Stem Cell Discovery
Vol.4 No.2(2014), Article ID:45327,17 pages DOI:10.4236/scd.2014.42004
Multipotentialmesenchymal Stromal Cells (MMSC) Ameliorate Graft versus Host Disease (GVHD) in a Mouse Model, But Major Suppression of GVHD Permits Leukemic Relapse
Barbara J. O’Kane1*, Marcel D. DeVetten2, John J. Jackson3, Jordan P. Lacy4, Tracy L. Farrell5, John G. Sharp6
1Department of Oral Biology, School of Dentistry, Creighton University, Omaha, USA
2The Nebraska Medical Center, Omaha, USA
3Institute for Regenerative Medicine, Wake Forest University, Winston-Salem, USA
4Department of Surgery, University of Nebraska Medical Center, Omaha, USA
5Department of Pharmacy Practice, University of Nebraska Medical Center, Omaha, USA
6Department of Genetics, Cell Biology & Anatomy, University of Nebraska Medical Center, Nebraska Medical Center, Omaha, USA
Email: *barbaraokane@creighton.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 12 March 2014; revised 9 April 2014; accepted 16 April 2014
ABSTRACT
A significant complication in allogeneic stem cell transplantation is graft versus host disease (GVHD). The use of multipotential mesenchymal stem cells (MMSC) for the amelioration of GVHD has shown promise as a therapeutic intervention. Given that MMSC can suppress allogeneic immune responses, there is a concern that using these cells may promote leukemic relapse. We describe a murine model of GVHD in the presence of leukemic cells (L1210). Acute GVHD was induced in DBA mice by transplanting bone marrow and spleen cells from C57Bl/6J mice with or without prior injection of L1210 cells. The recipient mice were monitored for signs of GVHD. The mice were then treated with primary MMSC or a C57Bl bone marrow derived cloned mesenchymal cell line (OMA-AD). The results without L1210 cells, demonstrated that mice treated with primary MMSC that had developed moderate GVHD had increased long-term survival when compared to controls. The group treated with OMA-AD cells showed minimal GVHD so cloned OMA-AD MMSC cells provided a significant protective effect against GVHD, and the survival rate was superior to that of animals treated with primary MMSC on the same day. In the presence of L1210, the control mice all died by day 11, and the mice receiving OMA-AD and L1210 cells died by day 9. Both had minimal GVHD. Only the mice receiving primary MMSC that developed moderate to severe GVHD survived long term. It appears that although MMSC and OMA-AD cells can ameliorate GVHD; the greater immunosuppressive effect of OMA-AD cells permitted the re-growth of the leukemic cells. In contrast, the moderate GVHD that remained after primary MMSC treatment eliminated the leukemia in the majority of mice. These studies demonstrated that in the mouse model, as in man, administration of primary or cloned MMSC ameliorated GVHD. However, complete suppression of GVHD permitted leukemic relapse.
Keywords:Mesenchymal Stem Cells, GVHD, Leukemia, Mouse Model

1. Introduction
Acute graft versus host disease (GVHD) is the major cause of non-relapse mortality following hematopoietic stem cell transplantation for patients with hematological malignancies [1] . Le Blanc and colleagues have pioneered the evaluation of multipotential mesenchymal stromal cells (MMSC) for amelioration of GVHD and they provided the initial evidence that this therapeutic approach had clinical benefits [2] -[4] , which led to a multicenter European trial [5] . Other groups, in largely anecdotal reports, have also observed clinical benefits in steroid resistant GVHD with minimal toxicities [6] -[9] although some more recent studies, e.g. von Bonin et al. [10] did not observe as good outcomes as some earlier studies [3] . Clinical, as well as preclinical, studies have been reviewed by Lin and Hogan [11] , Baron and Storb [12] and Blazer et al. [13] . Osiris has received approval for Prochymal™ in treatment of GVHD, and other products such as MultiStem™ are under evaluation [14] [15] , although complete mechanisms of their actions are unclear. Long term, it has been observed that in patients with malignancies, the occurrence of GVHD is associated with a lower relapse rate [16] , so a major concern of the use of MMSC to ameliorate GVHD is that this therapy will decrease the anti-tumor effectiveness of graft versus leukemia (GVL) processes [12] .
The properties of MMSC’s have been under investigation for many years [17] . These cells, originally derived from bone marrow, were evaluated by Friedenstein et al. [18] and termed precursors of mechanocytes. A primary property of these cells is their ability to form clonal colonies of cells in vitro (fibroblast colony forming cells, FCFC, or colony forming cells fibroblast, CFC-F). In addition, these cells exhibit multipotential differentiation under different culture conditions to fibroblasts, chondrocytes, osteoblasts and adipocytes [19] [20] . Owen and Friedenstein [21] provided a summary of the biology of these cells in a CIBA Foundation Symposium and coined the term marrow stromal cells (MSC). Caplan and colleagues [22] evaluated the differentiation potential of these cells, and patented a description of these cells as mesenchymal stem cells (MSC). A generally accepted criterion for stem cells is that they exhibit self-renewal as well as the ability to produce at least one lineage of differentiated progeny. Most MSC do not demonstrate this ability in vitro or in vivo [23] and it is likely the majority are committed transit amplifying cells of the connective tissue lineage or connective tissue precursors (CTP) described by Muschler and Midura [24] . A subcommittee of the International Society for Cellular Therapy described the properties of these cells and recommended they be termed multipotential mesenchymal stromal cells (MMSC) and this terminology is adopted for this study [25] . MMSC can be obtained from bone marrow [20] , liposuction aspirates [26] -[28] , Wharton’s jelly, placenta [29] [30] and potentially other tissues and appear related to pericytes of blood vessels [31] [32] . The extent of differences based on tissue of origin requires further evaluation [33] .
In addition to their ability to differentiate to various connective tissue cell types, these cells have been shown to suppress allogeneic lymphocyte interactions, specifically mixed lymphocyte reactivity [34] [35] prolong the survival of allografts [36] and mediate tissue growth and repair [37] . The mechanism of this effect has been claimed to involve cellular interactions as well as humoral effects [38] . Some current reports are contradictory and mechanisms are difficult to investigate in patients, although studies of changes in gene expression profiles have been reported [39] . The immunomodulatory effects of MMSC have been described by several groups of investigators, but they differ in their interpretation of the mechanism(s) of these effects. Le Blanc and Ringden [40] claimed that MMSC escape the immune system and recognition by CTLs and alloreactive NK cells [41] . This position was supported by Maitra et al. [42] , who noted human MMSC did not elicit T cell activation in vitro. In contrast, Nauta et al. [43] noted that allogeneic MMSCs are not intrinsically immunoprivileged and can induce a memory T cell response. Regarding their immunosuppressive effects, inhibition of T lymphocyte proliferation, at least in part by a soluble factor, has been reported [34] [40] [41] . Plumas et al. [44] described MMSC induction of apoptosis of activated T cells with no effect on resting T cells. Indeed the roles of multiple cell sub-populations as well as cytokines/chemokines in GVHD have been described [45] . Such claims of the involvement of multiple mechanisms emphasize the need for an animal model to attempt to resolve these contradictions and to evaluate postulated mechanisms of action. However, as noted by Baron and Storb [12] , previous attempts to demonstrate the efficacy of MMSC to ameliorate GVHD in mouse models have had limited success.
Graft versus host disease (GVHD) arises when immune effector cells in a cell therapy product infused into an immunosuppressed (high dose therapy treated) recipient react, immunologically, against cells and tissues of the recipient [46] [47] . This complication is usually moderated primarily by use of immunomodulation, specifically the use of cyclosporin and methotrexate (immunosuppression), mycophenylatemofitil, antithymocyte globulin and high-dose steroids. However, these agents are only partially effective and some cases of GVHD are resistant to steroids and other therapies, and this indicates poor outcomes [48] -[50] . The global incidence of GVHD ranges from approximately 25% - 30% (related donors) to upwards of 50% (unrelated donors) [51] although advances in technology, e.g. T cell depletion [52] have reduced this mortality. Given that MMSC can suppress allogeneic immune responses and facing an extreme case of GVHD in a child receiving a haploidentical allogeneic transplant for leukemia, Le Blanc et al. [2] prepared MMSC from the child’s mother and infused them to this individual with severe gut GVHD as a second line of therapy. The symptoms of GVHD rapidly resolved. This procedure was repeated to treat a recurrence of GVHD several months later and again was successful. Unfortunately, the patient succumbed to GVHD recurrence and infection [11] , raising a concern that, although infusion of MMSC can ameliorate GVHD, this procedure might induce profound immunosuppression. This additionally raises concerns that suppression of GVHD might permit tumor relapses [53] . It appears that GVL can be separated from GVHD [54] [55] although this requires additional evaluation [56] . MMSC have been shown to reduce the anti-tumor activity of cytokine-induced natural killer cells [57] . Investigation of increased rates of tumor relapse requires an animal model. It has also been reported that MMSC can transform to form sarcomas [58] . Evaluation of this concern also requires an appropriate mouse model, likely based on the use of immunodeficient mice.
In vitro, MMSC suppress lymphocyte proliferation [35] and produce immunomodulatory factors [59] . In vivo, MMSC are immunomodulatory and alter immune cell populations [60] [61] . These immune cell populations play significant roles in engraftment and GVHD [62] including in xenogeneic GVHD [63] . A more detailed analysis of immune changes in GVHD could be facilitated and manipulated using a validated animal model. The initial applications of MMSC were for treatment of GVHD [9] . Subsequently, attempts have been made to prevent GVHD and increase cell engraftment, either by co-transplantation of stem cells and MSC [35] [64] or prior transplant of MSC [65] . In general, these approaches have not significantly improved outcomes. Dazzi and Marelli-Berg [66] noted that “MSC can treat GVHD only if administered in the presence of active disease”. This appears to be the case [12] . Consequently, this study emphasized development of a mouse model that reflects the clinical situation. The applications of xenogeneic models, although they employ human cells, may not adequately reflect the complex immunological interactions that occur in syngeneic/allogeneic situations [45] [67] .
Even though additional clinical studies indicate that this therapeutic approach has benefits [45] , the in vivo mechanism(s) of action remain uncertain [68] -[70] and soluble factors [60] [69] [71] as well as secreted galectins [59] have been implicated. Additional mechanisms include effects of MSC on T cells [69] [72] , T cells and B cells [73] particularly T regulatory cells, which appear increased [72] [74] -[76] . Recently, the secretion of bioactive molecules by MMSC in the form of exosomes has been described and this is a potential mechanism of action [77] [78] . A relevant animal model could assist in the resolution of the relevance of these competing mechanisms. Information on mechanisms is critical to the rationale development of MMSC for treatment of GVHD, which has both acute and chronic components. Furthermore, this approach may potentially offer benefits in a broader range of immune diseases, including autoimmune diseases, such as multiple sclerosis [79] , as well as inflammatory conditions including hemorrhagic cystitis, pneumomediastinum and perforated colon [80] although not all reports are positive [81] .
Consequently, there is merit in continuing to develop and evaluate animal models [76] for investigation of mechanisms of MMSC amelioration of GVHD, as well as effects on the risks of promoting relapse of malignancies. Unfortunately, reports investigating whether MMSCs control GVHD in mouse models, present contradictory conclusions. Yanez et al. [82] claimed that infusion of mouse MSCs in mice transplanted with haploidentical hematopoietic grafts controlled lethal GVHD. In contrast, Sudres et al. [35] and Badillo et al. [83] concluded that in their mouse models, the data did not support a significant immunosuppressive effect of MMSC in vivo for treatment of GVHD. Similarly, negative results have been reported in rats [84] and dogs [12] [85] . Furthermore, although primary MMSC have been reported to be effective, use of cloned MMSC cells was reported to be unsuccessful [86] . One problem is that primary mouse MMSC differ between strains [87] , and can be difficult to grow and are variable in morphology and properties depending on the culture conditions [88] [89] . This suggested that use of an appropriate MMSC line [20] with more consistent properties might offer advantages in experimental systems. Such an approach would offer advantages of consistency, economy, and regulatory efficiency in clinical applications.
In an attempt to resolve these controversies, the objective of this research was to develop, evaluate and validate a basic mouse model using strain combinations not previously evaluated [12] to investigate the ability of MMSC including a cloned MMSC line [20] to ameliorate acute GVHD, to examine effects on major immune cell types and determine if this therapy compromised allograft versus leukemia cell therapeutic benefits.
2. Materials and Methods
2.1. MMSC Isolation, Cell Culture and Characterization
Multipotential mesenchymal stromal cells (MMSC) were obtained initially from the femurs of C57Bl/6J female mice (6 - 8 weeks, and approximately 20 g body weight) (Jackson Labs, Bar Harbor, ME). Briefly, bone marrow was harvested from the femurs by removing the proximal and distal ends and gently expressing medium through the diaphysis using a 1 cc syringe equipped with a 21 gauge needle. The bone marrow cells were then made into a single cell suspension and counted using a Beckman Coulter AcT diff. Analyzer. Ten million cells were placed into T-25 cell culture flasks containing 10 mL of Mesencult Basal Medium for Mouse Mesenchymal Stem Cells (Stem Cell Technologies). The cultures were then incubated at 37˚C and 5% CO2 in air. In addition, a C57Bl mouse derived MMSC line (OMA-AD) was also employed [20] . These cells were characterized phenotypically employing flow cytometric analysis with a panel of antibodies. In addition, MMSC isolated from the recipient (DBA mice) and third party MMSC (Balb/c mice) were similarly isolated and evaluated for efficacy compared to MMSC matching the bone marrow cell donors (C57Bl mice).
2.2. Induction of Graft versus Host Disease (GVHD) and Assessment of GVHD
GVHD was induced using female DBA (H2d) mice as recipients and C57Bl/6J (H2b) female mice as donors (Figure 1). The recipient mice were irradiated (9.5 Gy) using a Picker Cobalt 60 Irradiator (Day 0). The following day (Day 1) the mice were transplanted with donor bone marrow cells (5 × 106/mouse, or 3.0 × 108/kg) and initially variable numbers of spleen cells. The effects of a low number (1.0 × 106/mouse, or 6.0 × 107/kg), intermediate (3.0 × 106/mouse, or 1.8 × 108/kg) and high dose (6.0 × 106/mouse, or 3.6 × 108/kg) of splenic CD3 T cells on GVHD development were determined. These studies identified 6 × 106 spleen cells/mouse (3.6 × 108/kg) as the optimal spleen cell dose to induce GVHD in 100% of recipients by 7 - 10 days post-transplant. Animals were monitored daily and scored twice weekly for symptoms of GVHD which included weight loss, as well as ruffled fur, hunched posture and diarrhea and scored on a 0.5 (slight) or 1.0 (significant) scale for a total of 3.0.
2.3. MMSC and OMA-AD Cell Dose Responses
Recipient mice were slowly infused with escalating doses of donor MMSC or OMA-AD on days 7 - 10 posttransplant. The MMSC cell doses included 1 × 105/mouse (6.0 × 106/kg), 5 × 105/mouse (3.0 × 107/kg), or 1 × 106/mouse (6.0 × 108/kg). The MMSC were harvested from the culture flasks, adjusted to the appropriate cell concentration and injected into the lateral tail vein of the recipients. Outcomes when employing matching donor or mismatched recipient MSC were noted. This established 1 × 105 MMSC/recipient, i.e. 6.0 × 106/kg, regard-

Figure 1. Outcomes of post-irradiation (9.5 Gy total body) transplantation of bone marrow plus various numbers of spleen cells to induce GVHD of varying severities.
less of histocompatibility differences, as an adequate cell dose. OMA-AD cells were also employed at a cell dose of 1 × 105 cells/recipient (6.0 × 106/kg). To establish the bona fide properties of the MMSC, their differentiation potential was evaluated in vitro. Bone marrow cells were placed in culture conditions known to promote the growth of MMSC’s and subcultured by trypsinization for several passages. The MMSC’s were then harvested and placed in 12-well plates containing osteoblast or adipose differentiation medium. After ten days in culture, the coverslips were removed fSrom the wells and stained with Oil Red-O to identify adipocytes and Alazarin red to identify osteoblasts and bone respectively to demonstrate they met the criteria established for MMSC.
2.4. Tracking of Infused Cells
Attempts were made to track two populations of infused cells, MMSC and L1210 cells. Since MMSC were primarily derived from C57Bl mice, in order to track the distribution of these cells following infusion into recipients, MMSC derived from enhanced green fluorescence protein (EGFP) expressing mice were isolated and infused into non-EGFP expressing recipients. The distribution of cells in various tissues was determined with time after infusion.
L1210 cells were phenotyped and shown to be detectable by expression of CD90 (Thy1) in the absence of CD4 or CD8 expression (Figure 4). Attempts were also made to transduce the cells using a DS Red vector to identify these cells in the presence of EGFP expressing MMSC. However, DS Red expression was rapidly diluted in culture and difficult to detect in vivo.
2.5. Immunological Evaluations in Mice with GVHD Receiving MMSC
Spleen cells were harvested from recipients of MMSC or OMA-AD versus saline treated controls and analyzed by flow cytometry for B cells, CD3, CD4 and CD8 T cells, as well as CD4/CD25/FOXP3 regulatory T cells. Additionally, this information was obtained from transplanted mice receiving L1210 cells at one week post-transplant. Spleens were made into cell suspensions and nucleated cells enumerated. One million cells were stained with an antibody that detected donor (C57Bl, H2b) cells or recipient (DBA, H3d) cells in combination with an antibody to B cells (anti-B220) granulocytes (GR1), CD3, CD4 or CD8 T cells. In addition, cells were stained with CD4 and CD25, permeabilized and stained for FOXP3. All antibodies were purchased from eBioscience and Pharmingen. At least 1 × 106 cells were analyzed employing a Becton Dickenson FACS Aria and data was collected in list mode and analyzed employing FlowJo (TreeStar Inc., Ashland, Oregon) software. Data was obtained from mice post-transplantation, before administration of MMSC, and from survivors after MMSC and transplanted mice receiving L1210 cells.
2.6. Development and Application of the L1210 Leukemia Cell Model
L1210 cells were acquired from ATCC (American Type Culture Collection, Manassas, VA) and cultured according to the ATCC protocol. Based on CD90 (Thy1) expression, L1210 cells represented a T lymphoblastic leukemia cell model. Growth curves were generated and doubling time calculated in order to determine an appropriate cell number to establish an in vivo tumor model that resembled human leukemia. Additionally, in modeling the human situation, patients undergo high dose therapy before allogeneic bone marrow (stem) cell transplantation. In the mouse model this therapy was exposure to 9.5 Gy of total body Cobalt 60 irradiation, so the radiation sensitivity of the L1210 cells was analyzed. The survival and growth of 2 × 106 L1210 cells irradiated with 2 - 16 Gy of Cobalt 60 protons employing a Picker v90 Teletherapy irradiator was determined (Figure 5). This showed that at least 0.25 × 106 cells (12.5%) survived 8 Gy with a viability of 50% or greater. It was determined that if 5 × 103 L1210 leukemia cells were injected intravenously (on day 0) a sufficient number would survive 9.5 Gy total body irradiation on day 1, and bone marrow transplantation on day 2, to produce leukemia in mice over the next one to two weeks and in the absence of further therapies, would lead to death of the recipients.
3. Results
3.1. Induction and Characterization of GVHD
Consistent induction of GVHD in the mouse required infusion of a mixture of bone marrow cells (BMT), to effect lymphohematopoietic reconstitution and spleen cells primarily to provide immune T cells that induced GVHD. Allogeneic bone marrow alone repopulated (Figure 1) but did not induce significant GVHD, so no deaths were observed. Allogeneic spleen cells contributed to lymphohematopoietic reconstitution, but primarily induced GVHD. If a large dose of spleen cells was used (6 × 106 cells/mouse), GVHD was severe and all recipients died. Consequently, appropriate combinations of bone marrow and spleen cell doses were required. The optimal CD3 T cell doses were determined to be achieved by employing in the range of 1 - 3 × 106 spleen cells (Figure 1). When a higher spleen cell dose was employed, weight loss, hunched posture, diarrhea and death ensued rapidly and it was not possible to infuse MMSC (at 10 days) in time to ameliorate GVHD. If too low a spleen cell dose was employed, the severity of GVHD was significantly reduced and the beneficial impact of infusing MMSC was difficult to demonstrate. In subsequent studies, the day of MMSC infusion was advanced to day 7 to improve survival.
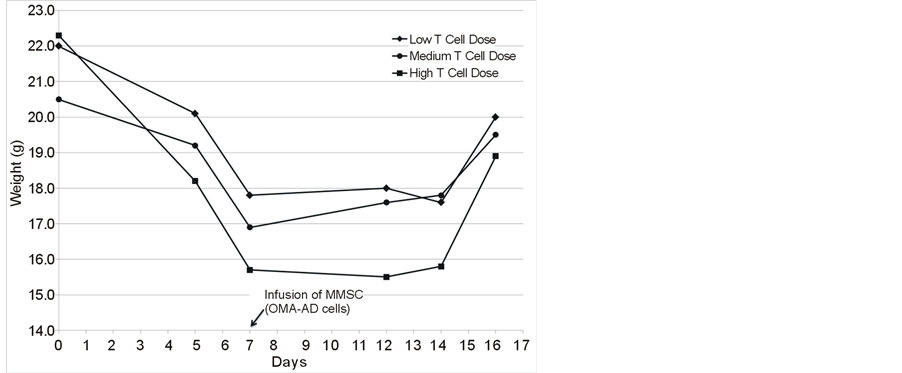
In order to refine this analysis, a titration of infused allogeneic spleen cells containing CD3 T cells was performed employing weight loss and GVHD score to evaluate the severity of GVHD and assess the ability of a MMSC cell line OMA-AD to ameliorate GVHD (Figures 2(A)-(C)). Following BMT and T cells infusions, mice lost weight (Figure 2(A)), and the extent of weight loss tracked with increasing numbers of infused allogeneic T cells. The GVHD score increased in proportion to the number of T cells infused (Figure 2(B)). The GVHD score correlated well with the extent of weight loss (Figure 2(C)). The severity of GVHD could be arbitrarily classified as minimal (GVHD score < 0.62, weight loss > 2.5 g), moderate (GVHD score 0.63 to 1.24, weight loss > 2.5 g, < 5.0 g) and severe (GVHD score > 1.25, weight loss > 5.0 g).
3.2. Characterization of MMSC Cell Populations
Primary MMSC were evaluated for ability to differentiate in culture into osteoblasts or adipocytes (Figures 3(A)-(C)) and for phenotype. The cells were Stro1+ and expressed low but variable levels of hematopoietic cell associated markers (Figure 3(D)). A MMSC cell line isolated from C57Bl mice has been evaluated and appears similar and does not express a marker that is specifically useful for isolation of MMSC. The primary distinguishing characteristics of these cells is the ability to differentiate in culture to predicted progeny of MMSC (chondrocytes, osteoblasts and adipocytes) [20] .
MMSC derived from mice matching the donor, the recipient and a third party were assessed. The ability to ameliorate GVHD did not appear to be significantly related to the immunological disparity between MMSC and the donor or recipient, and the differing doses of MMSC produced similar levels of survival (43% ± 4%, range 37% - 50%)
 (A)
(A) 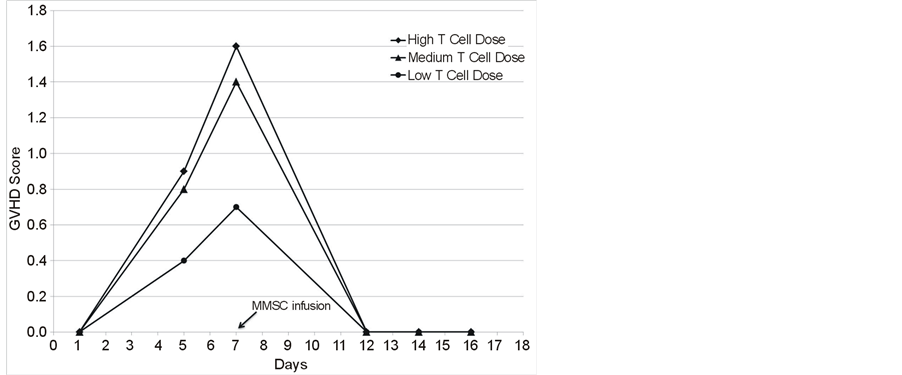 (B)
(B) 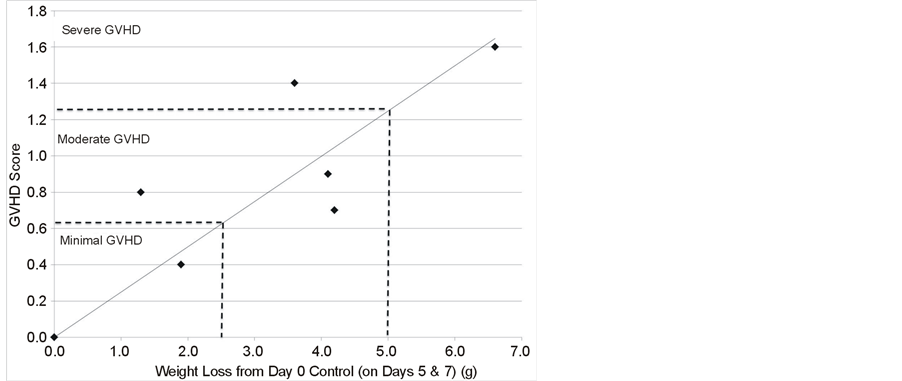 (C)
(C)
Figure 2. (A) Mouse body weight during development of GVHD and its amelioration by MMSC infusion; (B) GVHD Score in mice developing GVHD and its amelioration with MMSC; (C) Correlation of GVHD score with mouse weight loss.
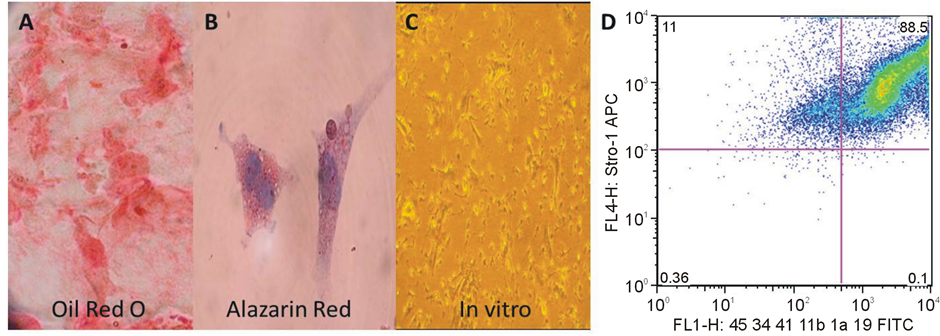
Figure 3. Characteristics of Primary MMSC (for characteristics of OMA-AD see Tuljupurkar et al., 2011); Differentiated to adipocytes (A); Differentiated to bone (B); Unmanipulated Control OMA-AD (Phase Contrast) (C); (D): The cells were Stro1+ and expressed low but variable levels of hematopoietic cell associated markers.
MMSC derived from transgene green fluorescent protein (GFP) expressing donors were injected and the recipients necropsied at various, relatively short term, time points post-injection. Most of the GFP expressing cells disappeared quickly following injection (data not shown), but a very low number <1% persisted beyond 72 hours (data not shown). MMSC are likely heterogeneous but the differences, if any, between MMSC that are cleared versus those that persist are not known.
3.3. Are MMSC Anti-Tumor (GVL) Effects Based on Cellular or Humoral Factors?
To determine if a soluble product of MMSC was produced and was as effective as cells as an anti-tumor mechanism, media was collected from OMA-AD cells, and concentrated and then injected into allografted mice in place of MMSC.
Media did not effectively ameliorate GVHD (data not shown) suggesting that cellular MMSC were required for anti-GVHD effects. However, the detailed mechanisms of all of these effects remain unclear at this time and these experiments did not eliminate the potential for biological effects of MMSC derived exosomes which would have minimal numbers in media but could persist longer than the cells in vivo. Overall, MMSC ameliorated GVHD but major suppression of GVHD might compromise anti-tumor responses. Note that a generally similar conclusion was reported by Truitt et al. [90] .
3.4. Immunological Effects of Infusion of MMSC or OMA-AD Cells into BMT Recipients
Surviving MMSC or OMA-AD infused mice were necropsied after 2 weeks and spleens evaluated for donor versus recipient immune cells and compared to post-transplant controls. Only low numbers of DBA cells were observed (less than 5%) indicating that the mice were chimeric and largely repopulated (90% +) by C57Bl cells. Compared to saline treated controls, the MMSC/OMA-AD treated mice had similar numbers of B cells and granulocytes and slightly lower numbers of CD3 and CD4 cells, especially OMA-AD recipients and slightly higher CD8 and T reg numbers (Table 1). In mice receiving L1210 cells, the leukemia cells dominated and significantly decreased the properties of immune cells in the spleen. This indicated that the L1210 cells survived the preparative regimen and grew in the mice.
3.5. Characterization of L1210 Leukemia Cells
The L1210 cells were predominantly Thy (CD90) positive (Figure 4(A), Figure 4(B)). The doubling time of L1210 cells was determined in order to estimate the number of cells to be infused to permit leukemia to develop and allow time to perform a therapeutic intervention (irradiation followed by BMT) followed by MMSC infusion at originally 10, but later at 7, days after BMT. Based on in vitro growth curves of 3 cell concentrations, the doubling time was estimated at 13 - 14 hours after a lag period of about 24 hours (Figure 4(C)). Note also that the growth of the cells increased and doubling times decreased based on the number of cells plated, suggesting that L1210 cells appear autostimulatory (Figure 4(D)). These data suggested that 5 × 103 L1210 cells was a suitable cell dose to model human leukemia.
An additional consideration was the impact of the 9.5 Gy dose of total body irradiation, employed as the preparative regimen, on the growth of L1210 cells. An attempt was undertaken to construct a radiation survival curve for L1210 cells. The results (Figure 5) suggested two components to a survival curve with a sensitive population of cells (Do of the order of 1 Gy) and a very resistant population that survived even in the face of irradiation with 8 - 64 Gy. This permitted re-growth of L1210 cells at 48 - 72 hours even following high doses of irradiation (Figure 5). Potentially, L1210 contains radiation sensitive proliferating cells in addition to quiescent radiation resistant cells, although this requires further evaluation. Overall, the data indicated that 9.5 Gy of total body irradiation did not eliminate L1210 cells (Figure 5), and following BMT, these cells could rapidly re-grow and kill the host unless additional anti-tumor mechanisms are active, e.g. graft versus leukemia (GVL) effects.

Table 1. Immune cell populations in receipient spleens following BMT only, survivors of BMT and MMSC, and BMT in mice bearing L1210 cells. ND = Not done, NA = Not applicable.
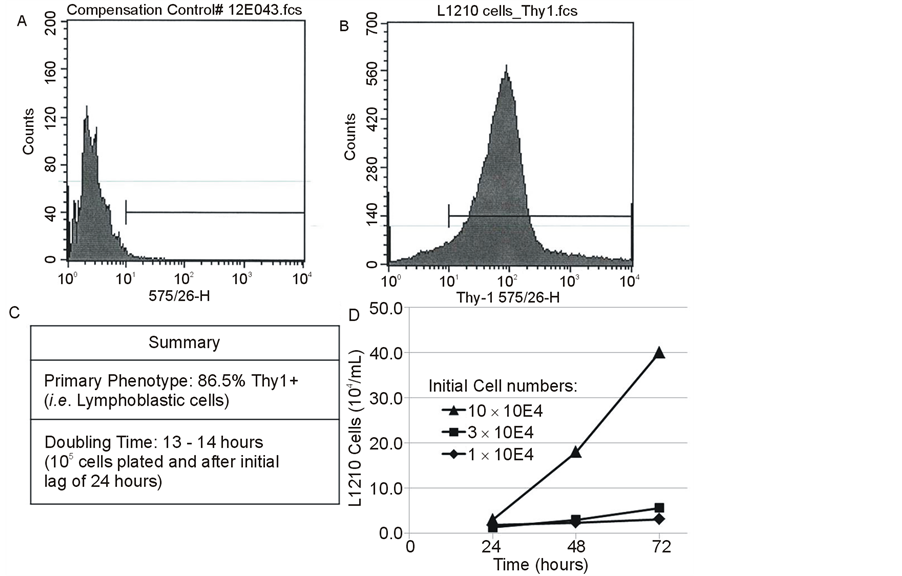
Figure 4. Characterization of L1210 cells: Phenotype (A,B), Growth (C) and Summary (D).
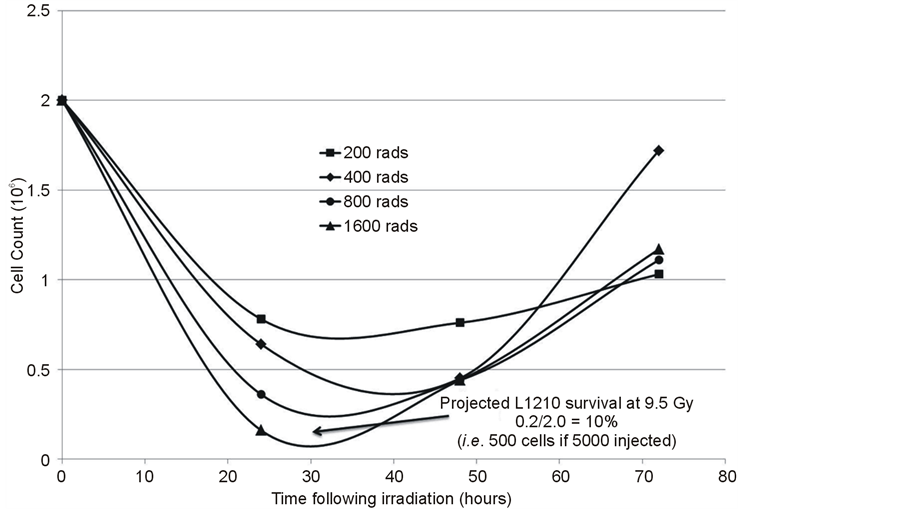
Figure 5. Characterization of effects of radiation on L1210 cells to select L1210 cell numbers required to survive 9.5 Gy TBI employed for BMT, but also allow for appropriate leukemia cell numbers based on doubling time (Figure 6) for evaluation of MMSC infusion on mouse survival.
In these experiments, 5 × 103 L1210 leukemia cells required to cause the leukemia in all intact syngeneic mice were infused and the mice were transplanted with bone marrow and spleen cells, followed by MMSC, and followed for development of GVHD or leukemia. Subsequently this experiment was repeated and seven days post-transplant the mice received either saline or OMA-AD cells iv and the effects on GVHD and leukemia incidence was noted (Figure 6). L1210 cells alone lead to death by 11 days which was a little slower than that observed in a non-therapeutic situation [91] . If primary MMSC were infused, significant GVHD ensued and long term survival was 80%. In contrast, if cell line, OMA-AD cells were infused, minimal GVHD was observed and the mice died within 9 days with evidence of leukemia.
Overall, these data indicate that MMSC ameliorate GVHD. The MMSC cell line (OMA-AD) was better at suppressing GVHD than primary MMSC. However, in the presence of L1210 leukemia cells, suppression of GVHD by OMA-AD cells lead to leukemic relapse and death by day 9. In contrast, although primary MMSC allowed development of moderate to severe GVHD, this provided long term survival (80%) suggesting effects of GVL in this situation.
4. Discussion
This research has developed, evaluated and validated an animal (mouse) model of GVHD that can be employed to investigate the mechanisms of MMSC amelioration of GVHD. This model employed the infusion of bone marrow cells, primarily to effect engraftment and regeneration of lymphohematopoietic tissues, with infusion of spleen cells to generate GVHD. The provision of allogeneic T cells in increasing numbers caused GVHD of increasing degrees of severity. Infusion of MMSC ameliorated GVHD that developed. Leukemia cells (L1210) were administered to determine if amelioration of GVHD compromised GVL effects. The resulting data indicated that partial suppression of GVHD did not suppress GVL, however complete suppression of GVHD with a MMSC cell line did suppress GVL sufficiently to permit leukemic cell re-growth. Therefore, although MMSC can suppress GVHD, they are currently a relatively crude tool that needs additional refinement and/or targeting.
To effect GVHD, allogeneic T cells in spleen cell suspensions were added to the bone marrow cell inoculum. Depending on spleen, and therefore CD3, T cell number, this combination induced minimal to relatively severe GVHD manifested as weight loss, diarrhea, hunched posture and histopathological changes and death within 7 - 14 days. It should be noted that housing conditions might also influence the course of GVHD. Studies of

Figure 6. Effects of MMSC on survival of mice with L1210 cell leukemia undergoing BMT.
animals maintained under gnotobiotic (germ-free) conditions demonstrated that this reduced the incidence/severity of GVHD [92] . The mice in this study were maintained in individual microisolators. These conditions have been shown to delay deaths of lethally irradiated mice (to 20 - 30 days post-irradiation) compared to conventional housing conditions (deaths 12 - 18 days post-irradiation). This might have a quantitative influence on the results. If mice exhibiting the initial signs and symptoms of GVHD at about day seven were infused with 1 × 105 MMSC cells, initially at day 10, and subsequently at day 7, the signs and symptoms of GVHD were ameliorated and 40% to 80% of the mice survived acute GVHD, in part depending on the day of MMSC infusion as well as T cell dose. These observations might reflect a role for Toll receptors in the pathogenesis of GVHD [93] [94] .
Essentially similar effects were observed when MMSC matching the genotype of the donor versus the recipient mice or third parties were infused. This result differs from that of Joo et al. [75] who observed a positive dose response for MSC inhibition of GVHD. A minimal cell dose to achieve an effect was not defined. However, since there was no evidence of toxicity of the lowest MMSC doses employed (105/mouse or 5 × 106/kg body weight), it appeared unnecessary to evaluate lower cell doses that might increase the risk of failure of the infused cells to ameliorate GVHD.
Clinically, there is an attraction to the use of a generic mesenchymal cell population as reviewed by Lin and Hogan [11] , since this would simplify regulatory issues and especially since the origin of the MMSC cells seems to be unimportant (see above). The MMSC were phenotyped and the ability of a MMSC cell line OMA-AD [20] previously described to ameliorate GVHD was assessed. When the ability of the cell line OMA-AD to suppress GVHD was compared to that of primary MMSC (Figure 5, Figure 6), the results showed that the cell line appeared to be more effective at ameliorating GVHD. However, this might be a disadvantage in the situation when there are surviving leukemic cells in the recipients (see below). Titration of MMSC dose might not matter for amelioration of GVHD, but it might be critically important in minimizing the risk of leukemic relapse. This requires further evaluation.
Since infusion of the MMSC OMA-AD cell line appeared at least as effective as primary MMSC cells (Figure 5) and maybe more efficacious, the use of a regulatory authority approvable MMSC cell line appears to be a viable and simpler option than generating primary MMSC for each patient. However, these data suggest that if MMSC suppression of GVHD is major, such that no or only minimal GVHD develops, the greater immunosuppressive effect may facilitate survival and relapse of the leukemic cells.
The doubling time of L1210 (CCL-219) cells reported at 8 - 10 hours (ATCC data sheet) was slightly longer in this study and was estimated at 13 - 14 hours. The radiation sensitivity assay suggested that at the irradiation dose employed (9.5 Gy), a sufficient number of L1210 cells would survive to cause death in the absence of therapy. This proved to be the case with 100% mortality within 11 days (Figure 6). This is a slightly longer period than approximately 8 days observed by Aldred et al. [91] in a non-therapeutic situation. This suggests that the preparative regimen had some minor therapeutic effect, although the leukemic cells rapidly become a dominant cell population in the spleens of the mice (Table 1). It should be noted that the symptoms associated with death due to the leukemia were similar to and overlapping with those observed due to GVHD in the absence of leukemia [91] which indicated that based on symptomatology, GVHD and leukemia symptoms were not readily distinguished.
The survival of bone marrow and spleen cell transplanted mice that had also been infused with L1210 leukemia cells (Figure 6) showed that the leukemia was fatal within two weeks. Transplanted mice infused with OMA-AD cells to ameliorate GVHD had moderate GVHD but 80% survival long term. This was also the case for transplanted mice infused with both L1210 leukemia cells and with primary MMSC. These mice developed significant GVHD but the majority (80%) did not develop leukemia and survived long term. In contrast, the transplanted mice infused with L1210 leukemia cells and OMA-AD MMSC, only developed minimal GVHD and succumbed rapidly to the leukemia and all died by day 9. This experiment suggested that although MMSC can suppress lethal GVHD, the generation of at least moderate GVHD was necessary to prevent leukemic cell re-growth and relapse. If GVHD was suppressed to a major extent, then leukemic relapse was rapid and death ensued. Consequently, investigators need to be aware that the use of MMSC to ameliorate GVHD in the presence of leukemia cells appears to be a two-edged sword. Too great a success of suppression of GVHD permitted leukemic relapse. This can be inhibited or prevented if some GVHD develops, but this will have other negative consequences for the patient.
Clearly, the mechanism(s) of action of MMSC must involve immune modulation although actual mechanisms are controversial [38] [41] [44] [45] [71] . The evaluation of immune cell populations, including regulatory cells, showed minor differences with slightly increased Treg and CD8 cells and decreased CD3 and CD4 cells. A further evaluation of immunological effects is needed. Also, the duration of survival of MMSC in the recipients was relatively short and it is unknown, at this time, if a specific subpopulation of MMSC survives and what the immunological effects of these cells might be. Alternatively, exosomes produced by MMSC are a candidate mechanism of effect. This could explain why the tissue matching of MMSC is unimportant and is compatible with media alone being ineffective (too few exosomes) as well as not requiring the MMSC themselves to persist in the recipients. This is worthy of additional evaluation.
It should be noted that this study only models acute GVHD and additional models are needed for evaluation of chronic GVHD, autoimmune diseases, the risk of tumor relapse and the de novo generation of sarcomas by the infused MMSC [58] . Indeed, the effects of MMSC in such situations may differ from those in acute GVHD. If the immunological effects of MMSC affect the initial processes of immune activation, for which there is evidence [61] [66] , then amelioration of acute GVHD might not translate into similar effects in chronic GVHD or autoimmune diseases where autoreactive clones of immune cells may have been generated a considerable time previously. In fact, there is a suggestion that this may be the case, since a majority of patients with acute GVHD treated with MMSC responded completely but one with chronic GVHD had only a transient and incomplete response [3] . Very early administration of MMSC to prophylactically inhibit GVHD was also not successful, suggesting immune activation and/or potentially some initial tissue damage may be required and it is these processes that the MMSC (or their products) target. Supernatants of cultured MMSC were not effective. However, soluble products may have a short half-life and/or inadequate concentrations. Also, in a collagen induced arthritis model in mice, infused MMSC did not confer any benefit [95] . Consequently, additional efforts are required to develop appropriate animal models in which to evaluate the therapeutic efficacy of MMSC in chronic GVHD and autoimmune diseases.
Overall, these results describe a mouse model of acute GVHD employing strain combinations not previously evaluated in which the amelioration of GVHD by infusion of MMSC was clearly demonstrated. The infusion of MMSC appears to be safe, although the optimal methods to prepare these cells are an active area of investigation [11] [12] . Furthermore, this model demonstrated that MMSC from donor, recipient, third party or an “off the shelf” cell line ameliorated GVHD, similar to the situation in man. It appears that amelioration of acute GVHD can be accomplished without abrogating allograft versus leukemia (GVL) effects, but at the same time, complete abrogation of GVHD may also abrogate GVL and permit leukemic relapse. The impact of a greater degree of acute GVHD suppression on the severe infections and subsequent development of chronic GVHD is unclear. The phenotype of effective MMSC has not been identified nor have the immunological mechanisms been defined by which this potentially important cellular therapy works [37] [96] , although an increase in regulatory T cells appears to be important. At this time, in the absence of an understanding of mechanisms, a major problem in the use of MMSC for amelioration of GVHD is the inability to predict the extent of immunosuppression that will be induced in recipients. This animal model will permit future investigations of these unanswered questions and contribute significantly to the translation of MMSC for therapy of GVHD and, potentially, other immune diseases, including autoimmune diseases, into clinical reality, as well as determining if fine tuning of amelioration of GVHD without promoting the survival or re-emergence of minimal residual malignant disease can be reliably accomplished or whether alternative approaches to targeting effector cells [97] -[99] will be necessary.
Acknowledgements
The authors thank Sue Brusnahan and Robyn Smith for technical assistance and the Flow Cytometry facilities at UNMC (Dr. Charles Kuszynski) and Creighton University (Dr. Greg Perry). This research was supported by the UNMC Fred and Pamela Buffet Cancer Center as well as with funds from the Gala of Hope and Cattleman’s Ball. This support is gratefully acknowledged.
References
- Cotliar, J.A. (2013) Diagnosing and Treating Acute Graft vs Host Disease. The ASCO Post, 4, 87.
- Le Blanc, K., Rasmusson, I., Sundberg, B., et al. (2004) Treatment of Severe Acute Graft-versus-Host Disease with Third Party Haploidentical Mesenchymal Stem Cells. Lancet, 363, 1439-1441. http://dx.doi.org/10.1016/S0140-6736(04)16104-7
- Ringden, O., Uzunel, M., Rasmusson, I., et al. (2006) Mesenchymal Stem Cells for Treatment of Therapy-Resistant Graft-versus-Host Disease. Transplantation, 81, 1390-1397. http://dx.doi.org/10.1097/01.tp.0000214462.63943.14
- Le Blanc, K. and Ringden, O. (2006) Mesenchymal Stem Cells: Properties and Role in Clinical Bone Marrow Transplantation. Current Opinion in Immunology, 18, 586-591. http://dx.doi.org/10.1016/j.coi.2006.07.004
- Le Blanc, K., Frassoni, F., Ball, L., et al. (2008) Mesenchymal Stem Cells for Treatment of Steroid-Resistant, Severe, Acute Graft-versus-Host Disease: A Phase II Study. Lancet, 371, 1579-1586. http://dx.doi.org/10.1016/S0140-6736(08)60690-X
- Lee, S.T., Jang, J.H., Cheong, J.W., et al. (2002) Treatment of High-Risk Acute Myelogenous Leukaemia by Myeloablative Chemoradiotherapy Followed by Co-Infusion of T Cell-Depleted Haematopoietic Stem Cells and Culture-Expanded Marrow Mesenchymal Stem Cells from a Related Donor with One Fully Mismatched Human Leucocyte Antigen Haplotype. British Journal of Haematology, 118, 1128-1131. http://dx.doi.org/10.1046/j.1365-2141.2002.03767.x
- Lazarus, H.M., Koc, O.N., Devine, S.M., et al. (2005) Cotransplantation of HLA-Identical Sibling Culture-Expanded Mesenchymal Stem Cells and Hematopoietic Stem Cells in Hematologic Malignancy Patients. Biology of Blood and Marrow Transplantation, 11, 389-398. http://dx.doi.org/10.1016/j.bbmt.2005.02.001
- Fang, B., Song, Y.P., Liao, L.M., Han, Q. and Zhao, R.C. (2006) Treatment of Severe Therapy-Resistant Acute Graft-versus-Host Disease with Human Adipose Tissue-Derived Mesenchymal Stem Cells. Bone Marrow Transplant, 38, 389-390. http://dx.doi.org/10.1038/sj.bmt.1705457
- Kebriaei, P. and Robinson, S. (2011) Treatment of Graft-versus-Host-Disease with Mesenchymal Stromal Cells. Cytotherapy, 13, 262-268. http://dx.doi.org/10.3109/14653249.2010.549688
- von Bonin, M., Stolzel, F., Goedecke, A., et al. (2009) Treatment of Refractory Acute GVHD with Third-Party MSC Expanded in Platelet Lysate-Containing Medium. Bone Marrow Transplant, 43, 245-251. http://dx.doi.org/10.1038/bmt.2008.316
- Lin, Y. and Hogan, W.J. (2011) Clinical Application of Mesenchymal Stem Cells in the Treatment and Prevention of Graft-versus-Host Disease. Advances in Hematology, 2011, Article ID: 427863.
- Baron, F. and Storb, R. (2012) Mesenchymal Stromal Cells: A New Tool against Graft-versus-Host Disease? Biology of Blood and Marrow Transplantation, 18, 822-840. http://dx.doi.org/10.1016/j.bbmt.2011.09.003
- Blazar, B.R., Murphy, W.J. and Abedi, M. (2012) Advances in Graft-versus-Host Disease Biology and Therapy. Nature Reviews Immunology, 12, 443-458. http://dx.doi.org/10.1038/nri3212
- Van Bokkelen, G. (2011) Company Profile: Athersys. Regenerative Medicine, 6, 39-43. http://dx.doi.org/10.2217/rme.10.90
- Vaes, B., Hof, W.V., Deans, R. and Pinxteren, J. (2012) Application of MultiStem((R)) Allogeneic Cells for Immunomodulatory Therapy: Clinical Progress and Pre-Clinical Challenges in Prophylaxis for Graft versus Host Disease. Frontiers in Immunology, 3, 345. http://dx.doi.org/10.3389/fimmu.2012.00345
- Pasquini, M.C. (2008) Impact of Graft-versus-Host Disease on Survival. Best Practice & Research Clinical Haematology, 21, 193-204. http://dx.doi.org/10.1016/j.beha.2008.02.011
- Charbord, P. (2010) Bone Marrow Mesenchymal Stem Cells: Historical Overview and Concepts. Human Gene Therapy, 21, 1045-1056. http://dx.doi.org/10.1089/hum.2010.115
- Friedenstein, A.J., Petrakova, K.V., Kurolesova, A.I. and Frolova, G.P. (1968) Heterotopic of Bone Marrow. Analysis of Precursor Cells for Osteogenic and Hematopoietic Tissues. Transplantation, 6, 230-247. http://dx.doi.org/10.1097/00007890-196803000-00009
- Sharp, J.G., Murphy, B.O., Jackson, J.D., Kessinger, A., Brusnahan, S.K. and Neff, J.R. (2005) Promises and Pitfalls of Stem Cell Therapy for Promotion of Bone Healing. Clinical Orthopaedics and Related Research, 435, 52-61.
- Tuljapurkar, S.R., Jackson, J.D., Brusnahan, S.K., O’Kane, B.J. and Sharp, J.G. (2012) Characterization of a Mesenchymal Stem Cell Line That Differentiates to Bone and Provides Niches Supporting Mouse and Human Hematopoietic Stem Cells. Stem Cell Discovery, 2, 5-14. http://dx.doi.org/10.4236/scd.2012.21002
- Owen, M. and Friedenstein, A.J. (1988) Stromal Stem Cells: Marrow-Derived Osteogenic Precursors. Ciba Foundation Symposium, 136, 42-60.
- Caplan, A.I. (1991) Mesenchymal Stem Cells. Journal of Orthopaedic Research, 9, 641-650. http://dx.doi.org/10.1002/jor.1100090504
- Javazon, E.H., Beggs, K.J. and Flake, A.W. (2004) Mesenchymal Stem Cells: Paradoxes of Passaging. Experimental Hematology, 32, 414-425. http://dx.doi.org/10.1016/j.exphem.2004.02.004
- Muschler, G.F. and Midura, R.J. (2002) Connective Tissue Progenitors: Practical Concepts for Clinical Applications. Clinical Orthopaedics and Related Research, 395, 66-80.
- Dominici, M., Le Blanc, K., Mueller, I., et al. (2006) Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy, 8, 315-317. http://dx.doi.org/10.1080/14653240600855905
- Moseley, T.A., Zhu, M. and Hedrick, M.H. (2006) Adipose-Derived Stem and Progenitor Cells as Fillers in Plastic and Reconstructive Surgery. Plastic & Reconstructive Surgery, 118, 121S-128S. http://dx.doi.org/10.1097/01.prs.0000234609.74811.2e
- Lindroos, B., Suuronen, R. and Miettinen, S. (2011) The Potential of Adipose Stem Cells in Regenerative Medicine. Stem Cell Reviews and Reports, 7, 269-291. http://dx.doi.org/10.1007/s12015-010-9193-7
- Philips, B.J., Marra, K.G. and Rubin, J.P. (2012) Adipose Stem Cell-Based Soft Tissue Regeneration. Expert Opinion on Biological Therapy, 12, 155-163. http://dx.doi.org/10.1517/14712598.2012.644533
- McGuirk, J.P. and Weiss, M.L. (2011) Promising Cellular Therapeutics for Prevention or Management of Graft-versusHost Disease (a Review). Placenta, 32, S304-S310. http://dx.doi.org/10.1016/j.placenta.2011.04.013
- Zhang, Q.Z., Nguyen, A.L., Yu, W.H. and Le, A.D. (2012) Human Oral Mucosa and Gingiva: A Unique Reservoir for Mesenchymal Stem Cells. Journal of Dental Research, 91, 1011-1018. http://dx.doi.org/10.1177/0022034512461016
- Pontikoglou, C., Deschaseaux, F., Sensebé, L. and Papadaki, H.A. (2011) Bone Marrow Mesenchymal Stem Cells: Biological Properties and Their Role in Hematopoiesis and Hematopoietic Stem Cell Transplantation. Stem Cell Reviews and Reports, 7, 569-589. http://dx.doi.org/10.1007/s12015-011-9228-8
- Corselli, M., Crisan, M., Murray, I.R., et al. (2013) Identification of Perivascular Mesenchymal Stromal/Stem Cells by Flow Cytometry. Cytometry Part A, 83, 714-720. http://dx.doi.org/10.1002/cyto.a.22313
- Wegmeyer, H., Bröske, A.M., Leddin, M., et al. (2013) Mesenchymal Stromal Cell Characteristics Vary Depending on Their Origin. Stem Cells and Development, 22, 2606-2618. http://dx.doi.org/10.1089/scd.2013.0016
- Aggarwal, S. and Pittenger, M.F. (2005) Human Mesenchymal Stem Cells Modulate Allogeneic Immune Cell Responses. Blood, 105, 1815-1822. http://dx.doi.org/10.1182/blood-2004-04-1559
- Sudres, M., Norol, F., Trenado, A., et al. (2006) Bone Marrow Mesenchymal Stem Cells Suppress Lymphocyte Proliferation in Vitro but Fail to Prevent Graft-versus-Host Disease in Mice. Journal of Immunology, 176, 7761-7767.
- Kuo, Y.R., Chen, C.C., Goto, S., Lin, P.Y., Wei, F.C. and Chen, C.L. (2012) Mesenchymal Stem Cells as Immunomodulators in a Vascularized Composite Allotransplantation. Clinical and Developmental Immunology, 2012, Article ID: 854846. http://dx.doi.org/10.1155/2012/854846
- Boregowda, S.V. and Phinney, D.G. (2012) Therapeutic Applications of Mesenchymal Stem Cells: Current Outlook. BioDrugs, 26, 201-208. http://dx.doi.org/10.1007/BF03261879
- Zhang, Q., Shi, S., Liu, Y., et al. (2009) Mesenchymal Stem Cells Derived from Human Gingiva Are Capable of Immunomodulatory Functions and Ameliorate Inflammation-Related Tissue Destruction in Experimental Colitis. The Journal of Immunology, 183, 7787-7798. http://dx.doi.org/10.4049/jimmunol.0902318
- Sadeghi, B., Al-Chaqmaqchi, H., Al-Hashmi, S., et al. (2013) Early-Phase GVHD Gene Expression Profile in Target Versus Non-Target Tissues: Kidney, a Possible Target? Bone Marrow Transplant, 48, 284-293. http://dx.doi.org/10.1038/bmt.2012.120
- Le Blanc, K. and Ringdén, O. (2005) Immunobiology of Human Mesenchymal Stem Cells and Future Use in Hematopoietic Stem Cell Transplantation. Biology of Blood Marrow Transplantation, 11, 321-334. http://dx.doi.org/10.1016/j.bbmt.2005.01.005
- Rasmusson, I., Ringden, O., Sundberg, B. and Le Blanc, K. (2003) Mesenchymal Stem Cells Inhibit the Formation of Cytotoxic T Lymphocytes, but Not Activated Cytotoxic T Lymphocytes or Natural Killer Cells. Transplantation, 76, 1208-1213. http://dx.doi.org/10.1097/01.TP.0000082540.43730.80
- Maitra, B., Szekely, E., Gjini, K., et al. (2004) Human Mesenchymal Stem Cells Support Unrelated Donor Hematopoietic Stem Cells and Suppress T-Cell Activation. Bone Marrow Transplant, 33, 597-604. http://dx.doi.org/10.1038/sj.bmt.1704400
- Nauta, A.J., Westerhuis, G., Kruisselbrink, A.B., Lurvink, E.G., Willemze, R. and Fibbe, W.E. (2006) Donor-Derived Mesenchymal Stem Cells Are Immunogenic in an Allogeneic Host and Stimulate Donor Graft Rejection in a Nonmyeloablative Setting. Blood, 108, 2114-2120. http://dx.doi.org/10.1182/blood-2005-11-011650
- Plumas, J., Chaperot, L., Richard, M.J., Molens, J.P., Bensa, J.C. and Favrot, M.C. (2005) Mesenchymal Stem Cells Induce Apoptosis of Activated T Cells. Leukemia, 19, 1597-1604. http://dx.doi.org/10.1038/sj.leu.2403871
- De Miguel, M.P., Fuentes-Julian, S., Blazquez-Martinez, A., et al. (2012) Immunosuppressive Properties of Mesenchymal Stem Cells: Advances and Applications. Current Molecular Medicine, 12, 574-591. http://dx.doi.org/10.2174/156652412800619950
- Choi, S. and Reddy, P. (2010) Graft-versus-Host Disease. Panminerva Medica, 52, 111-124.
- Ferrara, J.L., Levine, J.E., Reddy, P. and Holler, E. (2009) Graft-versus-Host Disease. The Lancet, 373, 1550-1561. http://dx.doi.org/10.1016/S0140-6736(09)60237-3
- Pavletic, S.Z., Kumar, S., Mohty, M., et al. (2010) NCI First International Workshop on the Biology, Prevention, and Treatment of Relapse after Allogeneic Hematopoietic Stem Cell Transplantation: Report from the Committee on the Epidemiology and Natural History of Relapse Following Allogeneic Cell Transplantation. Biology of Blood and Marrow Transplantation, 16, 871-890. http://dx.doi.org/10.1016/j.bbmt.2010.04.004
- Bhatia, S., Davies, S.M., Baker, K.S., Pulsipher, M.A. and Hansen, J.A. (2011) NCI, NHLBI First International Consensus Conference on Late Effects after Pediatric Hematopoietic Cell Transplantation: Etiology and Pathogenesis of Late Effects after HCT Performed in Childhood—Methodologic Challenges. Biology of Blood and Marrow Transplantation, 17, 1428-1435. http://dx.doi.org/10.1016/j.bbmt.2011.07.005
- Storb, R., Gyurkocza, B., Storer, B.E., et al. (2013) Graft-versus-Host Disease and Graft-versus-Tumor Effects after Allogeneic Hematopoietic Cell Transplantation. Journal of Clinical Oncology, 31, 1530-1538. http://dx.doi.org/10.1200/JCO.2012.45.0247
- Jacobsohn, D.A. and Vogelsang, G.B. (2007) Acute Graft versus Host Disease. Orphanet Journal of Rare Diseases, 2, 35. http://dx.doi.org/10.1186/1750-1172-2-35
- Pavletic, S.Z., Carter, S.L., Kernan, N.A., et al. (2005) Influence of T-Cell Depletion on Chronic Graft-versus-Host Disease: Results of a Multicenter Randomized Trial in Unrelated Marrow Donor Transplantation. Blood, 106, 3308- 3313. http://dx.doi.org/10.1182/blood-2005-04-1614
- Ning, H., Yang, F., Jiang, M., et al. (2008) The Correlation between Cotransplantation of Mesenchymal Stem Cells and Higher Recurrence Rate in Hematologic Malignancy Patients: Outcome of a Pilot Clinical Study. Leukemia, 22, 593-599. http://dx.doi.org/10.1038/sj.leu.2405090
- Chester, S.J., Esparza, A.R. and Albala, M.M. (1975) Graft versus Leukemia without Fatal Graft-versus-Host Disease in AKR Mice. Cancer Research, 35, 637-639.
- Chester, S.J., Esparza, A.R., Flinton, L.J., Simon, J.D., Kelley, R.J. and Albala, M.M. (1977) Further Development of a Successful Protocol of Graft versus Leukemia without Fatal Graft-versus-Host Disease in AKR Mice. Cancer Research, 37, 3494-3496.
- Bacigalupo, A. (2007) Management of Acute Graft-versus-Host Disease. British Journal of Haematology, 137, 87-98. http://dx.doi.org/10.1111/j.1365-2141.2007.06533.x
- Li, Y., Qu, Y.H., Wu, Y.F., et al. (2011) Bone Marrow Mesenchymal Stem Cells Reduce the Antitumor Activity of Cytokine-Induced Killer/Natural Killer Cells in K562 NOD/SCID Mice. Annals of Hematology, 90, 873-885. http://dx.doi.org/10.1007/s00277-011-1156-9
- Tolar, J., Nauta, A.J., Osborn, M.J., et al. (2007) Sarcoma Derived from Cultured Mesenchymal Stem Cells. Stem Cells, 25, 371-379. http://dx.doi.org/10.1634/stemcells.2005-0620
- Sioud, M. (2011) New Insights into Mesenchymal Stromal Cell-Mediated T-Cell Suppression through Galectins. Scandinavian Journal of Immunology, 73, 79-84. http://dx.doi.org/10.1111/j.1365-3083.2010.02491.x
- Yi, T. and Song, S.U. (2012) Immunomodulatory Properties of Mesenchymal Stem Cells and Their Therapeutic Applications. Archives of Pharmacal Research, 35, 213-221. http://dx.doi.org/10.1007/s12272-012-0202-z
- Anthony, B.A. and Hadley, G.A. (2012) Induction of Graft-versus-Host Disease and in Vivo T Cell Monitoring Using an MHC-Matched Murine Model. Journal of Visualized Experiments, 29, Article ID: e3697.
- Fricke, S., Rothe, K., Hilger. N., et al. (2012) Allogeneic Bone Marrow Grafts with High Levels of CD4+CD25+ FoxP3+ T Cells Can Lead to Engraftment Failure. Cytometry A, 81A, 476-488. http://dx.doi.org/10.1002/cyto.a.22061
- Ali, N., Flutter, B., Sanchez Rodriguez, R., et al. (2012) Xenogeneic Graft-versus-Host-Disease in NOD-scid IL- 2Rgammanull Mice Display a T-Effector Memory Phenotype. PLoS ONE, 7, Article ID: e44219. http://dx.doi.org/10.1371/journal.pone.0044219
- Baron, F., Lechanteur, C., Willems, E., et al. (2010) Cotransplantation of Mesenchymal Stem Cells Might Prevent Death from Graft-versus-Host Disease (GVHD) without Abrogating Graft-versus-Tumor Effects after HLA-Mismatched Allogeneic Transplantation Following Nonmyeloablative Conditioning. Biology of Blood and Marrow Transplantation, 16, 838-847. http://dx.doi.org/10.1016/j.bbmt.2010.01.011
- Itakura, S., Asari, S., Rawson, J., et al. (2007) Mesenchymal Stem Cells Facilitate the Induction of Mixed Hematopoietic Chimerism and islet Allograft Tolerance without GVHD in the Rat. American Journal of Transplantation, 7, 336- 346. http://dx.doi.org/10.1111/j.1600-6143.2006.01643.x
- Dazzi, F. and Marelli-Berg, F.M. (2008) Mesenchymal Stem Cells for Graft-versus-Host Disease: Close Encounters with T Cells. European Journal of Immunology, 38, 1479-1482. http://dx.doi.org/10.1002/eji.200838433
- van Rijn, R.S., Simonetti, E.R., Hagenbeek, A., et al. (2003) A New Xenograft Model for Graft-versus-Host Disease by Intravenous Transfer of Human Peripheral Blood Mononuclear Cells in RAG2-/- Gammac-/- Double-Mutant Mice. Blood, 102, 2522-2531. http://dx.doi.org/10.1182/blood-2002-10-3241
- Eisenberg, R.A. and Via, C.S. (2012) T Cells, Murine Chronic Graft-versus-Host Disease and Autoimmunity. Journal of Autoimmunity, 39, 240-247. http://dx.doi.org/10.1016/j.jaut.2012.05.017
- Sato, K., Ozaki, K., Mori, M., Muroi, K. and Ozawa, K. (2010) Mesenchymal Stromal Cells for Graft-versus-Host Disease: Basic Aspects and Clinical Outcomes. Journal of Clinical and Experimental Hematopathology, 50, 79-89. http://dx.doi.org/10.3960/jslrt.50.79
- Young, J.S., Wu, T., Chen, Y., et al. (2012) Donor B Cells in Transplants Augment Clonal Expansion and Survival of Pathogenic CD4+ T Cells That Mediate Autoimmune-Like Chronic Graft-versus-Host Disease. The Journal of Immunology, 189, 222-233. http://dx.doi.org/10.4049/jimmunol.1200677
- Koenecke, C., Lee, C.W., Thamm, K., et al. (2012) IFN-Gamma Production by Allogeneic Foxp3+ Regulatory T Cells Is Essential for Preventing Experimental Graft-versus-Host Disease The Journal of Immunology, 189, 2890-2896. http://dx.doi.org/10.4049/jimmunol.1200413
- Tian, Y., Deng, Y.B., Huang, Y.J. and Wang, Y. (2008) Bone Marrow-Derived Mesenchymal Stem Cells Decrease Acute Graft-versus-Host Disease after Allogeneic Hematopoietic Stem Cells Transplantation. Immunological Investigations, 37, 29-42.
- Weng, J.Y., Du, X., Geng, S.X., et al. (2010) Mesenchymal Stem Cell as Salvage Treatment for Refractory Chronic GVHD. Bone Marrow Transplantation, 45, 1732-1740. http://dx.doi.org/10.1038/bmt.2010.195
- Kuo, Y.R., Goto, S., Shih, H.S., et al. (2009) Mesenchymal Stem Cells Prolong Composite Tissue Allotransplant Survival in a Swine Model. Transplantation, 87, 1769-1777. http://dx.doi.org/10.1097/TP.0b013e3181a664f1
- Joo, S.Y., Cho, K.A., Jung, Y.J., et al. (2010) Mesenchymal Stromal Cells Inhibit Graft-versus-Host Disease of Mice in a Dose-Dependent Manner. Cytotherapy, 12, 361-370. http://dx.doi.org/10.3109/14653240903502712
- Tolar, J., Villeneuve, P. and Keating, A. (2011) Mesenchymal Stromal Cells for Graft-versus-Host Disease. Human Gene Therapy, 22, 257-262. http://dx.doi.org/10.1089/hum.2011.1104
- Kim, S., Shin, J., Kim, Y. and Kim, C. (2011) Exosomes from Mouse Bone Marrow-Derived Mesenchymal Stem Cells (mMSCs) Mediate a Potent Immunosuppressive Function. Journal of Immunology, 186, 15.
- Lai, R.C., Chen, T.S. and Lim, S.K. (2011) Mesenchymal Stem Cell Exosome: A Novel Stem Cell-Based Therapy for Cardiovascular Disease. Regenerative Medicine, 6, 481-492. http://dx.doi.org/10.2217/rme.11.35
- Connick, P., Kolappan, M., Crawley, C., et al. (2012) Autologous Mesenchymal Stem Cells for the Treatment of Secondary Progressive Multiple Sclerosis: An Open-Label Phase 2a Proof-of-Concept Study. The Lancet Neurology, 11, 150-156. http://dx.doi.org/10.1016/S1474-4422(11)70305-2
- Ringdén, O., Uzunel, M., Sundberg, B., et al. (2007) Tissue Repair Using Allogeneic Mesenchymal Stem Cells for Hemorrhagic Cystitis, Pneumomediastinum and Perforated Colon. Leukemia, 21, 2271-2276. http://dx.doi.org/10.1038/sj.leu.2404833
- Youd, M., Blickarz, C., Woodworth, L., et al. (2010) Allogeneic Mesenchymal Stem Cells Do Not Protect NZBxNZW F1 Mice from Developing Lupus Disease. Clinical & Experimental Immunology, 161, 176-186.
- Yañz, R., Lamana, M.L., Garcia-Castro, J., Colmenero, I., Ramirez, M. and Bueren, J.A. (2006) Adipose Tissue-Derived Mesenchymal Stem Cells Have in Vivo Immunosuppressive Properties Applicable for the Control of the Graftversus-Host disease. Stem Cells, 24, 2582-2591. http://dx.doi.org/10.1634/stemcells.2006-0228
- Badillo, A.T., Peranteau, W.H., Heaton, T.E., Quinn, C. and Flake, A.W. (2008) Murine Bone Marrow Derived Stromal Progenitor Cells Fail to Prevent or Treat Acute Graft-versus-Host Disease. British Journal of Haematology, 141, 224-234. http://dx.doi.org/10.1111/j.1365-2141.2008.07040.x
- Zinöcker, S., Wang, M.Y., Rolstad, B. and Vaage, J.T. (2012) Mesenchymal Stromal Cells Fail to Alleviate Experimental Graft-versus-Host Disease in Rats Transplanted with Major Histocompatibility Complex-Mismatched Bone Marrow. Scandinavian Journal of Immunology, 76, 464-470. http://dx.doi.org/10.1111/j.1365-3083.2012.02758.x
- Mielcarek, M., Storb, R., Georges, G.E., et al. (2011) Mesenchymal Stromal Cells Fail to Prevent Acute Graft-versusHost Disease and Graft Rejection after Dog Leukocyte Antigen-Haploidentical Bone Marrow Transplantation. Biology of Blood and Marrow Transplantation, 17, 214-225. http://dx.doi.org/10.1016/j.bbmt.2010.08.015
- Kitazawa, Y., Li, X.K., Xie, L., Zhu, P., Kimura, H. and Takahara, S. (2012) Bone Marrow-Derived Conventional, but Not Cloned, Mesenchymal Stem Cells Suppress Lymphocyte Proliferation and Prevent Graft-versus-Host Disease in Rats. Cell Transplantation, 21, 581-590.
- Peister, A., Mellad, J.A., Larson, B.L., Hall, B.M., Gibson, L.F. and Prockop, D.J. (2004) Adult Stem Cells from Bone Marrow (MSCs) Isolated from Different Strains of Inbred Mice Vary in Surface Epitopes, Rates of Proliferation, and Differentiation Potential. Blood, 103, 1662-1668. http://dx.doi.org/10.1182/blood-2003-09-3070
- Solchaga, L.A., Johnstone, B., Yoo, J.U., Goldberg, V.M. and Caplan, A.I. (1999) High Variability in Rabbit Bone Marrow-Derived Mesenchymal Cell Preparations. Cell Transplantation, 8, 511-519.
- Ho, A.D., Wagner, W. and Franke, W. (2008) Heterogeneity of Mesenchymal Stromal Cell Preparations. Cytotherapy, 10, 320-330. http://dx.doi.org/10.1080/14653240802217011
- Truitt, R.L., Shih, C.C. and LeFever, A.V. (1986) Manipulation of Graft-versus-Host Disease for a Graft-versus-Leukemia Effect after Allogeneic Bone Marrow Transplantation in AKR Mice with Spontaneous Leukemia/Lymphoma. Transplantation, 41, 301-310. http://dx.doi.org/10.1097/00007890-198603000-00005
- Aldred, A.J., Cha, M.C. and Meckling-Gill, K.A. (2002) Determination of a Humane Endpoint in the L1210 Model of Murine Leukemia. Contemporary Topics in Laboratory Animal Science, 41, 24-27.
- Heidt, P.J. and Vossen, J.M. (1992) Experimental and Clinical Gnotobiotics: Influence of the Microflora on Graftversus-Host Disease after Allogeneic Bone Marrow Transplantation. Journal of Medicine, 23, 161-173.
- Calcaterra, C., Sfondrini, L., Rossini, A., et al. (2008) Critical Role of TLR9 in Acute Graft-versus-Host Disease. The Journal of Immunology, 181, 6132-6139.
- Zhao, Y., Liu, Q., Yang, L., et al. (2013) TLR4 Inactivation Protects from graft-versus-Host Disease after Allogeneic Hematopoietic Stem Cell Transplantation. Cellular & Molecular Immunology, 10, 165-175. http://dx.doi.org/10.1038/cmi.2012.58
- Djouad, F., Fritz, V., Apparailly, F., et al. (2005) Reversal of the Immunosuppressive Properties of Mesenchymal Stem Cells by Tumor Necrosis Factor Alpha in Collagen-Induced Arthritis. Arthritis & Rheumatism, 52, 1595-1603. http://dx.doi.org/10.1002/art.21012
- Phinney, D.G. (2012) Functional Heterogeneity of Mesenchymal Stem Cells: Implications for Cell Therapy. Journal of Cellular Biochemistry, 113, 2806-2812. http://dx.doi.org/10.1002/jcb.24166
- Fanning, S.L., Zilberberg, J., Stein, J., et al. (2013) Unraveling Graft-versus-Host Disease and Graft-versus-Leukemia Responses Using TCR Vbeta Spectratype Analysis in a Murine Bone Marrow Transplantation Model. The Journal of Immunology, 190, 447-457. http://dx.doi.org/10.4049/jimmunol.1201641
- Hatano, R., Ohnuma, K., Yamamoto, J., Dang, N.H., Yamada, T. and Morimoto, C. (2013) Prevention of Acute Graftversus-Host Disease by Humanized Anti-CD26 Monoclonal Antibody. British Journal of Haematology, 162, 263-277. http://dx.doi.org/10.1111/bjh.12378
- Yolcu, E.S., Kaminitz, A., Mizrahi, K., et al. (2013) Immunomodulation with Donor Regulatory T Cells Armed with Fas-Ligand Alleviates Graft-versus-Host Disease. Experimental Hematology, 41, 903-911. http://dx.doi.org/10.1016/j.exphem.2013.04.016
NOTES

*Corresponding author.