Open Journal of Statistics
Vol.05 No.05(2015), Article ID:58483,12 pages
10.4236/ojs.2015.55038
Identification of Global Gene Expression Shifts Using Microarray Data from Different Biological Conditions
José Rafael Tovar Cuevas1, Liliana López Kleine2, José Alejandro Ordoñez1
1Escuela de Estadística, Universidad del Valle, Santiago de Cali, Colombia
2Departamento de Estadstica, Universidad Nacional de Colombia, Bogotá, Colombia
Email: rtovar34@hotmail.com, llopezk@unal.edu.co, ordonezjosealejandro@gmail.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 21 May 2015; accepted 28 July 2015; published 31 July 2015
ABSTRACT
Gene expression data have been very useful during the past two decades for the detection of differentially expressed genes when two (or more) biological conditions are compared. Studies seeking for differentially expressed genes are based on testing gene by gene for a mean differential expression between two conditions. Nevertheless, the global shift in gene expression when taking into account all genes present on a microarray experiment, has not yet been investigated and could provide different information on genes that could be affected by the condition under research. Such a global approach would help identifying a gene expression threshold, characteristic of a certain condition and therefore could be used for diagnosis together with the list of differentially expressed genes detected by classical methods. Moreover, characterizing genes below or above such a threshold could give new insights into the molecular mechanisms implicated functionally in each condition. Here, we present a simple methodology, based on heuristics, gene filtering, variable transformation and descriptive statistics in order to identify such global gene expression shifts and the characteristic threshold so the same can be applied by any professional that works with expression gene data and not only by statisticians. Our procedure is illustrated on a real gene expression data set comparing pathogen inoculated tomatoes with non-inoculated tomatoes. This methodology can be used for the identification of the threshold values when we have continuous variable data sets from two populations with overlapped distributional forms (histograms) in most of their percentiles.
Keywords:
Gene Expression Shift, Filtering, Transformation, Threshold, Biological Conditions

1. Introduction
Recent advances in genomic and post-genomic technologies have provided the opportunity to analyze genomic data publicly available in databases. Several molecular mechanisms can be understood better through the analysis of genomic data such as gene expression data. One of the molecular mechanisms, that need to be better understood in order to reduce losses caused by plant pathogens, is plant immunity. Losses caused by plant pathogens represent one of the most important limitations in crop production, and these losses can compromise food supply. Plant immunity depends on the recognition of conserved Microbial Associated Molecular Patterns (MAMPs) or strain-specific effectors by Pattern Recognition Receptors (PPRs) or resistance (R) proteins, triggering MTI (MAMP Triggered-Immunity) and ETI (Effector Triggered-Immunity), respectively. Upon recognition plants activate a complex network of responses that include signal transduction pathways, novel protein interactions and coordinated changes in gene expression. Detailed information concerning specific and punctual interactions between effector and resistance proteins has been accumulated in the last years, and in some cases, a global picture for some of these interactions has been established [1] [2] . Some of these responses are triggered due to pathogen presence and others when the plant is exposed to chemical agents such as salicylic acid.
Although gene expression has been studied at a global and systemic level constructing gene networks [1] - [3] , differential gene expression has been used to identify individual genes that change their expression when two biological conditions are compared. An example of identification of gene expression changes in plants exposed to pathogenes is the work of Lopez et al. [4] . This means that, until now, it has been investigated if some genes or gene groups are repressed or activated when comparing one condition to the other. It would be interesting to detect a global shift in gene expression taking all genes together. This shift could be an overall change of the gene expression distribution or a global change in gene expression levels, i.e. a change of the gene distribution on the real line indicating a global change and not a local change implying just some genes. Once identified, it would be possible to classify genes in two groups based on their gene expression level: 1) Genes participating in the gene expression shift or being responsible for the shift and 2) Genes that do not participate in the shift. This kind of classification can then be used as a functional prediction of genes participating in any molecular mechanism that could be related to the biological condition of interest revealing a normally non-explored aspect of gene expression modification. This kind of shift can only be obtained for gene expression comparing two conditions and for which replicates are available for each condition, which is the case for most of the microarray studies undertaken until now. In order to investigate this kind of global gene expression shift, we propose an algorithm in six steps: 1) Describing gene expression distribution for each condition, 2) Transforming gene expression data to obtain distributions as symmetric as possible, 3) Filtering very variable genes and detecting outliers, 4) Computing mean of gene expression over all replicates of each condition, 5) Identifying a charac- teristic gene expression level (threshold for classification) for each condition and 6) Detecting genes responsible for the shift based on their expression level.
In the current work we present the overall methodology for detecting a global gene expression shift and we apply it to a real data set. The first experiment we considered here is available at the tomato expression data-set at http://ted.bti.cornell.edu/ and compares two conditions in the field using the two channel microarray technique: healthy cherry tomato and P. infestans inoculated tomato at 4 time points. In a previous work we have detected differentially expressed genes on this data set and detected only gene activations in the inoculated tomato [4] . The third time-point (36 hours after inoculation) showed the highest number of differentially expressed genes. We concentrated on this time-point and, based on the proposed methodology, we were able to detect an overall change in gene expression that indicates that the overall shape of the gene expression distribution remains unchanged, but that there was a shift in position and that gene expression is lower in P. infestans inoculated tomatoes. We identified 316 genes whose gene expression is characteristic of the inoculated condition. Only 14 of these genes turned out to be resistance genes and are all different from the previously differentially expressed genes [4] indicating that both methodologies are complementary and detect different aspects of gene expression changes.
2. Methods
The procedure to identify a gene expression shift and select genes involved in the shift can be summarized in the following main steps, which are also illustrated in Figure 1:

Figure 1. Graphical summary of the procedures proposed to identify a global shift in gene expression data.
・ Describing gene expression distribution for each condition (the data for each experimental condition was treated separately): in this step we obtained the descriptive statistics for all replicates involved in the analysis focusing especially on the percentiles in order to identify extreme values or outliers and check the distri- bution shape. Genes were labeled as belonging to the inoculated condition or to the non-inoculated condition depending on the data set. Before filtering, genes in each group were the same.
・ Transforming gene expression data seeking to reduce the asymmetry of the distribution in each data base separately: When the distribution has a very pronounced asymmetry, which is common in gene expression data, it is necessary to choose an adequate transformation that modifies the shape of the distribution in a way that reduces asymmetry. If the distribution of the transformed data does not have the expected shape it is possible to make a new modification on the transformed data to obtain it or a shape very similar with the expected. Here, logarithmic or generalized logarithm transformations can be used [5] .
・ Filter very variable genes: Here we propose to compute de coefficient of variation within the replicates of each condition and evaluate the intern consistency using a a biological criterion All these sample points with cv higher than the established value as criterion should be eliminated in order to avoid noise. At this point different genes from each data base (inoculated/non-inoculated) are eliminated.
・ Computing mean of gene expression over all replicates of each condition.
・ With the vector of means of transformed data, we obtained the descriptive statistics and boxplots and checked again for outliers using the following criterion [6] : an outlying value is a value x as such
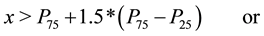 (1)
(1)
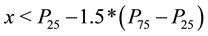
where the
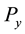 indicates the
indicates the
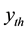 percentile. For extreme outliers the range is changed from 1.5 to 3 in (1).
percentile. For extreme outliers the range is changed from 1.5 to 3 in (1).
・ With the vector of means of the transformed data the distributional shape is investigated and filtered again, and the transformation is repeated if necessary.
・ Identifying a characteristic gene expression level (threshold for classification) for each condition: This gene expression level will be used as a classification threshold. Having a lower (or higher) expression level as this threshold will be understood as being responsible for the gene expression shift and therefore potentially involved in a key function of the biological condition of interest.
3. Example of Application on Tomato Microarray Data
3.1. Microarray Data
The first data sets was obtained from the Tomato Expression Database website http://ted.bti.cornell.edu/). In this study we used experiments that were carried out using the TOM1 DNA chip available at (http://ted.bti.cornell.edu/cgi-bin/TFGD/miame/experiment.cgi?ID=E022). We focused on the experiments carried out by Christine Smart and collaborators (Accession number E022) where gene expression profiling of infection of tomato by P. infestans in the field was studied. The goal of this experiment was to gain insight into the molecular basis of the compatible interaction between P. infestans and its hosts (with a major emphasis on the role of gene suppression). We used the data from that experiment separately for inoculated plants (condition I) vs. non inoculated plants (condition NI) in the field. For this comparison four time points were available at 0, 12, 36 and 60 hours with 8 replicates of each condition (32 experiments). We focused on the third time point (36 hours), for which the most differentially expressed genes had been detected [4] .
・ Gene expression distribution: we constructed a histogram of the gene distribution for each condition. Initially we had a data set with 13,440 records of eight gene replicates for each experimental form (inoculated and non-inoculated tomato plants). We computed the mean of the replicates and we obtained 13340 means for which we draw the histogram, the boxplot and computed several descriptive statistics as explained in the materials and methods section. To obtain more symmetric distributional forms, we used classical methods developed in the statistical literature to evaluate the fit of the a data set to the normal distribution, however, other procedures could be useful to fulfill that purpose. The distributional plots showed an important right asymmetry and overlapping of both data sets, which did not allow to establish a direct difference of the gen distribution between inoculated and non-inoculated plants. A box plot of the two data sets showed a large number of outliers (Figure 2) and on the quintile plot most points were far from the reference line of a normal distribution (Figure 3 and Figure 4). On the other hand, we obtained the descriptive statistics (mean, median, variance and coefficient of variation) for each row of the data set (eight replicates) and we observed high variability (CV between 9.1% and 279% in both data sets).
・ We identified a split of the gene distributions to identify those genes associated with each condition (ino- culated and non-inoculated). In order to achieve a clear split it was necessary to apply a data transformation using a shape parameter of the gene distribution. As it is common with microarray data, we decided to apply the natural logarithm on gene expression replicates and then compute the mean of the logarithms as repre- sentative data of each condition. The log-transformed data showed less asymmetry but we did not have enough symmetry (even when the q-q plot of normal distribution shows a better performance respect to the fit to normal distribution used as reference to evaluate the shape) (Figure 5 and Figure 6) and the variability between experiments remained higher that the expected value (Table 1).
Since we had no yet the desired distributional form we decided to undertake gene filtering for outlier re- duction and further transformations as follows:
・ Filter very variable genes: We established as selection criterion to eliminate all records with a coefficient of variation less than or equal to 10%. In that way, we obtained a data set with 7858 genes in each group. With the new data sets, we constructed again the histogram and the box plot with the outliers per condition. Even though, we had achieved less variability, the gene expression distributions were asymmetric and they showed many extreme points.
・ Distributional shape: we decided to undertake a second transformation step and we transformed the data using the Box Cox transformation as follows:
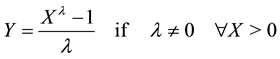
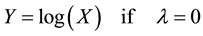 (2)
(2)

Figure 2. Boxplot of initial values of gene expression in P. infestans inoculated and non- inoculated tomatoes.

Figure 3. Q-Q-plot of gene expression of P. infestans inoculated tomatoes.
We tested different values of the lambda parameter for the Box Cox transformation in the same way as Osborne 2010, optimizing the reduction of outliers and a q-q normal plot. Finally, with a
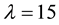 we obtained the expected distributional shapes that allowed separating gene expression of inoculated and non-inoculated
we obtained the expected distributional shapes that allowed separating gene expression of inoculated and non-inoculated

Figure 4. Q-Q-plot of gene expression of non-inoculated tomatoes.

Figure 5. Q-Q-plot of log-transformed values of gene expression in P. infestans inoculated tomatoes.
plants using 7802 gene records of non-inoculated tomatoes and 7751 genes of inoculated tomato (Figure 7). According to the distributional forms, although the distributions are overlapped, they show less outliers (Figure 8) and a more symmetry shape (Figure 9 and Figure 10). On the other hand, the obtained forms indicate that the

Figure 6. Q-Q-plot of log-transformed values gene expression of P. infestans non-inoculated tomatoes.

Figure 7. Histogram and adjusted density of the distribution for each condition after Box-Cox transformation showing a light shift in distribution.

Table 1. Descriptive summaries for non-transformed (original) and log-transformed data.
SD: standard deviation; CV: coefficient of variation.
inoculated condition does not modify the gene expression drastically.
・ Identify a characteristic gene expression level: to find the threshold value, we obtain six percentile values for both gene distributions (see Table 2). In accordance with the results showed in Table 2, the gene expression in the inoculated tomatoes take values lower than those observed in the non-inoculated tomatoes for all percentiles and the minimum expression gen. To obtain the threshold value, we need to establish the gene expression in the inoculated group whose value is lower than the minimum observed in the non-inoculated distribution (reference distribution). From Table 2, it is possible to see that this value is lower than the P5 in the expression gen distribution of the inoculated tomatoes. To establish the exact value, we developed an algorithm that may be implemented in an excel sheet as follows.
- List the gene expression data of the non-reference distribution with value less than or equal to identified percentile (in this case the P5 so that, we have 390 values of gene expressions of inoculated tomatoes) and order the values from the lowest to highest.
- For each gene expression compute the percentile value using the associated position (q) of the datum (x), as follows:
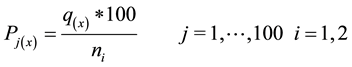 (3)
(3)
- Let
 be a gene expression value in the reference distribution,
be a gene expression value in the reference distribution,
 be a gene expression in the other distribution and
be a gene expression in the other distribution and
 minimum (
minimum ( ), then, compute the difference
), then, compute the difference .
.
- Check the differences and the desired value will be that where the
 value changes its sign. In our case, the threshold value was 0.79011 located in the percentile 4.07689 of the inoculated gene expression dis- tribution and we obtained 316 gene expressions associated only with inoculated tomatoes.
value changes its sign. In our case, the threshold value was 0.79011 located in the percentile 4.07689 of the inoculated gene expression dis- tribution and we obtained 316 gene expressions associated only with inoculated tomatoes.
None of these 316 genes had been detected as differentially expressed genes in previous analysis of these data [4] . Fourteen of them are known resistance genes. This is a very low number indicating that the genes re- sponsible for the gene expression shift are not necessarily genes directly related to the investigated biological condition but affected in their overall gene expression level by the phenomenon that is investigated.

Figure 8. Boxplots of Box-Cox-transformed values of gene expression in P. infestans ino- culated and non-inoculated tomatoes.

Figure 9. Q-Q-plot of Box-Cox-transformed values of gene expression of P. infestans ino- culated tomatoes.

Figure 10. Q-Q-plot of Box-Cox-transformed values of gene expression of non-inoculated tomatoes.
Table 2. Gene distribution using the percentiles.
4. Bootsrap and Simulation Study
With the purpose to evaluate the performance of our procedure, we used the bootstrap method with our vectors of gene expressions in inoculated and non-inoculated tomatoes. We took samples with replacement of each vector of gene expressions. With each bootstrap sample we obtained the position in the distribution of inoculated tomatoes associated to minimum value of the gene expression in the non-inoculated tomatoes distribution. We repeated this procedure 1000 times and we obtained the histogram of the observed positions, see Figure 11. In accordance with our results, most of positions have taken values between 290 and 330 with a median of 316 and a range o values taking values between 253 and 378. The initial position (with the real sample) was 316 which agree with the median of the observed positions in the bootstrap samples.
In addition to bootstrap procedure, we simulated data under two different conditions. To obtain asymmetric distributions, we generated with help of the R package a vector of 10,000 values with distribution Gamma (a = 1.5, b = 10) beginning in the value of 35. The vector simulating the second condition was generated with data under the same distribution but now, the minimum simulated value was 36. We repeated 10,000 times our procedure transforming the data, removing outliers and separating distributions to obtain the positions in a distribution associated to the minimal value in the other one. To establish if the procedure works, we obtained the vector of positions and the minimal values and we observed that in all development we obtained the values established initially. Figure 12 and Figure 13 show the performance of the simulated sample of pairs of data and the final result obtained. The red line indicates the location of the position that divide both distributions.
5. Discussion
Classification methods developed for diagnostic purposes generally use the information obtained from obser- vations of variables measured in a continuous scale and applied strategies to establish a specific value to be used as a cut point that allows classifying individuals as belonging to a category depending on the variable value. In this sense, it is very easy to distinguish individuals with the characteristic because they take higher values of the measured variable while those that do not have the characteristic take values lower than the cut point. Even

Figure 11. Distribution of positions obtained using bootsrapping.

Figure 12. Distribution of simulated data assuming two different conditions.

Figure 13. Distributions of simulated data after to apply the procedure to obtain the shift position.
when the two distributions overlap, in the upper tails those individuals without the characteristic take a value lower than the maximum value taken by the elements with the characteristic. Generally, the method used to obtain the cut point is the ROC curve (receiving operating curve) and it is assumed that the distributions of the measured variable in both populations fit to the normal distribution (See Sullivan, 2004 [7] , among many others). In this paper, we propose a classification methodology based only on heuristics, descriptive statistics, data trans- formation and filtering of gene expression data that does not depend strictly on the fit to some theoretic dis- tribution in situations for which we have overlapped sample data distributions of measured variables with marked asymmetry and the possible cut point is unknown. In our application data, this cut point even turned out to be a lower value.
Although it has been investigated if individual genes change their expression and generally, the procedures developed to establish differences between populations using the replicates obtained for each experimental condition [4] , a global shift comprising the whole distribution of this kind had not yet been reported to our knowledge. The proposed methodology uses the information contained in the complete data set of gene expression regardless the individual behaviour of the specific gen, which is from our perspective, a novel approach to address a problem that can be common in microarray data sets. Microarray samples could be classified into one of two conditions based only on their gene expression level. Moreover, genes that are below/above the identified threshold could be used as list of gene candidates potentially implicated in the function of interest regarded from a global point of view that takes into account global gene expression of all genes and not only of one, which is not the case for t-test based detection of differentially expressed genes as was applied before [4] .
The presented methodology could also be used for the analysis of other type of samples comparing individuals belonging to two conditions that are characterized using a continuous variable. This could be the level of a substance in blood or another measure like blood pressure, weight, body mass index, etc. In that case in lieu of genes, measures would be available on individuals for two conditions (healthy and diseased, for example) and the values of the measured variable would be transformed, individuals filtered in order to identify a shift. We used a data set of gene expression in tomatoes to illustrate our classification procedure when two biological conditions are compared. The results obtained indicate that an overall shift in gene expression occurs when tomatoes are inoculated with a pathogen.
It is important to point out that, the proposed procedure uses the methods developed to fit data sets to normal distribution of probabilities to evaluate the changes in the symmetry of the distributions in order to get a better view of the behaviour of the data, which does not imply that we need to adjust the data to the normal distribution to reach our goal. Because the procedure is a non-parametric approach, we use the test developed in the literature to evaluate the fit of data to normal distribution just to check how changes of the shape of the distribution in the data and the distributions are separated, however, other methods developed to address that kind of situations could be used. On the other hand, the proposed procedure can be applied regardless the procedure used in the laboratory to obtain the gene expression data. A limitation of the method, in situations where is necessary to apply the Box Cox transformation, could be the difficulty to obtain the estimate of the lambda parameter given that, procedures are not implemented in all statistical packages but general methods have been proposed [8] .
Cite this paper
José Rafael TovarCuevas,Liliana LópezKleine,José AlejandroOrdoñez, (2015) Identification of Global Gene Expression Shifts Using Microarray Data from Different Biological Conditions. Open Journal of Statistics,05,360-372. doi: 10.4236/ojs.2015.55038
References
- 1. Pop, A., Huttenhower, C., Iyer-Pascussi, A., Benfey, P.N. and Troyanskaya, O.G. (2010) Integrated Functional Networks of Process, Tissue, and Developmental Stage Specific Interactions in Arabidopsis thaliana. BMC Systems Biology, 4, 180. http://dx.doi.org/10.1186/1752-0509-4-180
- 2. Pritchard, L. and Birch, P. (2011) A Systems Biology Perspective on Plant-Microbe Interactions: Biochemical and Structural Targets of Pathogen Effectors. Plant Science, 180, 584-603. http://dx.doi.org/10.1016/j.plantsci.2010.12.008
- 3. López-Kleine, L., Torres-Avilés, F., Tejedor, F.H. and Gordillo L.A. (2012) Virulence Factor Prediction in Streptococcus pyogenes Using Classification and Clustering Based on Microarray Data. Applied Microbiology and Biotechnology, 93, 2091-2098.
- 4. López-Kleine, L., Pinzón, A., Chaves, D., Restrepo, S. and Riaño-Pachón, D.M. (2013) Chromosome 10 in the Tomato Plant Carries Clusters of Genes Responsible for Field Resistance/Defence to Phytophthora infestans. Genomics, 101, 249-255. http://dx.doi.org/10.1016/j.ygeno.2013.02.001
- 5. Huber, W., Heydebreck, A., Sültmann, H., Poustka, A. and Vingron, M. (2002) Variance Stabilization Applied to Microarray Data Calibration and to the Quantification of Differential Expression. Bioinformatics, 18, S96-S104.
- 6. Rosner, B. (1986) Fundamentals of Biostatistics. Cengage Learning, Sidney.
- 7. Sullivan, M. (2003) The Statistical Evaluation of Medical Tests for Classification and Prediction. Oxford Statistical Science Series 31, Oxford University Press, New York.
- 8. Osborne, J.W. (2010) Improving Your Data Transformations: Applying the Box-Cox Transformation. Practical Assessment, Research and Evaluation, 15, 1-9. http://pareonline.net/pdf/v15n12.pdf