Open Journal of Physical Chemistry
Vol. 2 No. 2 (2012) , Article ID: 19160 , 9 pages DOI:10.4236/ojpc.2012.22012
Hydroxyalkylation of Cyclic Imides with Oxiranes Part I. Kinetics of Reaction in Presence of Triethylamine as Catalyst
Faculty of Chemistry, Rzeszów University of Technology, Rzeszów, Poland
Email: jml@prz.edu.pl
Received February 6, 2012; revised March 7, 2012; accepted April 9, 2012
Keywords: Imides; Oxiranes; Kinetic; Reactivity; Influence of Solvents
ABSTRACT
Literature describes kinetics of reactions of alcohols, phenols, carboxylic acids, amines and amides with oxiranes such as ethylene oxide and propylene oxide. However, there is no information regarding kinetic of reaction of imides with oxiranes. In this article the kinetics of the reaction of cyclic monoimides: succinimide, phtalimide, and glutarimide, with ethylene and propylene oxides in presence of triethylamine in aprotic solvent was studied. The rate laws for those processes were established based upon on dilatometric measurements. I was said that cyclic monoimides react with oxiranes in presence of triethylamine to give N-(2-hydroxyalkyl)imides as major product. This product react further with oxiranes in consecutive reaction. The kinetics of the reaction of cyclic mono-imides with oxiranes obey the following rate law: V = k1/2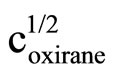
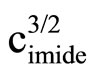
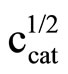 . Based upon kinetic data the following orders of reactivity of imides and oxiranes were obtained: phtalimide ≥ succinimide > glutarimide and ethylene oxide > propylene oxide. The solvent (DMF, DMSO and dioxane) effect was also studied. From temperature dependences the thermodynamic parameters: activation energy, enthalpy and entropy from linear Eyring plots were obtained.
. Based upon kinetic data the following orders of reactivity of imides and oxiranes were obtained: phtalimide ≥ succinimide > glutarimide and ethylene oxide > propylene oxide. The solvent (DMF, DMSO and dioxane) effect was also studied. From temperature dependences the thermodynamic parameters: activation energy, enthalpy and entropy from linear Eyring plots were obtained.
1. Introduction
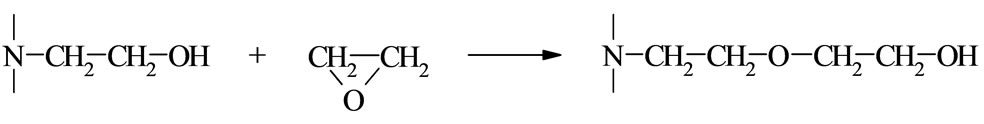
Cyclic imides (I) reveal weak acidity of the O= C-NH-C=O group. Among those the succinimide (SI, R = (-CH2-)2), glutarimide (GI, R = (-CH2-)3), phtalimide (PI, R = Ph), and maleimide (MI, R = -CH=CH-) are common compounds [1]. The first three form N-(hydroxyalkyl) imides (II) in reactions with oxiranes [2] according to the following scheme:
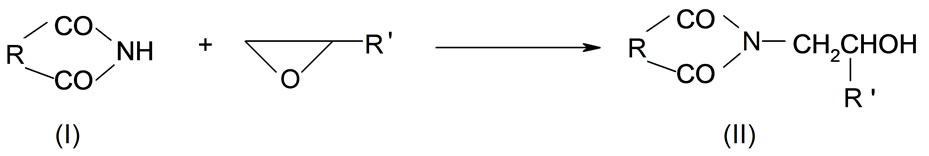 (1)
(1)
where: R’ = -H, -CH3, -CH2Cl The N-(hydroxyalkyl)imides can also be obtained by straightforward reaction of corresponding anhydride with aminoalcohol [3]. The products of the reaction of cyclic monoimides with ethylene carbonate and ethylene oxide were described [4,5]. These products have alcohol character which upon esterification with fatty acids appeared very early to be useful plastificators of polymers [6], while those obtained from multifunctional imides, like isocyanuric acid and ethylene oxide (EO) or propylene oxide (PO) were used for synthesis of polyurethanes, mainly to give polyurethane foams of remarkable thermal resistance [7].
The hydroxyalkylation of cyclic monoimides with oxiranes or formaldehyde was the matter of patents [2,8,9]. However the kinetics and mechanisms for those processes were not studied in detail. In contrary, the kinetics and mechanisms were described for the reactions of oxiranes with alcohols [10,11], phenols [12], thiols [13,14], carboxylic acids [15-17], and amines [18,19] depending on solvent, acidity of substrates and products and a catalyst used [18,20-25]. The reactions of amides with oxiranes were studied by Knaggs [26] and Grosmann [27]. Knaggs have shown that oxirane reacts with hydroxyl group of N-hydroxyalkyl substituent instead of amide group [26]. Grosmann has demonstrated the product distribution by preparative and analytical methods [27]. The mechanism of isomerization of imides substituted with oxirane and oxetane groups on nitrogen was also described [28].
In this paper the kinetics and products of the reaction of cyclic monoimides with oxiranes in presence of triethylamine (TEA) as catalyst were studied in order to establish the mechanism of the reaction. These processes can provide the kinetic models mimicking initial steps of the synthesis of polyetherols from oxiranes and azacyclic substrates containing few imide groups, for example isocyanuric acid [29], barbituric acid [30] or 6-amino-uracyl [31]. These azacyclic substrates are sparingly soluble in organic solvents and therefore their reactivities cannot be studied in this class of solvents.
The method used for following the progress of the reactions studied in this work deserves a short comment. Our preliminary results indicated that determination of epoxide groups based on alkalimmetric titration of excess hydrochloric acid in dioxane in these systems failed due to the presence of amine catalyst. Therefore we have used the dilatometric method [32] by measuring the volume contraction that occurs with advancing reaction. Our preliminary studies indicated that the contraction of volume was proportional to the consumption of oxirane (cox). This was achieved by the kinetic studies of the reaction of imide with variable amount of EO or PO at the level of 0.2 to 1.0 equivalent and determination of the final volume of contraction (V∞) to give a linear V∞ = f(cox).
2. Experimental
2.1. Starting Materials
SI, PI, GI (pure, Sigma-Aldrich) were recrystallized from ethanol (reagent grade, POCH, Poland). Dimethylsulfoxide (DMSO, pure, Avocado, Germany) was dried over molecular sieves 4A (POCH, Poland), while EO and PO (racemate) (pure, Fluka, Switzerland), TEA (pure, Avocado, Germany) were used as received. Dimethyl-formamide (DMF, reagent grade, Merck) was purified by azeotropic distillation with water/benzene system and stored over molecular sieves 4A. Dioxane (reagent grade, Merck) was purified by 3 hours heating with 10 weight % concentrated hydrochloric acid, shaking with solid KOH and distillation from over the sodium.
2.2. Synthesis of N-hydroxyalkyl Derivatives of Cyclic Imides
N-(hydroxymethyl)succinimide (HMSI)
The HMSI was synthesized according to published protocol [9]. The crude product precipitated from reaction mixture, which was further purified by recrystallization from benzene.
HMSI: Yield—77%; mp.ref. 64˚C - 66˚C [9], mp.found 66˚C; elemental analysis—% Calcd.: C 46.51; H 5.43; N 10.85; % Found: C 46.30; H 5.33; N 10.80; IR (in KBr pellet) [cm–1], 3450 (OH), 1740 (C=O), 1440 - 1400 (CH2), 1200 (-C-O-), 1100 (OH); 1H-NMR (d6-DMSO) [ppm], 4.6 (H, s, OH), 4.95 (2H, s, N-CH2-) 2.70 (4H, s, CH2CO).
N-(2-hydroxyethyl)succinimide (HESI) and N-(2-hydroxyethyl)phtalimide (HEPI)
HESI and HEPI were synthesized according to [4]. The products were purified by recrystallization from benzene or ethyl alcohol.
HESI: Yield—85%; mp.ref 60˚C [4], mp.found 60˚C; elemental analysis—% Calcd.: C 50.35; H 6.29; N 9.79; % Found: C 50.50; H 6.43; N 9.80; IR (in CHCl3) [cm–1], 3450 (OH), 1740 (C=O), 1440 - 1400 (CH2), 1144 (C-O), 1100 (OH); 1H-NMR (d6-DMSO) [ppm], 4.2 ( H, s, OH), 3.73 (2H, m, -CH2-O-), 3.60 (2H, t, N-CH2-), 2.78 (4H, s, CH2CO).
HEPI: Yield—82%; mp.ref. 128˚C [4], mp.found 127˚C; elemental analysis—% Calcd.: C 62.83; H 4.71; N 7.33; % Found: C 62.63; H 4.53; N 7.51; IR (in CHCl3) [cm–1], 3350 (OH), 1720 (C=O), 1440 - 1400 (CH2), 1225 (C-H in Ph), 1125 (C-O-), 1050 (OH); 1H-NMR (d6-DMSO) [ppm], 7.80 (4H, m, in Ph), 4. 25 (H, s, OH) 3.70 (2H, m, -CH2-O-), 3.60 (2H, t, N-CH2-).
2.3. Kinetic Measurements
Weighed sample of imide was placed in 50 cm3 volumetric flask and dissolved in ca 30 cm3 DMSO. After dissolving of imide and TEA as catalyst the temperature of the solution was raised to 25˚C, 30˚C, 35˚C, 40˚C, 45˚C or 50˚C (±0.05˚C). Oxiranes (EO or PO) as liquids stored in refrigerator were transferred into the flask, which was immediately closed to avoid evaporation of boiling oxirane and weighed. The concentration of oxirane was calculated based on mass of substrates used. The solutions prepared in that way were then rapidly transferred into thermostated dilatometer.
The influence of catalyst concentration on the rate of reaction was studied at constant concentration of imide and EO (1.0 mol/dm3). The TEA concentration was varied within 0.1 - 0.5 mol/dm3 region with 0.1 mol/dm3 step. The influence of imide concentration on the rate of reaction was studied at fixed concentration of EO (0.25 mol/dm3) and constant concentration of catalyst (0.5 mol/dm3) with variable imide concentration within 1 - 3 mol/dm3 region. The change of rate constant in the course of reaction and order for oxirane, calculated for the system with excess of imide is exemplified in Table 1.
The influence of oxirane concentration on the rate of reaction was studied varying its concentration within 0.25 - 1.00 mol/dm3 with 0.25 mol/dm3 step at constant concentration of imide (1.0 mol/dm3) and TEA (0.5 mol/dm3). Additionally, the kinetics was studied at variable concentration of imide (at 0.25 - 1.00 mol/dm3) at fixed EO concentration (1.0 mol/dm3). The results are given in Tables 3 and 4.
The temperature dependence was obtained for the system with 1.0 mol/dm3 imide and oxirane and 0.5 mol/dm3 TEA concentrations within 30˚C - 45˚C for the reaction with EO and 35˚C - 50˚C for the reaction with PO temperature range with 5˚C step. The temperature range for kinetic measurements is limited from lower side by long reaction time (2 - 4 weeks for EO to 2 - 3 months for PO), which precludes the good accuracy of measurement, and from the higher side by boiling of oxiranes in reaction mixture (above 40˚ for EO and 50˚ for PO).
DMSO, DMF and dioxane were used as solvents. Due to low solubility of some imides and catalyst in dioxane, the imide and oxirane concentrations were 0.5 mol/dm3 and that of TEA was 0.25 mol/dm3.
The glass dilatometer of total volume of ca. 45 cm3 was used equipped with 40 cm long capillary of inner diameter 1 mm. Before filled in it was kept at reaction temperature for at least 15 min. The reaction time was measured from the moment of mixing the reactants. The first reading of meniscus level was usually made after 10 min. needed to fill in the dilatometer and bringing the mixture to reaction temperature. The next readings were made after constantly increasing time intervals until the meniscus level stabilized. The initial level of meniscus was calculated by extrapolating the readouts to the time t = 0. Further reading were made in increasing time intervals until constant level of meniscus in capillary was attained.
Relative concentration of oxirane (b) was defined as:
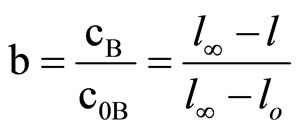 (2)
(2)
where: c0B, cB—corresponding to initial and instantaneous concentrations of oxirane, mol/dm3. l - l(t)—the meniscus level at time t and 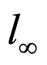 and
and 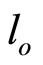 are the levels at the end and beginning of experiment, respectively.
are the levels at the end and beginning of experiment, respectively.
In order to establish the rate low, the substitution method was applied. The values of relative concentration of oxirane, b obtained for 0.2 - 0.8 region with 0.05 step and reaction time were put into the kinetic law equations of various orders (Table 1) to establish the order of reaction for oxirane. Standard deviation of k (determined 13 - 15 times in single kinetic run) and reproducibility (every determination of k was obtained in three independent kinetic runs) were within 6% - 7%, in some cases they were 10%.
Considering the system with EO as standard, the relative reactivity of PO (rPO) was used as the ratio of the rate constants at the same temperature:
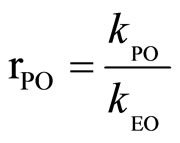 (3)
(3)
where: kPO, kEO—rate constants for PO and EO, respecttively.
Relative reactivity of imides was analyzed in analogous manner using succinimide as standard.
The activation energy (ΔG≠) was calculated from [33]:
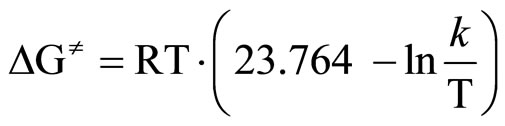 (4)
(4)
where: R—gas constant [J/mol·K]T—temperature of measurement, [K]k—rate constant.
By plotting 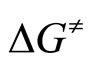 versus T a straight line was obtained with activation enthalpy and entropy of the reaction as the coefficients of the line [33]:
versus T a straight line was obtained with activation enthalpy and entropy of the reaction as the coefficients of the line [33]:
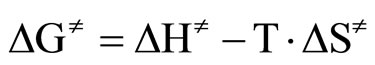 (5)
(5)
2.4. Product Analysis
The products of reactions were isolated after full kinetic run. The DMSO was distilled off under reduced pressure. The oily N-(hydroxyalkyl)imides were extracted and purified by crystallization from ethanol or benzene (N-(hydroxyalkyl)imides of SI and PI) or by distillation under reduced pressure (N-(hydroxyalkyl)imides of GI). Pure products were characterized by elemental analysis (EA 1108, Carlo-Erba, Italy) and melting point, as well as the IR and 1H-NMR spectra. The yield was estimated based on chromatographic analysis.
HESI: Yield—75.4%; mp.ref. 60˚C [4], mp.found 59˚C - 60˚C; elemental analysis - % Calcd.: C 50.35; H 6.29; N 9.79; % Found: C 50.15; H 6.12; N 9.79; (the IR and 1H-NMR spectra are analogous to those in Synthesis of N-hy-droxyalkyl derivatives of cyclic imides section).
HEPI: Yield—79.5%; mp.ref. 128˚C [4], mp.found 127˚C; elemental analysis - % Calcd.: C 62.83; H 4.71; N 7.33; % Found: C 62.94; H 4.58; N 7.22. % (the IR and 1HNMR spectra are analogous to those in Synthesis of N-hydroxyalkyl derivatives of cyclic imides section).
N-(2-hydroxypropyl)succinimide (HPSI): Yield—78.5%; mp.found 82˚C - 83˚C; elemental analysis - % Calcd.: C 53.50; H 7.01; N 8.92; % Found: C 53.25; H 6.92; N 8.91. IR (in CHCl3) [cm–1], 3500 (OH), 1700 (C=O), 1450 (CH2), 1345 (CH3) 1180 (-C-O-), 1120 (OH); 1H-NMR (d6- DMSO) [ppm], 5.1 (H, s, OH), 4.10 (H, m CH-O), 3.75 (2H, d, N-CH2-), 2.78 (4H, s, CH2CO), 1.10 (3H, d, CH3).
N-(2-hydroxypropyl)phtalimide (HPPI): Yield—79.9, mp.found 81˚C; elemental analysis—% Calcd.: C 64.39; H 5.37; N 6.83; % Found: C 64.28; H 5.27; N 6.94; IR (in CHCl3) [cm–1], 3280 (OH), 1720 (C=O), 1450 (CH2), 1380 (CH3), 1200 (-C-O-), 1050 (OH); 1H-NMR (d6- DMSO) [ppm], 7.85 (4H, m, in Ph), 4.25 (H, s, OH), 4.10 (H, m, -CH-O-), 3.80 (2H, d, N-CH2-), 1.10 (3H, d, CH3).
N-(2-hydroxyethyl)glutarimide HEGI: Yield—74.9%; elemental analysis—% Calcd.: C 53.50; H 7.01; N 8.92; % Found: C 53.35; H 6.83; N 8.80; IR (in CHCl3) [cm–1], 3490 (OH), 1720 (C=O), 1460 (CH2), 1100 (-C-O-, OH); 1H-NMR (d6-DMSO) [ppm], 5.1 ( H, s OH), 3.70 (2H, m CH2-O), 3.60 (2H, t, N-CH2-), 2.45 (4H, t, CH2CO), 1.85 (2H, m, CH2- CH2- CH2-).
N-(2-hydroxypropyl)glutarimide (HPGI): Yield—75.4%; elemental analysis—% Calcd.: C 56.14; H 7.60; N 8.19; % Found: C 56.25; H 7.92; N 8.34; IR (in CHCl3) [cm–1], 3400 (OH), 1720 (C=O), 1460 (CH2), 1370 (CH3) 1175 (C-O), 1130 (OH); 1H-NMR (d6-DMSO) [ppm], 5.1 ( H, s, OH), 4.10 (H, m CH-O), 3.75 (2H, d, N-CH2-) 2.42 (4H, s, CH2CO), 1.85 (2H, m, CH2- CH2- CH2-), 1.12 (3H, d, CH3).
In some cases the post-reaction mixtures were analyzed chromatographically (KB 5901 chromatograph, CORRABID, Poland) using: UV detector working at 253, 7 mm, 6 atm pressure, room temperature, steel columns of 100 mm length and 4 mm diameter, silicagel, 60%/40% v/v hexane/dioxane, 1 cm/min eluent flow. The system was calibrated using the imides and appropriate N-(hydroxyalkyl)imides, namely: HESI, HEPI, HPSI, HPPI, HEGI, HPGI as standards. The standards were synthesized from succinic or phthalic anhydrides and ethanoloamine (HESI, HEPI) [3] or SI, PI or GI and appropriate oxiranes or alkylene carbonates according to the procedures used before [4,5], and purified by recrystallization from benzene or ethanol and identified by elemental analysis and NMR spectroscopy.
Some reactions of imides with EO or PO in acetone in presence of TEA were also performed and the products were analyzed in order to isolate and determine quantitatively the products in solvent of different polarity.
IR spectra of N-(hydroxyalkyl)imides were recorded in KBr pellets or in CHCl3, while the spectra of post-reaction mixtures were taken in films (spectrometer PARAGON 1000 FT IR, Perkin Elmer). 1H-NMR spectra of (N-hydroxyalkyl)imides were recorded on a spectrometer BS586A, 80 MHz, TESLA, Czechoslovakia (in d6- DMSO or in CHCl3) with HMDS internal standard.
3. Results and Discussion
3.1. Kinetics
The rate law for the reaction of SI with EO was obtained by substituting the instantaneous values of relative oxirane concentration, b (Equation (2)) and reaction time into kinetic equations of various orders (Table 1). In presence of 12-fold excess of imide (as well as lower excess of imide) the following rate law was found:
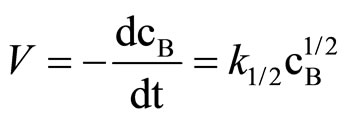 (6)
(6)
where cB is instantaneous concentration of oxirane, mol/dm3.
In columns 3 and the rate constants calculated for closest orders (zero and first) are shawn for comparison. In column 3 (zero order fit) the rate constant constantly decreased, while in column 5 (first order fit) it increases constantly, therefore the best fit was for 1/2 order. The rate constant depends on the catalyst concentration (ck) according to the formula:
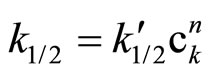 (7)
(7)
Plotting 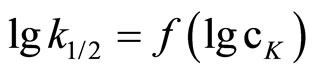 the n ≈ 0.5 was obtained (Figure 1).
the n ≈ 0.5 was obtained (Figure 1).
The reaction of monoimide with oxirane is composed of two steps: the reaction of imide groups AH with oxirane (reaction 1), and the reaction of hydroxyalkyl groups with remaining oxirane:
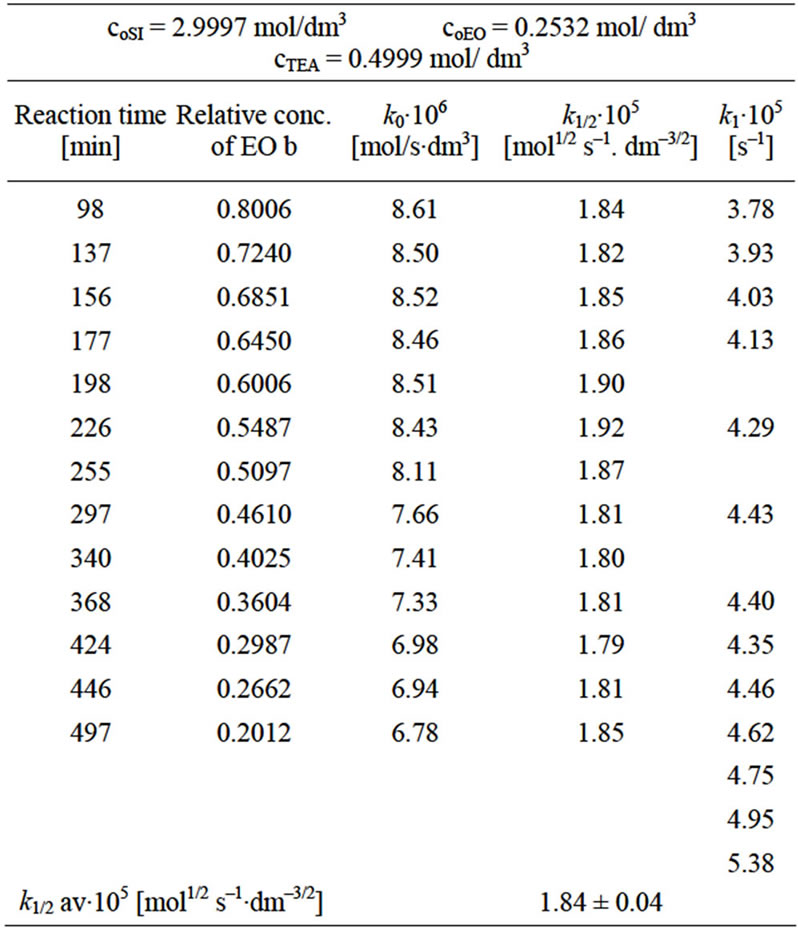
Table 1. Rate constants for the reaction of SI with EO at 313 K (40˚C) calculated for various kinetic laws.
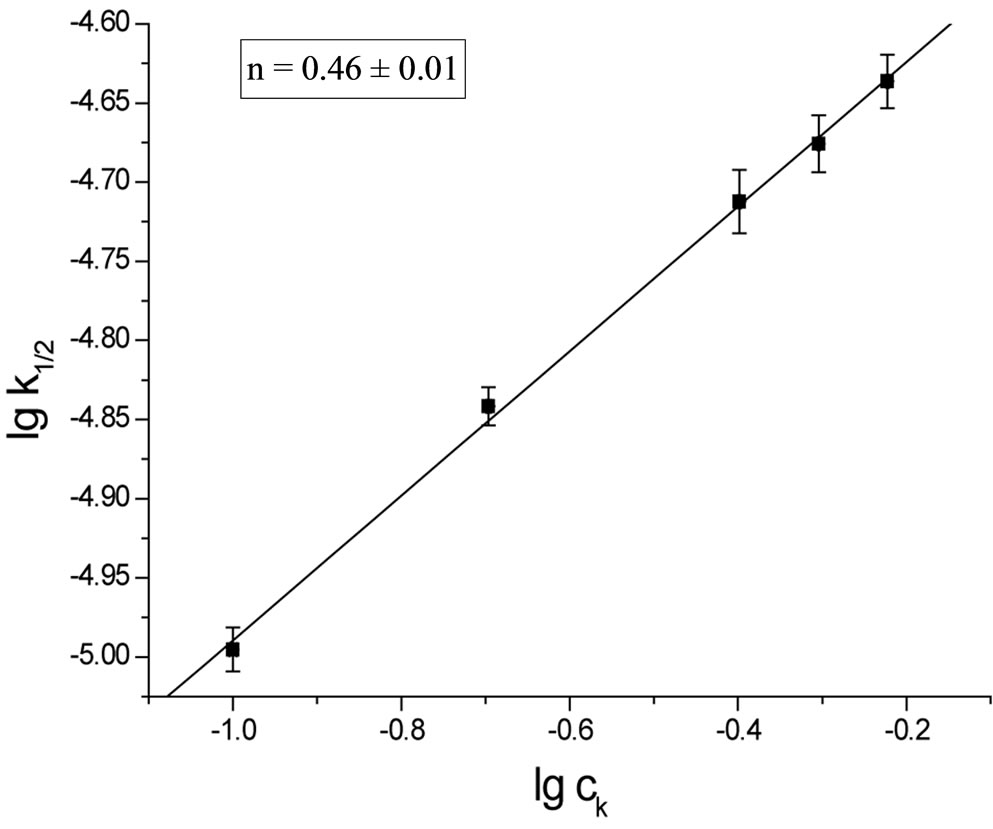
Figure 1. The plot of rate constant versus catalyst concentration for the reaction of SI with EO.
 (8)
(8)
In order to avoid the second process, the reaction was performed at the excess of reactive groups of imide. This imposed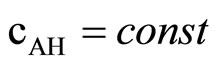 . In such a condition the dependence:
. In such a condition the dependence:
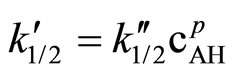 (9)
(9)
operates. According to kinetic measurements the power of the imide group concentration was p ≈ 3/2 (Figure 2). Thus the kinetic law describing the reaction of SI with EO is:
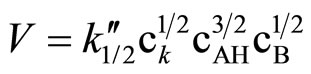 (10)
(10)
Other imides (PI, GI) and also HMSI react with EO and PO in similar manner indicating 1/2 order on oxirane concentration (Table 2).
If the Equation (10) is valid, the k1/2” should stay in agreement with the k calculated from the formula (11), in which the constant concentration of imide groups AH is not assumed, but the real momentary concentration is considered, according to the formula:
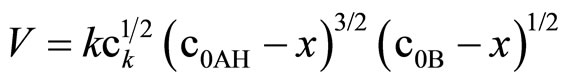 (11)
(11)
where: c0—initial concentration of imide or oxirane, mol/dm3, x—the decrease of oxirane concentration due to reaction with imide (consumption of oxirane due to side reactions was neglected), where:
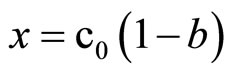 (12)
(12)
Integration of (11) led to:
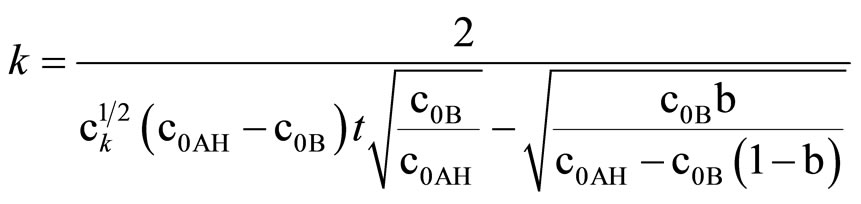 (13)
(13)
This equation can be used only if oxirane reacts with imide groups, i.e. when excess imide is present. The results on rate constants obtained from (10) and (11) are collected in Table 3. Satisfactory agreement was obtained when at least 6-fold imide excess related to oxirane was applied.
The involvement of consecutive reactions was critical importance for presented kinetic studies of reaction of imide group with oxirane. The hydroxyl groups of N-(hydroxyalkyl)imide was involved in consecutive reaction with oxirane. The latter react as soon as the product of the first step appears. Thus, both reactions compete in further progress of reaction. Therefore the products were analyzed carefully.
1) It has been found the products of reaction of imide with oxiranes were oils, while N-hydroxyalkyl derivatives of studied imides (SI, PI) are solids of defined melting point. The N-hydroxyalkyl derivatives crystallized from post-reaction mixtures upon long time. The consecutive products are oily compounds. These were isolated and identified in the reaction of imides with oxiranes used in twice excess. From the comparison of the IR spectra of reaction mixtures with those of pure N-(hydroxyalkyl) imides one can see the difference in the intensity of the band centered at ca 1050 cm–1, which is slightly higher for mixtures. This indicates the formation of ether bonds, i.e. the consecutive products. Moreover, the bands attributed to carbonyl groups (1700 cm–1), and hydroxyl
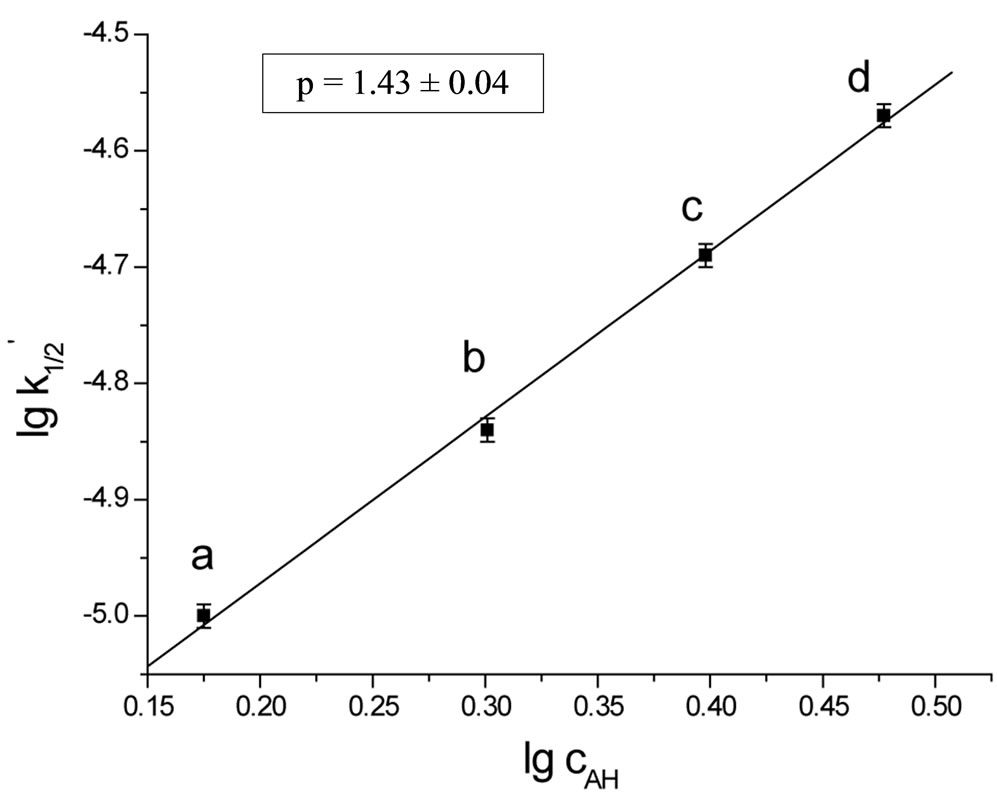
Figure 2. The plot of rate constant 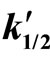 versus concentration of imide groups AH(c0AH/c0B = 6 (a), 8 (b), 10 (c), 12 (d)).
versus concentration of imide groups AH(c0AH/c0B = 6 (a), 8 (b), 10 (c), 12 (d)).
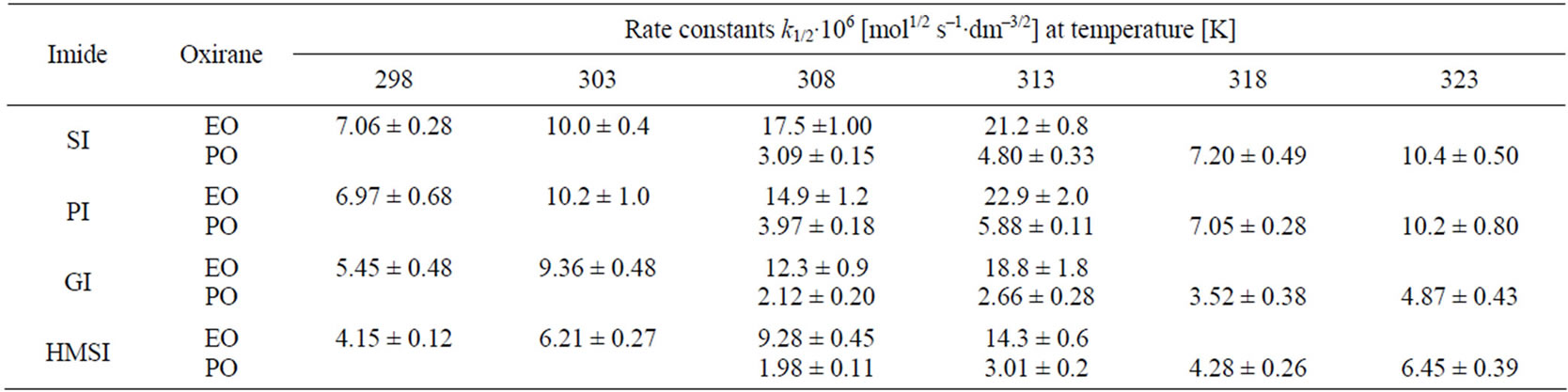
Table 2. Rate constants k1/2∙106 of the reactions of oxiranes with imides in presence of TEA catalyst.
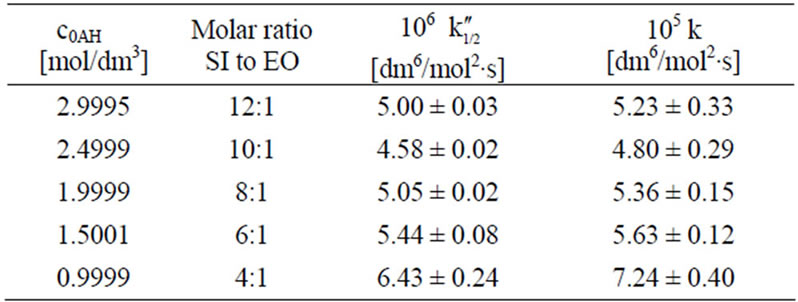
Table 3. Comparison of rate constants for the SI + EO system at imide excess condition calculated from kinetic Equations (10) and (11) (cB = 0.2530 ± 0.0006; cTEA = 0.4997 ± 0.0003).
groups (3400, 1250 and 1100 cm–1) were also observed.
2) Chromatographic analysis of oily product mixtures indicated that crude product consists of 75% - 80% N- (hydroxyalkyl)imides and 5% - 15% unreacted imide. The yields, elemental analysis and IR and 1H-NMR spectra of products isolated after kinetic measurements are specified in Product analysis section.
3) For comparison the products were also readily isolated from the post-reaction mixtures for the systems with SI or PI with oxiranes in acetone. N-(hydroxyalkyl)imides were isolated by extraction with benzene or ethanol to give 75% - 80% pure products and 10% - 15% imides. The analysis of mixtures combined with yield indicated that consecutive products were formed with the yield not larger than 5% - 10% weight.
4) Control experiments of N-(hydroxyalkyl)imides reactivity towards oxiranes were performed using HESI or HEPI with EO at 1 mol/dm3 imide derivative and oxirane and 0.5 mol/dm3 TEA concentrations at 40˚C. The following rate constants were obtained: (3.98 ± 0.12) 10–6 s–1 and (4.16 ± 0.23) 10–6·s–1, respectively. The order of reaction was first for oxirane. In contrary the order of reaction was 1/2 for oxirane and imide both at excess imide and equimolar conditions. If excess of oxirane was used (i.e. in conditions preferable for formation of consecutive products), the order of reaction changed to two for oxirane (Table 4), indicating clearly the change of a mechanism. In such condition the alcohol product prevails and the rate of oxirane consumption is determined by addition of alcohol to oxirane.
Due to slight involvement of consecutive reactions in equimolar imide and oxirane mixtures the negligible deviations of rate constants at late stages of reactions were found. Initially the rate constants do not depend on changes of oxirane concentration, while at the 60% - 70% of progress of reaction (b ≤ 0.4 - 0.3) the value of calculated constant decreases continuously (Table 1). Analogous dependence was observed for the reactions of PI, GI, and HMSI with oxiranes. Those deviations were less than 10%. However, in order to avoid the consecutive reactions which might influence the accuracy of obtained rate constants for N-hydroxyalkylation reaction, only the kinetic data for 20% - 60% progress of the process were taken for calculations (0.8 ≤ b ≤ 0.4, Table 2).
3.2. Reactivity of Monoimides and Oxiranes
Considering the electronic structure of oxiranes, the reactivity of these towards monoimides in DMSO should depend on the electropositive character of carbon 1:
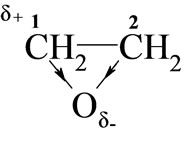
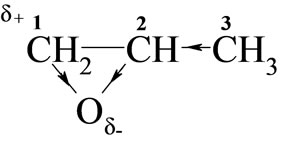
Higher reactivity of EO than PO was expected, because electrophilicity of C-1 in PO is weakened due to electro-donation from methyl group. Indeed, rate constants (Table 2) for EO with all studied imides were higher than those for PO.
Comparing relative reactivity of imides (Table 2) one can see that the rate constants for SI and PI were similar, while GI and HMSI showed lower reactivity than SI and PI. In case of HMSI, the hydroxymethyl group instead of imide group reacted with oxiranes. Its reactivity was therefore lower. In order to compare the relative reactivity of SI and PI the reactions with oxiranes with large excess of those substrates were run. It has been found out that PI is slightly more reactive than SI (Table 5) with EO, while the reactivity towards PO is the same. This will be discussed in details in Part II.
Based upon kinetic data the following orders of reactivity of imides and oxiranes were obtained:
PI ≥ SI > GI > HMSI and EO > PO
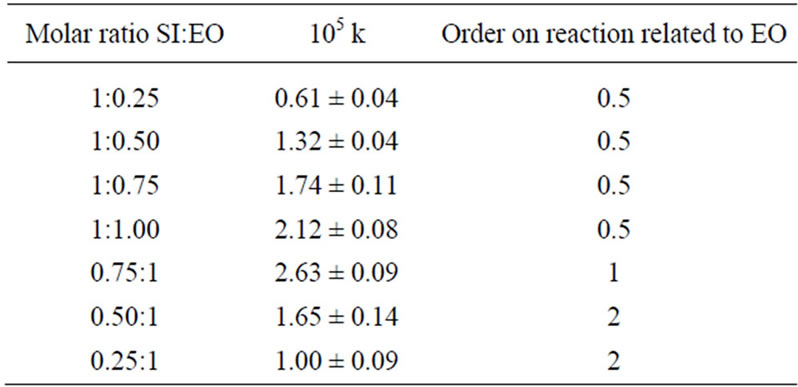
Table 4. Rate constants and reaction order for the SI + EO reaction at variable initial molar ratio at 313 K (40˚C).
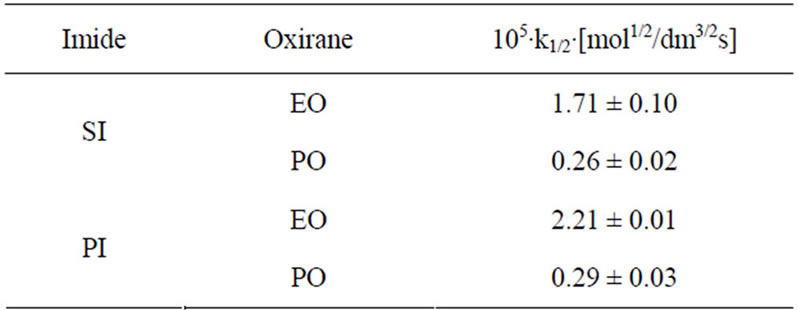
Table 5. Rate constants for the SI and PI + oxiranes at 8- fold imide excess in DMSO at 40˚C. (cAH = 2.0000 ± 0.0050; cB = 0.2500 ± 0.0020; cK = 0.5000 ± 0.0012).
Activation energy (ΔG≠) of the reactions were obtained from linear Eyring plots obtained from temperature dependence (Figure 3). Mean values of enthalpy and entropy of activation are collected in Table 6. From the values of entropy of activation one can deduce that the transition state in case of the reaction of PI and GI with PO is more ordered, probably because of rotational free-
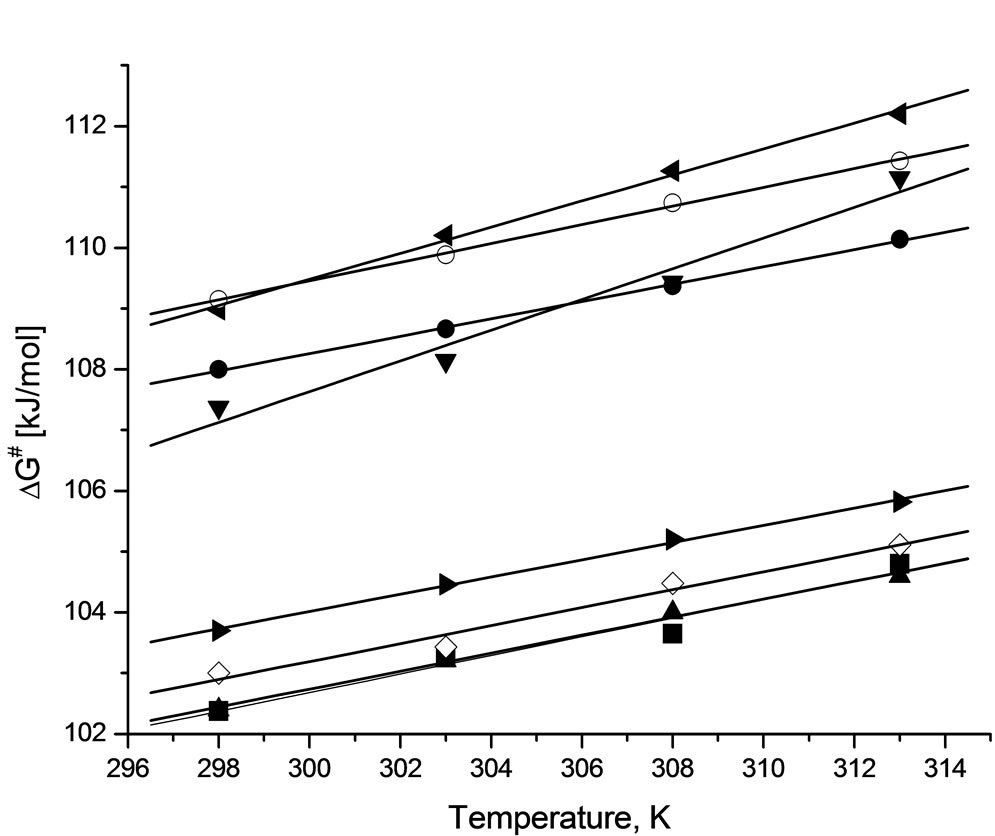
Figure 3. The changes of Gibbs energy of activation in function of temperature for the following reactions: SI + EO (■ r = 0.9839), SI + PO (● r = 0.9994), PI + EO (▲ r = 0.9977), PI + PO (▼r = 0.9961), GI + EO (◊ r = 0.9884), GI + PO (◄r = 0.9983), HMSI + EO (► r = 0.9989), HMSI + PO (○ r = 0.9992); r-correlation coefficient.
dom in case of the GI and PO system in comparison with other systems. This will be discussed later in Part II.
The discussed reactions were conducted in DMSO. The same reactions were also performed in DMF and dioxane and the results of kinetic measurements are collected in Table 7. The 1/2 order of concentration of EO and PO were also found in these solvents, which suggested the same mechanism operating in these solvents. The decreasing values of rate constants were found in the order: DMF > DMSO > dioxane. The highest reaction rate in DMF is presumably consistent with its basic character related to the presence of nitrogen atom, which might play the same catalytic role as TEA, i.e. enabling the proton transfer from imide into oxirane (cf. mechanism in Part II). The least polar dioxane cannot be involved in this step and therefore the rates of reactions were the least. Based upon rate constants the relative reactivity of substrates in all solvents was obtained (Table 8).
According to these data the relative reactivity of EO and PO depends only slightly on the solvent used. In polar solvents (DMF or DMSO) the subtle difference of reactivity of PI and SI with oxiranes was found; PI is about 1.1 - 1.2 times more reactive in DMF than is SI. In DMSO their reactivity depends on the kind of oxirane used, while in dioxane the PI is less reactive. The differences in reactivity can be explained probably in terms of solvation of transition state PI-oxiranes with polar solvent. The solvation of transition state is strong in DMF and DMSO causes the increase of the reactivity of PI.
4. Summary and Conclusions
1) Cyclic monoimides react with oxiranes in presence of triethylamine to give N-(2-hydroxyalkyl)imides as major product. This product react further with oxiranes in consecutive reaction.
2) The kinetics of the reaction of cyclic monoimides with oxiranes obey the following rate law:
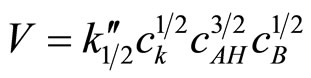
where cK, cAH and cB stay for concentrations of catalyst, imide, and oxirane, respectively.
3) In polar solvents the reactivity of imides and oxiranes decreases in the raw:
PI ≥ SI > GI > HMSI and EO > PO.
4) The decreasing values of rate constants were found in the order: DMF > DMSO > dioxane. The highest reaction rate in DMF is presumably consistent with its basic character related to the presence of nitrogen atom, which might play the same catalytic role as TEA, i.e. enabling the proton transfer from imide into oxirane The least po-
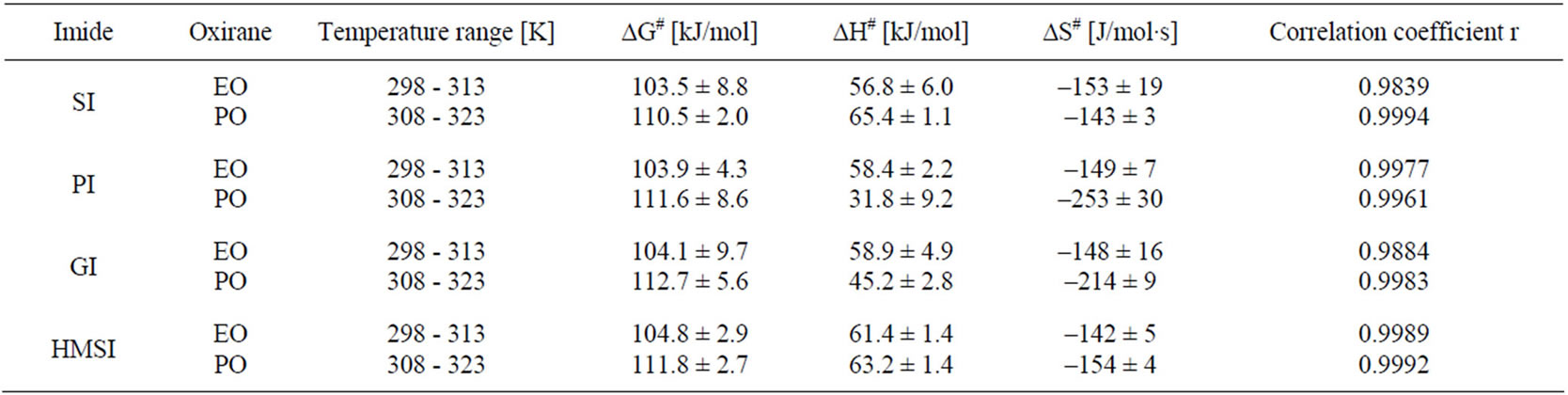
Table 6. Activation parameters for the reactions of imides with oxiranes.
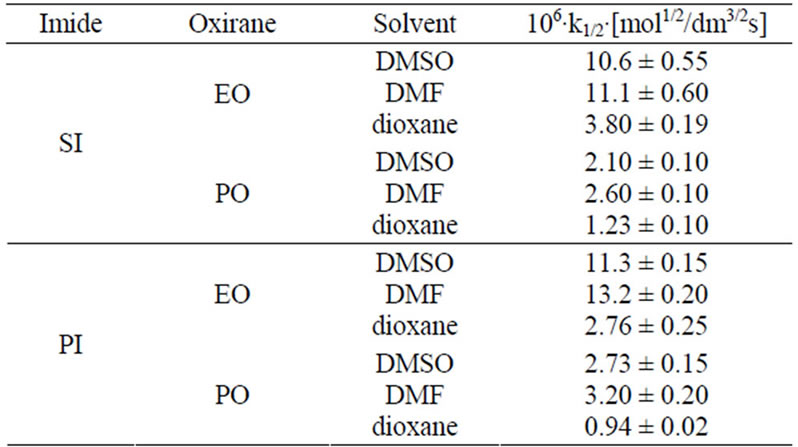
Table 7. The solvent dependence of rate constants for the reaction of imides with oxiranes at various solvents at 40˚C.
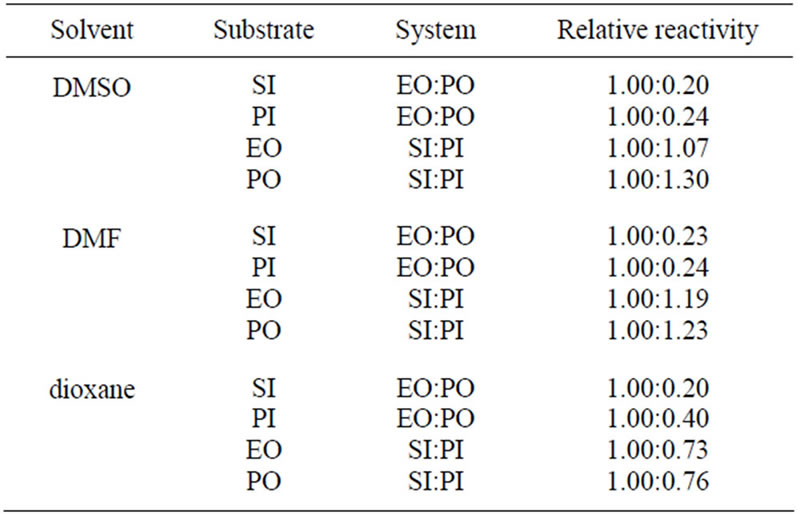
Table 8. Relative reactivity of imides and oxiranes at 40˚C at various solvents.
lar dioxane cannot be involved in this step and therefore the rates of reactions were the least.
5) The reaction of N-(hydroxyalkyl)imides with oxiranes was first order for oxirane. In contrary the order of reaction was 1/2 for oxirane and imide both at excess imide and equimolar conditions. If excess of oxirane was used, the order of reaction changed to two for oxirane indicating clearly the change of a mechanism. In such condition the alcohol product prevails and the rate of oxirane consumption is determined by addition of alcohol to oxirane.
REFERENCES
- A. Kirrmann, J. Cantacuzene and P. Duhamel, “Chimie Organique, Fonctions Simples,” WNT, Warsaw, Vol. 2, 1980, pp. 225-226.
- R. Steele, A. Katzakian, J. Scigliano and E. Hamel, “Imide Oxirane Reactions,” US Patent No. 3962182, 1976.
- J. Billman and E. Parker, “Amino Acids. I. Glycine,” Journal of the American Chemical Society, Vol. 65, No. 5, 1943, pp. 761-762. doi:10.1021/ja01245a008
- K. Yanagi and S. Akiyoshi, “Notes: Hydroxyethylation of Imides,” The Journal of Organic Chemistry, Vol. 24, No. 8, 1959, pp. 1122-1123. doi:10.1021/jo01090a601
- N. Schoenfeldt, “Grenzflaechenactive Aethylenoxid— Addukte,” Wissenschaftliche Verlagsgesellschaft MBH, Stuttgart, 1976.
- R. Cornwell, “Composition Capable of Being Molded and Being Cast into Films,” US Patent No. 2487105, 1949.
- M. Kucharski and J. Lubczak, “Polyurethane Foams Based on Reaction Products of Ethylene Oxide with 1,3,5-Tris(Hydroxymethyl) Isocyanurate,” Polimery, Vol. 30, No. 9, 1985, pp. 354-361.
- P. Tawney, “N-methylol maleimide,” US Patent No. 2526517, 1950.
- G.Weitzel, F. Schneider, A. Fretzdorf, K. Seynsche and H. Finger, “Futher Tumor Inhibiting Compounds. I. Cytostatic Effects of Nand S-Hydroxy-Methyl Compounds,” Zeitung Physiologische Chemie, No. 334, 1963, pp. 1-25.
- S. Kucharski, “Nonionic Surfactants. I. Preparation of Poly(Oxyethylene) Alcohols,” Wiadomości Chemiczne, Vol. 25, No. 8, 1971, pp. 579-594.
- N. Lebedev and I. Baranov, “Reaction Involving α-Oxides. X. Kinetics and Mechanism of the Reaction of Ethylene Oxide and Alcohols under the Conditions of Base Catalysis,” Kinetika i Kataliz, Vol. 7, No. 4, 1966, pp. 619-626.
- N. Lebedev and V. Shvets, “Mechanism of Ethylene Oxide Reactions with Phenols,” Kinetika i Kataliz, Vol. 9, No. 3, 1968, pp. 504-510.
- V. Shvets and I. Y. Lykov, “Kinetics and Mechanism of the Reaction of Ethylene Oxide with n-Decyl Mercaptan during Basic Catalysis,” Kinetika i Kataliz, Vol. 12, No. 2, 1971, pp. 347-355.
- V. Shvets and I. Y. Lykov, “Kinetics and Mechanism of the Noncatalytic Reaction of Ethylene Oxide with n-Decyl Mercaptan,” Kinetika i Kataliz, Vol. 12, No. 4, 1971, pp. 883-888.
- V. Shvets, A. Romashkin and V. Yudina, “Kinetics and Mechanism of the Reaction of Ethylene Oxide with Terephthalic Acid during Catalysis by Tetraal-Kylammonium Halides,” Kinetika i Kataliz, Vol. 14, No. 4, 1973, pp. 928-932.
- N. Lebedev and K. Guskov, “Reaction of α-Oxides. II. Kinetics of the Reactions of Ethylene Oxides with Acetic and Monochloroacetic Acids,” Kinetika i Kataliz, Vol. 5, No. 4, 1963, pp. 116-127.
- L. Shechter and J. Wynstra, “Glycidyl Ether Reactions with Alcohols, Phenols, Carboxylic Acids, and Acid Anhydrides,” Industrial and Engineering Chemistry, Vol. 48, No. 1, 1956, pp. 86-93. doi:10.1021/ie50553a028
- N. Lebedev and M. Smirnova, “Mechanism of the Acid Catalysis of the Reaction of Ethylene Oxide with Amines,” Zhurnau Obshchei Khimii, Vol. 39, No. 12, 1969, pp. 2732-2737.
- A. Weindenbacher, S. Serban and E. Weindenbacher, “Kinetics of Ethoxylation Reactions in Series of Primary Aliphatic-Amines,” Revista de Chimie, Vol. 29, No. 3, 1978, pp. 204-209.
- N. Iranpoor, I. Mohammadpoor-Baltork and F. S. Zardaloo, “Ceric Ammonium Nitrate an Efficient Catalyst for Mild and Selective Opening of Epoxides in the Presence of Water Thiols and Acetic Acid,” Tetrahedron, Vol. 47, No. 47, 1991, pp. 9861-9866. doi:10.1016/S0040-4020(01)80723-7
- C. Moberg, L. Rakos, and L. Tottie, “Stereospecific Lewis Acid Catalyzed Methanolysis of Styrene Oxide,” Tetrahedron Letters, Vol. 33, No. 16, 1992, pp. 2191-2194. doi:10.1016/0040-4039(92)88174-4
- C. C. Price and D. D. Carmelite, “Reactions of Epoxides in Dimethyl Sulfoxide Catalyzed by Potassium t-Butoxide,” Journal of the American Chemical Society, Vol. 88, No. 17, 1966, pp. 4039-4044. doi:10.1021/ja00969a026
- A. Bukowska and W. Bukowski, “A Highly Regio-Selective Catalyst of Epichlorohydrin Acidolysis,” Journal of Chemical Technology & Biotechnology, Vol. 73, No. 4, 1998, pp. 341-344. doi:10.1002/(SICI)1097-4660(199812)73:4<3C341::AID-JCTB960>3.0CO;2-6
- A. Guy, J. Doussot, C. Ferroud, R. Garreau and A. Godefroy-Falguieres, “Regioselective Ring Opening of Epoxides with Lithium Azide,” Synthesis, Vol. 9, 1992, pp. 821-822. doi:10.1055/s-1992-26233
- M. Brunner, L. Mussmann and D. Vogt, “Kinetic Resolution of Oxiranes by Use of Chiral Lewis Acid Catalysts,” Synlett, Vol. 12, 1993, pp. 893-894. doi:10.1055/s-1993-22641
- E. Knaggs, “Alkanoloamides in Soft Detergents,” Soap Chemical Specialties, Vol. 40, No. 12, 1964, pp. 79-83.
- H. Grossmann, “Die Beeinflussung der Reaktion bei der Oxaethylierung von Fettsaeuremonoaethanolamiden,” Fette, Seifen, Anstrichmittel, Vol. 74, No. 1, 1972, pp. 58-63. doi:10.1002/lipi.19720740111
- S. Kanoh, M. Naka, T. Nishimura and M. Motoi, “Isomerization of Cyclic Ethers Having a Carbonyl Functional Group: New Entries into Different Heterocyclic Compounds,” Tetrahedron, Vol. 58, No. 35, 2002, pp. 7049-7064. doi:10.1016/S0040-4020(02)00701-9
- K. Frisch, D. Tummers and A. Nijenhuis, “Tris-(N-β-Hydroxypropyl) Isocyanurate,” Europe Patent No. 3620, 1979.
- A. Ślączka and J. Lubczak, “Hydroxyalkylation of Barbituric Acid. II. Synthesis of Polyetherols with Pyrimidine Ring,” Journal of Applied Polymer Science, Vol. 106, No. 6, 2007, pp. 4067-4074. doi:10.1002/app.26742
- E. Chmiel-Szukiewicz, “Polyetherols Obtained from 6- Aminouracil and Oxiranes,” Journal of Applied Polymer Science, Vol. 103, No. 3, 2007, pp. 1466-1472. doi:10.1002/app.25100
- J. Lubczak, “Reactions in the System (Hydroxymethyl)Melamines-Oxiranes: Addition,” Indian Journal of Chemistry, Vol. 33B, 1994, pp. 125-131.
- K. Schwetlick, “Kinetische Methoden zur Unter-Suchung von Reactionsmechanismen,” VEB, Deutscher Verlag der Wissenschaften, Berlin, 1971.