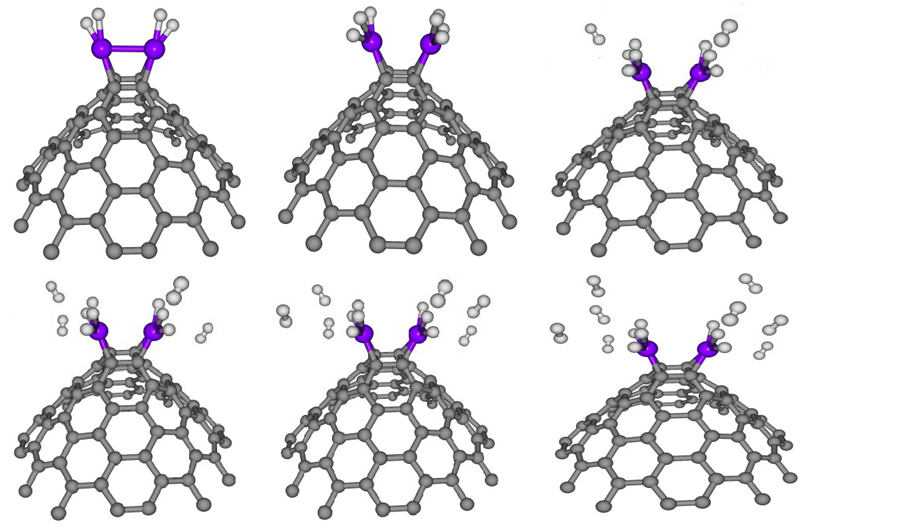
Figure 7. Optimized structure of one to six H2 adsorbed at nickel dimer doped CNC (nH2-Ni2-CNC) where n [1 - 6]. The circles in grey, blue and white denote carbon, nickel and hydrogen atoms, respectively. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
complexes to the CNC at 0.61 and 0.43 eV, respectively.
The expected gravimetric hydrogen storage capacities for nH2-Ni2-CNC (n = 2 - 12) complexes are listed in Table 5. For nH2-Ni2-CNC (n = 2, 4) complexes, the average adsorption energies of H2 are −1.057 and −0.820 eV, respectively, which is beyond the highest adsorption requirement (−0.60 eV). The hydrogen storage capacities of these two configurations are calculated to be 2.084% and 4.082%, respectively. The average adsorption energies of nH2-Ni2-CNC (n = 6, 8, 10, 12) complexes are −0.55, −0.50, −0.42 and −0.35 eV, respectively, which meet the DOE energy domain (−0.20 to −0.60 eV). The hydrogen storage capacities of these four configurations are expected to be 6.001%, 7.845%, 9.617%, and 11.323%, respectively. Consequently, these results indicate that the Ni-decorated C72 stories capable of storing 144 hydrogen molecules attached to 24 Ni atoms. Thus, the hydrogen storage capacity is up to 11.323 wt.%, which exceeds the DOE target for 2015 [5] .
3.5. Interaction of Oxygen with Ni Decoration
Decoration of different carbonaceous materials with metals has been investigated as an alternative to improve their capacity for hydrogen storage [9] [14] [19] [30] [32] [50] [51] . To follow experimental conditions, the effect of oxygen contamination from any residual atmosphere or pre-existing oxygen impurities on the nanotube surface, was examined. Rojas et al. [52] found that low quantities of oxygen present in the gas phase should yield the oxidation of the titanium atoms, even when hydrogen is stored in the system. They concluded that if the experimental system is exposed to air, titanium atoms on these surfaces are expected to oxidize to titanium dioxide. Felten et al. [53] studied the role of oxygen at the interface between titanium and carbon nanotubes. They observed that the presence of oxygen significantly weakened the Ti-CNT interaction and the Ti atoms at the surface preferentially bonded to oxygenated sites. Among these, oxygen molecules were found to be a strong inhibitor competitor for hydrogen adsorption.
To study the competition between O2 and H2 molecules at the Ni-CNC surface, we calculated the oxygen displacement energy that corresponded to the energy required to replace the adsorbed oxygen atom by nH2 adsorbed hydrogen molecules. This displacement energy was calculated according to
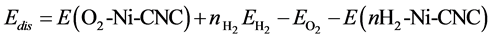 (13)
(13)
where E(nH2-Ni-CNC) and E(O2-Ni-CNC) denote the total energy of the substrate (Ni-CNC system) in the presence of nH2 hydrogen molecules and O2 oxygen molecules, respectively, n is the number of hydrogen molecules, and EO2 and EH2 are the energies of the isolated oxygen and hydrogen molecules respectively. In all cases, the values for Edis are shown in Table 6, which indicate that the replacement of oxygen by hydrogen is energetically impeded.
The results show that the two O atoms bind strongly to a single Ni atom (−3.799 eV) and Ni-O bonds of 1.76 Å, which significantly weakens the Ni-C interaction (bonds dilated to 2.02 Å). Consequently, the quantity of oxygen present in the initial surface layer crucially controls the Ni-CNC interaction. This is because initial Ni deposition preferentially forms Ni-O bonds, strongly reducing the interaction between the hydrogen molecules and Ni-CNC.
The average adsorption energy of H2 molecule at the surface of O2-Ni-CNC was calculated according to
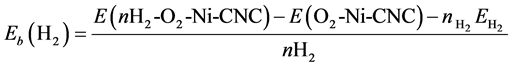 (14)
(14)
where E(nH2-O2-Ni-CNC) and E(O2-Ni-CNC) are defined earlier. The geometries obtained are shown in Figure 8. The results are reported in Table 6 and show that even at low oxygen concentrations, the interaction of the O2 molecule with the Ni decoration on CNC leads to the irreversible formation of nickel dioxide, and displaces the hydrogen molecule. The resulting nickel dioxide produces a lower storage capability than nickel at CNC. The formation of nickel dioxide was confirmed by further theoretical calculations [54] .
The destabilization of the O2 molecule by the adsorbed hydrogen molecule may be expressed as
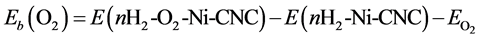 (15)
(15)
Eb(O2) is shown in the third column of Table 6, where it is found that the addition of hydrogen molecules exhibited a decrease in the binding energy of the oxygen molecules. However, O2 molecules remained strongly adsorbed on the surface, even in the presence six hydrogen molecules, with a binding energy of −2.633 eV.
Table 6. Edis Oxygen displacement energy corresponding to the energy required to replace the adsorbed oxygen atom by nH2 adsorbed hydrogen molecules. Eb(H2) adsorption energy (per hydrogen atom) of hydrogen species on a surface where one oxygen molecule is already adsorbed on the Ni atom. Eb(O2) binding energy of the oxygen molecule on a surface where nH2 hydrogen molecules are already adsorbed on the Ni atom.
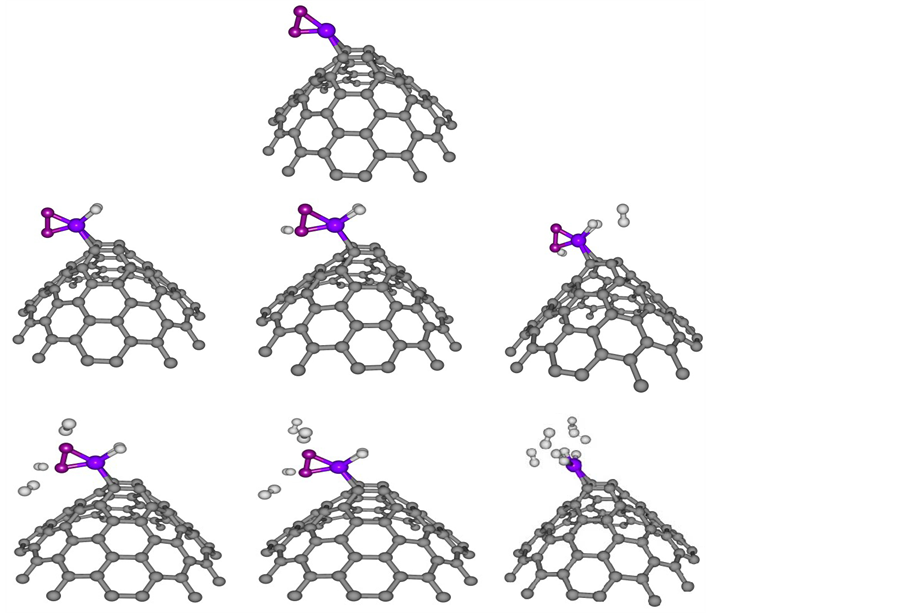
Figure 8. Optimized structure of competition between hydrogen and oxygen adsorption on the nickel doped CNC of one to six H2. Balls in grey, blue, red and white denote carbon, nickel, oxygen and hydrogen atoms respectively. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
Consequently, under normal conditions, oxygen interferes strongly with hydrogen adsorption.
The calculations show strong qualitative variations in the charge transfer behavior due to the adsorption of O2 molecule at Ni-CNC surface. From Table 1, the interaction of Ni atom on CNC shows a strong net charge transfer from the Ni to the CNC resulting in a Ni charge of +0.589 e, where the H2 molecules transfer charge to the decorated Ni atom. In contrast, the interaction of oxygen molecules with Ni-CNC causes the charge at the Ni atom to increase to +0.971 e. This is due to the O2 molecules being more electronegative and withdrawing electrons from the metal charge on the oxygen atoms (−0.655 e). Therefore, the dipole moment of O2-Ni-CNC and Ni-CNC is calculated to be 10.831 and 19.097, respectively. This is sufficient to reduce the van der Waal’s interaction between hydrogen and O2-Ni-CNC. Figure 9 confirms the results in Table 6 and the adsorption properties that were discussed earlier. Ao et al. [55] [56] showed that the application of a perpendicular electric field may act as a catalyst for the dissociative adsorption of hydrogen on pristine and nitrogen-doped graphene. Therefore, the application of an external field could be an alternative to favor the selective hydrogen adsorption. This subject will be a topic of future research.
4. Conclusions
Using DFT calculations, Ni-decorated CNC has been investigated for hydrogen storage applications. PDOS and NBO analysis have been performed to understand the H2 adsorption mechanism. The adsorption mechanism for H2 on Ni decorated CNC was primarily attributed to the polarization induced by electrostatic field of metal atoms on CNC and the hybridization between the Ni atom and hydrogen molecules. Two types of reactions (reversible and irresversible) were characterized in terms of (PDOS), electrophilicity, and statistical thermodynamic
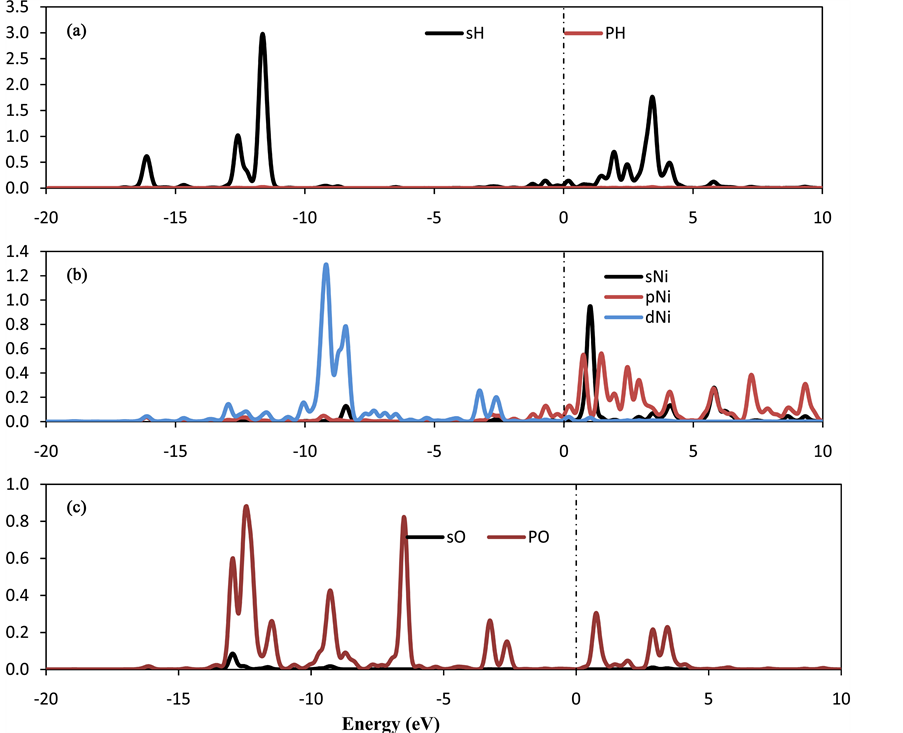
Figure 9. PDOS of (a) oxygen molecule (b) decorated Ni atoms in the (4H2-O2-Ni-CNC) system. The Fermi level is set to zero and indicated by a dotted line.
stability descriptors. The thermodynamics of the nH2-Ni-CNC (n = 3, 4) reactions with reference to the ultimate targets of the DOE for physisorption, gravimetric hydrogen storage capacity, minimum and maximum temperatures and pressures, and optimal reaction enthalpy, were analyzed in considerable detail.
The adsorption binding energy meets the ultimate values for achieving adsorption and desorption at near ambient conditions. However, due to lower migration barriers and strong metal-to-metal attraction, aggregation of metal atoms in the form of a metallic layer or cluster on typical experimental CNC seems inevitable. It was observed that the structure where Ni atoms cluster was −0.738 eV lower in energy than when they remained isolated. Ni2-CNC can bind six H2 molecules and their corresponding storage capacity is 11.323 wt.%, which is higher than the revised 2015 target of U.S. DOE. Consequently, Ni-CNC could be used as a high capacity hydrogen storage medium in onboard automobile applications. On the other hand, the present results show that the competitive adsorption between H2 and O2 molecules strongly favors oxygen. Therefore, O2 strongly reduces the interaction of hydrogen molecules with the Ni-CNC. Consequently, this interaction will inhibit the practical use for hydrogen storage.
Cite this paper
S. AbdelAal,A. S. Shalabi, K. A.Soliman, (2015) High Capacity Hydrogen Storage in Ni Decorated Carbon Nanocone: A First-Principles Study. Journal of Quantum Information Science,05,134-149. doi: 10.4236/jqis.2015.54016
References
- 1. Xu, W.C., Takahashi, K., Matsuo, Y., Hattori, Y., Kumagai, M., Ishiyama, S., et al. (2007) Investigation of Hydrogen Storage Capacity of Various Carbon Materials. International Journal of Hydrogen Energy, 32, 2504-2512.
http://dx.doi.org/10.1016/j.ijhydene.2006.11.012 - 2. Sakintuna, B., Lamari-Darkrim, F. and Hirscher, M. (2007) Metal Hydride Materials for Solid Hydrogen Storage: A Review. International Journal of Hydrogen Energy, 32, 1121-1140.
http://dx.doi.org/10.1016/j.ijhydene.2006.11.022 - 3. Zhang, Z.W., Li, J.C. and Jiang, Q. (2010) Hydrogen Adsorption on Eu/SWCNT Systems: A DFT Study. The Journal of Physical Chemistry C, 114, 7733-7737.
http://dx.doi.org/10.1021/jp100017y - 4. Schlapbach, L. (2009) Technology: Hydrogen-Fuelled Vehicles. Nature, 460, 809-811.
http://dx.doi.org/10.1038/460809a - 5. U.S. Department of Energy.
http://www.eere.energy.gov/hydrogen and fuel cells/mypp/ - 6. Kuzmin, S. and Duley, W.W. (2010) Ab Initio Calculations of a New Type of Tubular Carbon Molecule Based on Multi-Layered Cyclic C6 Structures. Physics Letters A, 374, 1374-1379.
- 7. Baei, M.T., Peyghan, A.A., Bagheri, Z. and Tabar, M.B. (2012) B-Doping Makes the Carbon Nanocones Sensitive towards NO Molecules. Physics Letters A, 377, 107-111.
http://dx.doi.org/10.1016/j.physleta.2012.11.006 - 8. Iijima, S. (1991) Helical Microtubules of Graphitic Carbon. Nature, 354, 56-58.
http://dx.doi.org/10.1038/354056a0 - 9. Surya, V.J., Iyakutti, K., Venkataramanan, N., Mizuseki, H. and Kawazoe, Y. (2010) The Role of Li and Ni Metals in the Adsorbate Complex and Their Effect on the Hydrogen Storage Capacity of Single Walled Carbon Nanotubes Coated with Metal Hydrides, LiH and NiH2. International Journal of Hydrogen Energy, 35, 2368-2375.
http://dx.doi.org/10.1016/j.ijhydene.2010.01.001 - 10. Cui, S.S., Zhao, N.Q., Shi, C.S., Feng, C., He, C.N., Li, J.J. and Liu, E.Z. (2014) Effect of Hydrogen Molecule Dissociation on Hydrogen Storage Capacity of Graphene with Metal Atom Decorated. The Journal of Physical Chemistry C, 118, 839-844.
http://dx.doi.org/10.1021/jp409594r - 11. Wang, F.D., Zhang, N.N., Li, Y.H., Tang, S.W. and Sun, H. (2013) High-Capacity Hydrogen Storage of Na-Decorated Grapheme with Boron Substitution: First-Principles Calculations. Chemical Physics Letters, 555, 212-216.
http://dx.doi.org/10.1016/j.cplett.2012.11.015 - 12. Hussain, T., Pathak, B., Ramzan, M., Maark, T.A., Ahuja, R. and De Sarkar, A. (2012) Calcium Doped Graphane as a Hydrogen Storage Material. Applied Physics Letters, 100, Article ID: 183902.
http://dx.doi.org/10.1063/1.4710526 - 13. Reyhani, A., Mortazavi, S.Z., Mirershadi, S., Moshfegh, A.Z., Parvin, P. and Golikand, A.N. (2011) Hydrogen Storage in Decorated Multiwalled Carbon Nanotubes by Ca, Co, Fe, Ni, and Pd Nanoparticles under Ambient Conditions. The Journal of Physical Chemistry C, 115, 6994-7001.
http://dx.doi.org/10.1021/jp108797p - 14. Yildirim, T. and Ciraci, S. (2005) Titanium-Decorated Carbon Nanotubes as a Potential High-Capacity Hydrogen Storage Medium. Physical Review Letters, 94, Article ID: 175501.
http://dx.doi.org/10.1103/PhysRevLett.94.175501 - 15. Lopez-Corral, I., German, E., Juan, A., Volpe, M.A. and Brizuela, G.P. (2012) Hydrogen Adsorption on Palladium Dimer Decorated Graphene: A Bonding Study. International Journal of Hydrogen Energy, 37, 6653-6665.
http://dx.doi.org/10.1016/j.ijhydene.2012.01.039 - 16. Liu, Y., Brown, C.M., Neumann, D.A., Geohegan, D.B., Puretzky, A.A. and Rouleau, C.M. (2012) Metal-Assisted Hydrogen Storage on Pt-Decorated Single-Walled Carbon Nanohorns. Carbon, 50, 4953-4964.
http://dx.doi.org/10.1016/j.carbon.2012.06.028 - 17. Vinayan, B.P., Sethupathi, K. and Ramaprabhu, S. (2013) Facile Synthesis of Triangular Shaped Palladium Nanoparticles Decorated Nitrogen Doped Graphene and Their Catalytic Study for Renewable Energy Applications. International Journal of Hydrogen Energy, 38, 2240-2250.
http://dx.doi.org/10.1016/j.ijhydene.2012.11.091 - 18. Seenithurai, S., Pandyan, R.K., Kumar, S.V. and Mahendran, M. (2013) H2 Adsorption in Ni and Passivated Ni Doped 4 Å Single Walled Carbon Nanotube. International Journal of Hydrogen Energy, 38, 7376-7381.
http://dx.doi.org/10.1016/j.ijhydene.2013.04.085 - 19. Lee, J.W., Kim, H.S., Lee, J.Y. and Kanga, J.K. (2006) Hydrogen Storage and Desorption Properties of Ni-Dispersed Carbon Nanotubes. Applied Physics Letters, 88, Article ID: 143126.
http://dx.doi.org/10.1063/1.2189587 - 20. Iijima, S., Ichihashi, T. and Ando, Y. (1992) Pentagons, Heptagons and Negative Curvature in Graphite Microtubule Growth. Nature, 356, 776-778.
http://dx.doi.org/10.1038/356776a0 - 21. Ge, M. and Sattler, K. (1994) Observation of Fullerene Cones. Chemical Physics Letters, 220, 192-196.
http://dx.doi.org/10.1016/0009-2614(94)00167-7 - 22. Huang, C.J., Chih, Y.K., Hwang, J., Lee, A.P. and Kou, C.S. (2003) Field Emission from Amorphous-Carbon Nanotips on Copper. Journal of Applied Physics, 94, 6796.
http://dx.doi.org/10.1063/1.1620681 - 23. Matelloni, P., Grant, D.M. and Walker, G.S. (2009) Supporting Metal Catalysts on Modified Carbon Nanocones to Optimize Dispersion and Particle Size. MRS Proceedings, 1216. http://dx.doi.org/10.1557/PROC-1216-W02-02
- 24. Liao, M.-L. (2012) A Study on Hydrogen Adsorption Behaviors of Open-Tip Carbon Nanocones. Journal of Nanoparticle Research, 14, 837.
http://dx.doi.org/10.1007/s11051-012-0837-1 - 25. Gotzias, A., Heiberg-Andersen, H., Kainourgiakis, M. and Steriotis, T. (2011) A Grand Canonical Monte Carlo Study of Hydrogen Adsorption in Carbon Nanohorns and Nanocones at 77K. Carbon, 49, 2715-2724.
http://dx.doi.org/10.1016/j.carbon.2011.02.062 - 26. Wang, Q., Sun, Q., Jena, P. and Kawazoe, Y. (2009) Potential of AlN Nanostructures as Hydrogen Storage Materials. ACS Nano, 3, 621-626.
http://dx.doi.org/10.1021/nn800815e - 27. Shalabi, A.S., Taha, H.O., Soliman, K.A. and Abeld Aal, S. (2014) Hydrogen Storage Reactions on Titanium Decorated Carbon Nanocones Theoretical Study. Journal of Power Sources, 271, 32-41.
http://dx.doi.org/10.1016/j.jpowsour.2014.07.158 - 28. Yang, J., Sudik, A., Wolverton, C. and Siegelw, D.J. (2010) High Capacity Hydrogenstorage Materials: Attributes for Automotive Applications and Techniques for Materials Discovery. Chemical Society Reviews, 39, 656-675.
http://dx.doi.org/10.1039/B802882F - 29. Kubas, G.J. and Organo, J. (2001) Metal-Dihydrogen and σ-Bond Coordination: The Consummate Extension of the Dewar-Chatt-Duncanson Model for Metal-Olefin π Bonding. Journal of Organometallic Chemistry, 635, 37-68.
http://dx.doi.org/10.1016/S0022-328X(01)01066-X - 30. Durgun, E., Ciraci, S. and Yildirim, T. (2008) Functionalization of Carbon-Based Nanostructures with Light Transition-Metal Atoms for Hydrogen Storage. Physical Review B, 77, Article ID: 085405.
http://dx.doi.org/10.1103/PhysRevB.77.085405 - 31. Samolia, M. and Kumar, T.J.D. (2014) Hydrogen Sorption Efficiency of Titanium-Functionalized Mg-BN Framework. The Journal of Physical Chemistry C, 118, 10859-10866.
http://dx.doi.org/10.1021/jp501722z - 32. Zielinski, M., Wojcieszak, R., Monteverdi, S., Mercy, M. and Bettahar, M.M. (2007) Hydrogen Storage in Nickel Catalysts Supported on Activated Carbon. International Journal of Hydrogen Energy, 32, 1024-1032.
http://dx.doi.org/10.1016/j.ijhydene.2006.07.004 - 33. Ayala, P., Freire, F.L., Gu, L., Smith, D.J., Solo′rzano, I.G. and Macedo, D.W. (2006) Decorating Carbon Nanotubes with Nanostructured Nickel Particles via Chemical Methods. Chemical Physics Letters, 431, 104-109.
http://dx.doi.org/10.1016/j.cplett.2006.09.039 - 34. Sun, Q., Wang, Q., Jena, P. and Kawazoe, Y. (2005) Clustering of Ti on a C60 Surface and Its Effect on Hydrogen Storage. Journal of the American Chemical Society, 127, 14582-14583.
http://dx.doi.org/10.1021/ja0550125 - 35. Becke, A.D. (1993) Density-Functional Thermochemistry. III. The Role of Exact Exchange. The Journal of Chemical Physics, 98, 5648.
http://dx.doi.org/10.1063/1.464913 - 36. Vosko, S.H., Wilk, L. and Nusair, M. (1980) Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Canadian Journal of Physics, 58, 1200-1211.
http://dx.doi.org/10.1139/p80-159 - 37. Becke, A.D. (1988) Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Physical Review A, 38, 3098-3100.
http://dx.doi.org/10.1103/PhysRevA.38.3098 - 38. Lee, C., Yang, W. and Parr, R.G. (1988) Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Physical Review B, 37, 785-789. http://dx.doi.org/10.1103/PhysRevB.37.785
- 39. Ricca, A. and Bauschlicher, C.W. (1994) Successive Binding Energies of Fe(CO)5+. The Journal of Physical Chemistry, 98, 12899-12903.
http://dx.doi.org/10.1021/j100100a015 - 40. Russo, T.V., Martin, R.I. and Hay, P.J. (1995) Application of Gradient-Corrected Density Functional Theory to the Structures and Thermochemistries of ScF3, TiF4, VF5, and CrF6. The Journal of Chemical Physics, 102, 8023.
http://dx.doi.org/10.1063/1.469000 - 41. Siegbahn, P.E. and Crabtree, R.H. (1997) Mechanism of C-H Activation by Diiron Methane Monooxygenases: Quantum Chemical Studies. Journal of the American Chemical Society, 119, 3103-3113.
http://dx.doi.org/10.1021/ja963939m - 42. Wolverton, C., Siegel, D.J., Akbarzadeh, A.R. and Ozolin, V. (2008) Discovery of Novel Hydrogen Storage Materials: An Atomic Scale Computational Approach. Journal of Physics: Condensed Matter, 20, Article ID: 064228.
http://dx.doi.org/10.1088/0953-8984/20/6/064228 - 43. Frisch, M.J., et al. (2009) Gaussian 09. Gaussian Inc., Pittsburgh.
- 44. O’Boyle, N.M., Tenderholt, A.L. and Langner, K.M. (2008) Cclib: A Library for Package-Independent Computational Chemistry Algorithms. Journal of Computational Chemistry, 29, 839-845.
http://dx.doi.org/10.1002/jcc.20823 - 45. Parr, R.G. and Yang, W. (1989) Density Functional Theory of Atoms and Molecules. Oxford University Press, New York.
- 46. Pearson, R.G. (1973) Hard and Soft Acids and Bases. Dowden, Hutchinson and Ross, Inc., Stroudsburg.
- 47. Yang, W. and Parr, R.G. (1985) Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis. Proceedings of the National Academy of Sciences of the United States of America, 82, 6723-6726.
http://dx.doi.org/10.1073/pnas.82.20.6723 - 48. Parr, R.G., Szentpály, L.V. and Liu, S.B. (1999) Electrophilicity Index. Journal of the American Chemical Society, 121, 1922-1924.
http://dx.doi.org/10.1021/ja983494x - 49. Griessen, R. and Riesterer, T. (1988) Heat of Formation Models. In: Schlapbach, L., Ed., Topics in Applied Physics: Hydrogen in Intermetallic Compounds I, Springer, Berlin, Heidelberg, 219-284.
- 50. Kim, H.S., Lee, H., Han, K.S., Kim, J.H., Song, M.S., Park, M.S., et al. (2005) Hydrogen Storage in Ni Nanoparticle-Dispersed Multiwalled Carbon Nanotubes. The Journal of Physical Chemistry B, 109, 8983-8986.
http://dx.doi.org/10.1021/jp044727b - 51. Ni, M., Huang, L., Guo, L. and Zeng, Z. (2010) Hydrogen Storage in Li-Doped Charged Single-Walled Carbon Nanotubes. International Journal of Hydrogen Energy, 35, 3546-3549.
http://dx.doi.org/10.1016/j.ijhydene.2010.01.103 - 52. Rojas, M.I. and Leiva, E.P.M. (2007) Density Functional Theory Study of a Graphene Sheet Modified with Titanium in Contact with Different Adsorbates. Physical Review B, 76, Article ID: 155415.
http://dx.doi.org/10.1103/PhysRevB.76.155415 - 53. Felten, A., Suarez-Martinez, I., Ke, X., Van Tendeloo, G., Ghijsen, J. and Pireaux, J.J. (2009) The Role of Oxygen at the Interface between Titanium and Carbon Nanotubes. ChemPhysChem, 10, 1799-1804.
http://dx.doi.org/10.1002/cphc.200900193 - 54. Sigal, A., Rojas, M.I. and Leiva, E.P.M. (2011) Interferents for Hydrogen Storage on a Graphene Sheet Decorated with Nickel: A DFT Study. International Journal of Hydrogen Energy, 36, 3537-3546.
http://dx.doi.org/10.1016/j.ijhydene.2010.12.024 - 55. Ao, Z.M. and Peeters, F.M. (2010) Electric Field Activated Hydrogen Dissociative Adsorption to Nitrogen-Doped Graphene. The Journal of Physical Chemistry C, 114, 14503-14509.
http://dx.doi.org/10.1021/jp103835k - 56. Ao, Z.M. and Peeters, F.M. (2010) Electric Field: A Catalyst for Hydrogenation of Grapheme. Applied Physics Letters, 96, Article ID: 253106.
http://dx.doi.org/10.1063/1.3456384
NOTES

*Corresponding author.