Stem Cell Discovery
Vol. 2 No. 1 (2012) , Article ID: 16681 , 10 pages DOI:10.4236/scd.2012.21002
Characterization of a mesenchymal stem cell line that differentiates to bone and provides niches supporting mouse and human hematopoietic stem cells
![]()
1Departments of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, USA
2Pathology and Microbiology, University of Nebraska Medical Center, Omaha, USA
3Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, USA
4Creighton University Medical Center, Omaha, USA; *Corresponding Author: jsharp@unmc.edu
Received 7 October 2011; revised 10 November 2011; accepted 21 November 2011
Keywords: Bone; Cell Therapy; Hematopoiesis; MSC; Regenerative Medicine; Stem Cells
ABSTRACT
Identification of mouse cell lines with properties of primary multipotential mesenchymal stromal cells (MSC) is required to facilitate the use of mouse models for evaluation of mechanisms in bone formation, hematopoiesis and cellular therapies for regenerative medicine. Primary murine MSC vary between strains, are difficult to grow in vitro and have inconsistent properties. The main aim of the study was to establish OMAAD cells as an appropriate model system to conduct studies on MSC, bone formation and hematopoiesis. OMA-AD cells were isolated by differential trypsinization of C57BL/6J mouse bone marrow (BM) cells. The cells were then repassaged, cloned and characterized. OMA-AD cells were immortal and non-tumorigenic, differentiated readily to all mesenchymal cell types including bone, supported mouse and human hematopoiesis and were immunosuppressive. Our results demonstrated that OMA-AD cells possessed the properties of primary MSC. In addition, these cells grew readily and consistently, thereby facilitating future studies of bone formation, hematopoiesis and mesenchymal cells for regenerative medicine.
1. INTRODUCTION
In recent years there has been increased interest in mesenchymal cells, their differentiation to cartilage and bone, their role in stem cell niches [1] and potential utility in regenerative medicine. Although, originally MSC were used therapeutically for skeletal problems [2] and gene therapy [3,4] multiple additional therapeutic applications have been evaluated, including promotion of hematopoietic recovery in stem cell transplant recipients [5,6] severe, acute graft-versus-host disease [7], autoimmune diseases [8], muscle repair [9], skin healing [10], intestine healing following irradiation [11], stroke [12], myocardial ischemia [13,14] as well as other diseases.
MSC were first identified in the bone marrow, where they are present with about ten-fold higher concentrations than the circulation and can be obtained from all tissues with varying frequencies [15,16]. In man, the number of viable, freshly isolated cells is limited due to dispensable bone marrow volume [17].
Consequently, animal studies are a powerful tool for investigations of diverse potential applications for MSC. Apart from the increasing applications of the functional properties of MSC, there is substantial interest in evaluation of the role of these cells in the stem cell niche and mouse models of diseases [18]. The numbers of MSC obtained in murine models are adequate and can be maneuvered to differentiate into several cell types. However, primary MSC from murine sources do not grow readily in culture, and exhibit considerable variability in their biological properties from different strains of mice [19]. Consequently, there is substantial merit in identifying mouse cell populations that grow consistently and at the same time exhibit characteristics of primary mouse MSC.
OMA-AD cells were obtained by differential trypsinization of whole bone marrow cells from C57BL/6J mice. The cells were passaged several times and only rare adherent cells that survived passaging were cloned and characterized further. Briefly, the cells were probed for their doubling time, phenotype and differentiation into various lineages. OMA-AD cells were seeded as the adherent layer in long-term cultures with BM cells and added to mixed lymphocyte reactions. In addition, OMAAD cells were injected into non-irradiated and lethally irradiated recipients. A preosteoblast cell line with apparently similar properties to OMA-AD cells has been described (D1 cells) and employed in orthopedic studies [20]. However, the D1 cell line is of Balb/c origin, whereas the OMA-AD cell line is of C57BL/6J origin therefore genetically compatible with most transgenic mouse models. OMA-AD cells have been employed and reported previously [21]. However, the isolation, derivation, differentiation to cartilage and bone, support of mouse and human hematopoiesis and overall properties of these cells have not been described previously and is the primary aim of this report.
2. MATERIALS AND METHODS
2.1. Isolation of OMA-AD Cell Line
C57BL/6J and C57BL/6J-EGFP (enhanced green fluorescent protein) expressing mice were purchased from Jackson Laboratories, or bred in the University of Nebraska Medical Center (UNMC) comparative medicine department. These mice were used in the studies as approved by the UNMC Institutional Animal Care and Use Committee (IACUC). The mice were housed in individual microisolator cages and provided food and water ad libitum. Bone marrow cells were gently aspirated from the femoral diaphysis and maintained in Fisher’s medium supplemented with 15% fetal bovine serum (FBS; Hyclone) and 15% horse serum (HS; Hyclone) The cultures were maintained at 33˚C in a mixture of 5% CO2 and air, as described previously for long term bone marrow cultures [22].
2.2. Cell Line Development
In order to obtain cell lines, the supernatants were discarded and the remaining cells were trypsinized repeatedly until only rare adherent cells remained. Fisher’s medium with 15% FBS and 15% HS was added and surviving cells were maintained in culture. The cells were observed for areas (colonies) of outgrowths of cells. Most of the cells in the majority of flasks differentiated and died. However, rare outgrowths were observed. These were trypsinized and re-passaged. The majority of re-passages failed to grow, but a few cells survived passaging. These cells were cloned in microtiter plates and progeny frozen for future studies. One of these clones, with the most robust and consistent patterns of growth, was designated as OMA-AD cell line and the properties of this cell line were characterized in more detail.
2.3. Morphology and Cell Growth Analysis
OMA-AD cells were plated in growth supporting medium [RPMI-1640 (Gibco Inc.), 10% FBS, 10% HS, 1% Penicillin-Streptomycin (Gibco Inc.), 1% L-glutamine (Gibco Inc.)] in T25 cm2 flasks (Corning Inc.) with a concentration of 1 × 105 cells or 4000 cells/cm2. For morphological analyses, the flasks were monitored for confluence and photographed using a Nikon phase contrast microscope and Nikon Coolpix P5000 10 MegaPixel camera. For doubling time analyses, at each defined time point, 3 flasks were trypsinized and cell counts were obtained using a cell counter (Beckman Coulter Inc.) The mean of the data points obtained were plotted (Sigmaplot Statistical Software 7.0) Doubling time was calculated from equation dt = t * ln2/ln(Ct/Co), where t = the time point in exponential growth of the plot, Ct = cell count at time “t”, Co = cell count when plated or initial time point on the exponential part of the growth curve.
2.4. Surface Phenotype
1 × 106 OMA-AD cells were placed into 12 × 75 mm test tubes, centrifuged for 2 minutes and resuspended in 200 μl staining buffer (Phospaphate Buffered Saline (PBS) with 0.2% Bovine Serum Albumin (BSA) 2 μl (0.4 µg/ml concentration) of antibody was added to cells and vortexed. A 20-minute incubation was performed on ice in the dark. The mixture was washed with 1 ml of staining buffer and centrifuged for 2 minutes using an immufuge (Baxter Corp). The cells were resuspended in 0.5 ml of staining buffer and 50 µl Vitalyse® fixative prior to analysis. Antibodies against mouse CD 3, 4, 8, 11b, 14, 19, 24, 25, 31, 34, 40, 44, 45, 48, 49d, 73, 80, 105, 133, 150, e-cadherin, SDF-1, Oct 3/4, B220, Mac-1, Gr-1, CXCR4, Thy 1.1 and 1.2, were purchased from BD Biosciences (San Jose, CA). Flow cytometric analysis was performed and analyzed using Cell Quest Pro (BD Biosciences) software.
2.5. Differentiation to Adipocytes, Osteoblasts and Chondrocytes
OMA-AD cells were seeded onto 3 - 6 coverslips in a 60 mm petridish with growth supporting medium (RPMI 1640 medium + 10% FBS + 10% HS + 1% Penicillin-Streptomycin + 1% L-glutamine) Upon 60% - 70% confluence, adipocyte differentiation medium (ADM) (DMEM with 10% FBS, dexamethasone 1 µM, isobutylmethylxanthine 0.5 µM, Insulin 1 µg/ml, indomethacin 100 mM) was added (day1) and changed every 2 days up to 2 weeks. The coverslips were mounted on slides using Permount solution on day 7. Adipogenic differentiation was monitored as lipid droplets formed inside the cells and as red droplets after Oil Red O staining at day 7. Osteoblast stimulating medium (OSM) (DMEM with 10% FBS, dexamethasone 100nM, beta-glycerophosphate 10 mM, L-ascorbic-2-phosphate 50 nM, penstrep 1%) was added upon 60% confluence (day1) 0.5 ml medium was added to the culture every 3rd day. After 12 days, coverslips were mounted on a slide and stained for alkaline phosphatase using a kit (Sigma-Aldrich) to identify the osteoblasts. Chondrocyte differentiation was carried out as per the protocol provided by Miltenyi Biotech. Briefly, 1 × 106 OMA-AD cells were centrifuged in a 15 ml conical polypropylene tube and formed into a compact pellet. Chondrocyte differentiation media (Miltenyi Biotech) was added on top of the pellet. The tubes were incubated at 37˚C, 5% CO2. Media was changed every other day for 24 days. The pellets were fixed in 3.7% neutral buffered formalin and subsequently embedded in paraffin. Chondrocytic differentiation was confirmed using Alcian blue-Periodic Schiff Staining. OMA-AD cells maintained undifferentiated in the growth supporting medium, were employed as controls and were confirmed by staining negative for Oil Red O, alkaline phosphatase and Alcian blue-PAS staining. All the slides were counterstained with wright giemsa stain (Fisher Scientific).
2.6. Support of Murine Hematopoiesis
The ability of OMA-AD cells to support mouse hematopoietic cells was evaluated by using OMA-AD cells as an adherent layer for long term bone marrow cultures. OMA-AD cells were seeded at 1 × 106 cells in a T25 culture flask (Greiner Biosciences). Upon 60% confluence, the flasks were seeded with whole bone marrow cells or bone marrow sorted side population (SP) cells from EGFP donors (EGFP expressing on background of C57BL/6J mice) so that any hematopoietic cells maintained and/or generated could be identified by fluorescence microscopy or by flow cytometric analyses. Cultures were maintained at 33˚C for 6 months with intermittent change of culture medium.
2.7. Support of Human Hematopoiesis
The ability of OMA-AD cells to maintain human hematopoiesis was determined by evaluating their ability to sustain human hematopoietic cell production and granulocyte-monocyte progenitor cells (GM-CFC) as assayed in a clonal colony forming cell assay. Human cell sources evaluated were monocyte depleted cord blood, T cell depleted cord blood and granulocyte colony stimulating factor (G-CSF) mobilized blood cells. The comparison adherent cells were grown with or without a pre-established, irradiated mouse bone marrow stromal layer. The cord blood cells were obtained from full term deliveries with IRB (Institutional Review Board) approval and informed consent of the donor. The cord blood samples were depleted of monocytes by employing 2 one hour adhesion incubations in culture dishes. After the first adhesion period, the non-adherent cells were removed and placed in the second culture dish. After the second adhesion period, the non-adherent cells were isolated and used in the experiments. T-cells were depleted employing magnetic bead separation. G-CSF mobilized blood depleted of T cells by anti-CD3 antibody were obtained with IRB approval and informed consent of a donor who received five consecutive daily subcutaneous injections of G-CSF (5 µg/kg) (Amgen Inc). The non-adherent cellularities of these co-cultures were determined weekly using an electronic cell counter. The GM-CFC assay was performed, as described previously [23], every two weeks, on non-adherent cells and, at the termination of the cocultures, on both the non-adherent and adherent cells.
2.8. Precursor Frequency Determination
For the G-CSF mobilized blood cells and human leukemic cell line; OMA-AML1 cells [24], a limiting dilution of cobblestone area formation was employed to determine precursor frequencies.
2.9. Radioprotection
One of two doses of total body irradiation were administered to C57BL/6J EGFP (to track the origin of cells) mice; a lethal dose (10 Gy) or a sub-lethal dose (7.5 Gy). Mice receiving the lethal dose (n = 7) and sub-lethal dose (n = 7) were injected with 1 × 106 OMAAD cells in 200 µl Hank’s Balance Salt Solution (HBSS; Gibco Inc). Controls (mice receiving the sublethal dose) were not injected with OMA-AD cells (n = 7). Circulating blood cell numbers, GFP expression and survival were tracked.
2.10. Tumorigenicity
1 × 105 OMA-AD cells were injected in 200 µl HBSS, subcutaneously (n = 3) and intraperitonealy (n = 3) subcutaneously in the flank (Ip and SQ are switched) of syngeneic C57BL/6J mice. The mice were examined twice weekly for 120 days and the site of injection palpated to detect any tumor growth. Any growths were examined histologically.
2.11. Immunosuppression
Spleen cells were obtained from DBA and C57BL/6J (referred to as B6) mice where DBA spleen cells were stimulators and B6 spleen cells were responders. 1 × 105 cells of each type were added to each well in a 96-well flat bottom plate (Corning Inc). OMA-AD cells were titrated into this assay which employed allogeneic mixtures of mouse spleen cells to generate a mixed lymphocyte reaction (MLR). Samples were run in quadruplicate. Spleen cells from DBA mice, appropriate numbers of spleen cells from B6 mice and OMA-AD cells were irradiated at 20 Gy. Irradiated DBA spleen cells along with non-irradiated B6 cells comprised the “experimental” group. Irradiated B6 cells along with non-irradiated B6 cells were the “control” group. Irradiated OMA-AD cells were plated 24 hrs prior to addition of other cell types to ensure adherence. The wells with OMA-AD cells along with irradiated DBA and non-irradiated B6 cells were termed “Experimental + OMA-AD” or control + OMAAD (irradiated B6 + non-irradiated B6 + OMA-AD). Cells were grown in RF 10 (RPMI-1640; Gibco, Fetal Bovine Serum 10%; Hyclone, Penicillin/Streptomycin; Gibco) for 4 days. The cells were pulsed at day 3 and day 4 with tritiated thymidine (1 microcurie/well) to measure cell proliferation. Upon 18 - 24 hrs incubation, cells were harvested using cell harvester (PHD cell harvester) and radioactivity determined using a liquid scintillation counter (Perkin Elmer, Shelton, CT). Stimulation of immune response or inhibition of stimulation was determined by measuring cell proliferation. Experimental groups were plotted against cell proliferation (counts/ minute) using SigmaPlot (7.0) software. Three sets of independent experiments were conducted and the data obtained was averaged and plotted in Figure 5.
2.12. Statistics
The statistical differences amongst groups were analyzed using unpaired Student’s t-test. Significance was set at a p-value of less than 0.05.
3. RESULTS
3.1. Morphology and Doubling Time
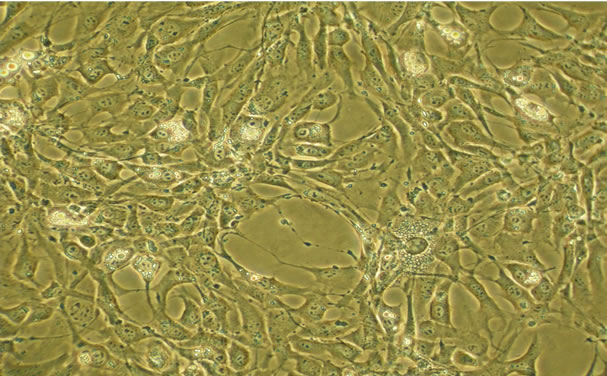
OMA-AD cells were isolated from a long-term (Dexter) bone marrow cultures by differential trypsinization and limiting dilution cloning. Despite originating from a single cell, cultured unmanipulated OMA-AD cells exhibited morphological heterogeneity (Figure 1(a)) with a doubling time of approximate 35 hrs (Figure 1(b)).
3.2. Phenotype
The phenotype of unmanipulated OMA-AD cells demonstrated a minor representation of several cell types including mesenchymal, endothelial and even differentiated hematopoietic cells (Table 1) but a predominant phenotypic marker was not identified and markers previously noted for MSC (CD 80, CD 90, CD 105) were not expressed at significant levels. Whether these data indicate that OMA-AD cells represent a primitive, entirely undifferentiated multipotential progenitor cell with a capacity to differentiate along various lineages or reflects epigenetically generated variations in progeny of a single cloned precursor cell is unclear. Note that these options are not necessarily exclusive.
3.3. Differentiation into Multiple Lineages
The primary characteristic of a bonafide MSC is their
 (a)
(a) (b)
(b)
Figure 1. In vitro characterization of OMA-AD cells. (a) OMAAD cells: Undifferentiated cells, display heterogeneity (Nikon Coolpix P5000, 20× Magnification); (b) Doubling time plot. OMA-AD cells had a doubling time of approximately 35 hours.

Table 1. Phenotypic characterization of OMA-AD cells. Based on the number of events (% positive OMA-AD cells), markers were segregated into biologically likely (>5% gated) and unlikely (<5% gated). The markers that were expressed <1% were considered as background. OMA-AD cells displayed heterogeneity in cell surface marker expression.
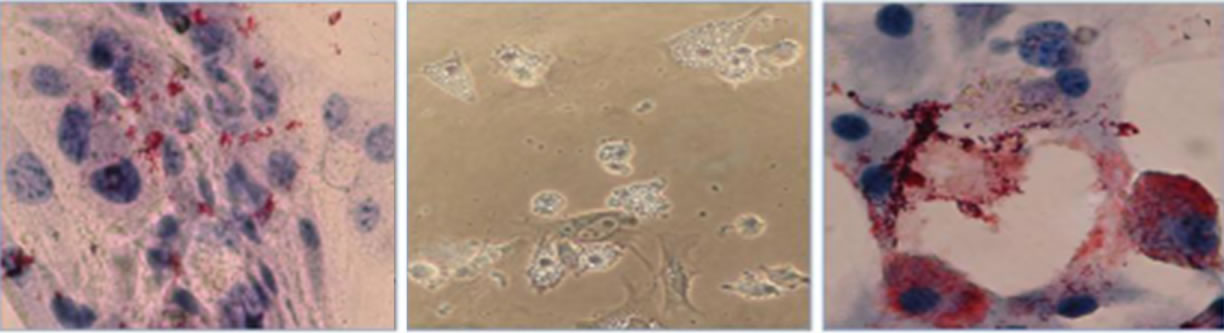
ability to differentiate into multiple mesenchymal lineages. OMA-AD cells were subjected to various differenttiation conditions to examine their ability to differentiate into adipocytic, osteoblastic and chondrocytic lineages. Upon incubation with ADM for 7 days, OMA-AD cells differentiated into adipocytes, with oil droplet accumulation observed at day 7 and confirmed by Oil Red O staining (Figures 2(b) and (c)) OMA-AD cells were incubated with OSM for 20 days, alkaline phosphatase staining at 11 days showed presence of osteoblasts, which was confirmed by calcium deposition detected by von Kassa staining at day 20 (Figures 2(e) and (f)) indicating an ability to form bone. OMA-AD cells were incubated for 24 days in chondrocyte differentiation medium and stained with Alcian blue-PAS to detect presence of chondrocytes (Figure 2(h)) OMA-AD cells maintained undifferentiated were confirmed by staining negative for Oil Red O, alkaline phosphatase and Alcian blue-PAS (Figures 2 (a), (d) and (g)).
 (a)
(a)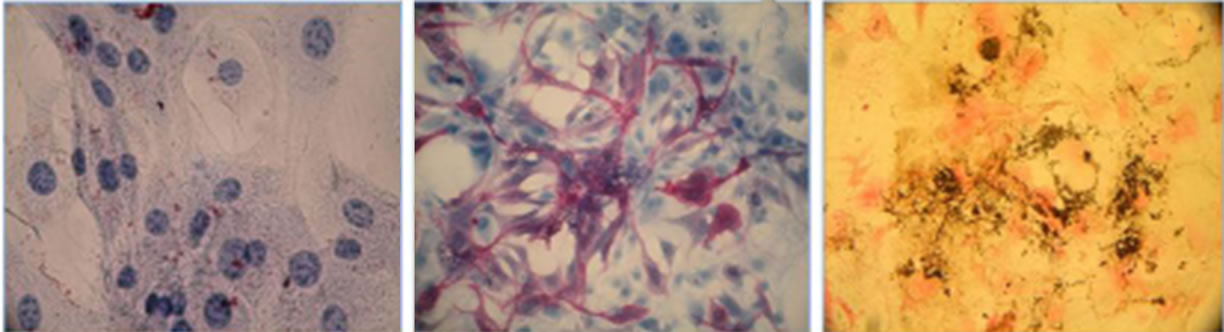 (b)
(b)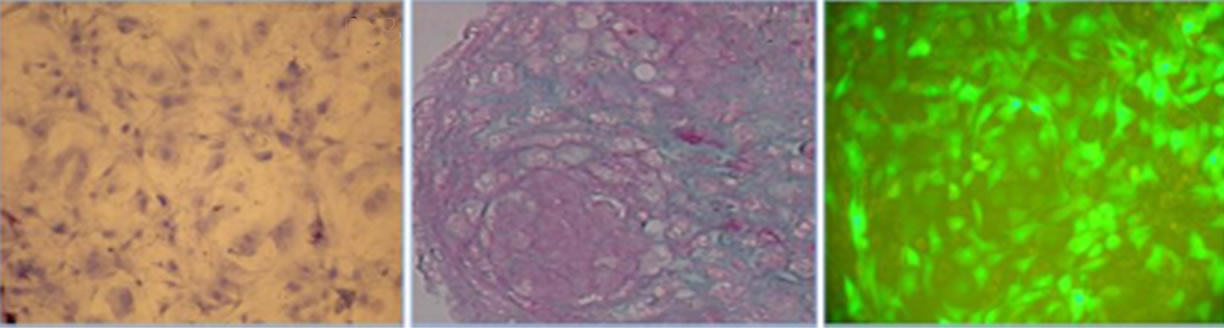 (c)
(c)
Figure 2. Differentiation and Hematopoietic Support. (a) Differentiation into adipocytes: Undifferentiated OMA-AD cells stain negative for Oil Red O (40× magnification); (b) Intracellular lipid accumulation observed adipocytes at day 7 (20× magnification); (c) Oil Red O positive adipocytes at day 7 (40× magnification); (d) Differentiation into Osteoblasts: Undifferentiated OMA-AD cells stain negative for alkaline phosphatase (20× magnification); (e) On day 11, OMA-AD cells differentiated into alkaline phosphatase positive osteoblasts (20× magnification); (f) Calcium deposition confirmed by von Kassa staining at day 20 (20× magnification); (g) Differentiation into chondroblasts: OMA-AD cells stained negative for Alcian blue-PAS staining (10× magnification); (h) OMA-AD cells differentiated to chondrocytes, Alcian blue-PAS staining (on day 24, 10× magnification); (i) OMA-AD cells support murine hematopoiesis in vitro. Bone marrow from GFP + C57BL/6J mouse were seeded upon adherent layer of OMA-AD cells. The cultures were photographed at 11 weeks (10× magnification). All photographs were taken using a Nikon microscope and Nikon Coolpix P5000 10 Megapixel camera.
3.4. Hematopoietic Support
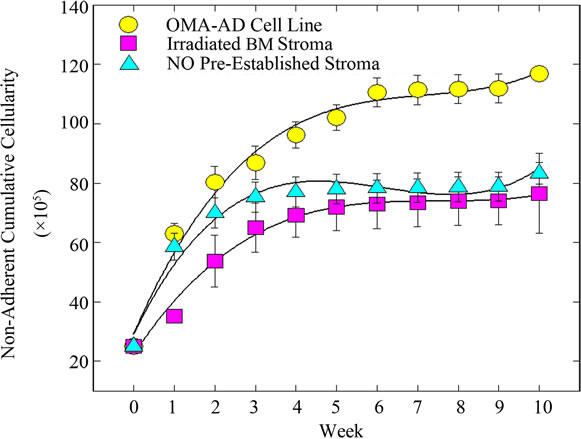
OMA-AD cells promoted survival of mouse hematopoietic stem cells and their differentiation for 3 months (as demonstrated in Figure 2(i)) OMA-AD cells provided stromal support to human hematopoietic cells compared to ones grown without stroma or irradiated BM stroma (Figure 3(a)) (p < 0.05). Similarly, the numbers of granulocytes and macrophages were significantly higher on OMA-AD supported stroma compared to ones grown without a stromal layer (Figure 3(b)) (p < 0.05) and pre-irradiated stroma (did not reach statistical significance) OMA-AD cells, grown as an adherent layer supported GM-CSF mobilized peripheral blood cells and human leukemic hematopoietic cells in culture (Figures 3(c) and (d)) OMA-AD cells supported human CFU-GM from T-cell and monocyte depleted cord blood cells (Tables 2 (a) and (b)) These results demonstrated that OMA-AD cells promoted survival of quiescent mouse pluripotent hematopoietic stem cells, normal and human leukemic cells and permitted determination of hematopoietic precursor frequencies thereby establishing their ability to provide niches for mouse and human stem cells.
3.5. Radioprotection
In preliminary experiments, lethally irradiated mice that received 106 OMA-AD cells intravenously survived about 10 days in apparent good health, then succumbed. Since there is no exclusive marker on OMA-AD cells, mice expressing the green fluorescent protein, i.e. GFP mice, were employed as the recipients. C57BL/6J mice were irradiated with a lethal (10 Gy) and sub lethal (7.5 Gy) doses of irradiation and subsequently injected intravenously with 1 × 106 OMA-AD cells. Control mice received sub lethal dose of irradiation (7.5 Gy) and were
 (a)
(a)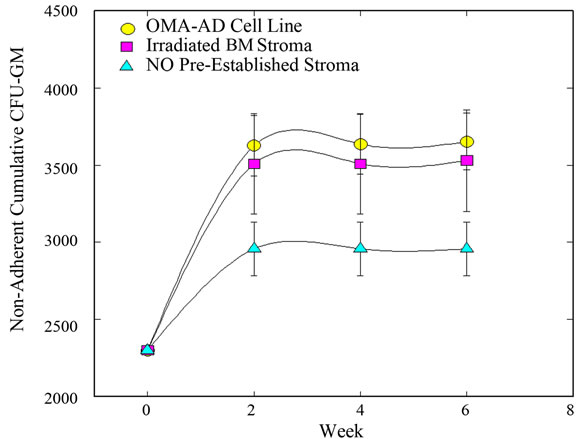 (b)
(b)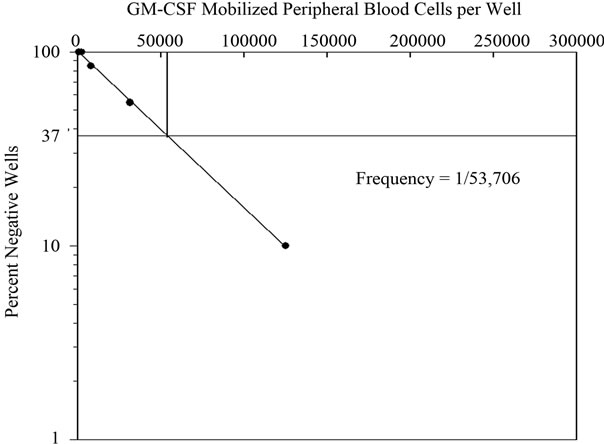 (c)
(c)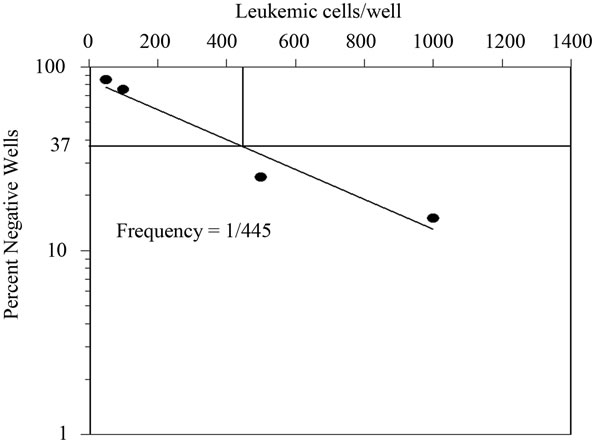 (d)
(d)
Figure 3. OMA-AD long-term support of human hematopoietic cells and long-term culture initiating cells (LTC-IC). (a) Hematopoietic cells grow better on an OMA-AD adherent layer than irradiated BM stroma (p < 0.05) or without stroma (p < 0.05); (b) Hematopoietic colony formation is improved in the presence of an OMA-AD adherent layer compared to controls i.e. without stroma (p < 0.05) and irradiated BM stroma (did not reach statistical significance); (c) Limiting dilution analysis of cobblestone area formation by GM-CSF mobilized blood cells with OMA-AD cells providing stromal support; (d) Limiting dilution analysis of cobblestone area formation by leukemic cells on OMA-AD cells as adherent layer.
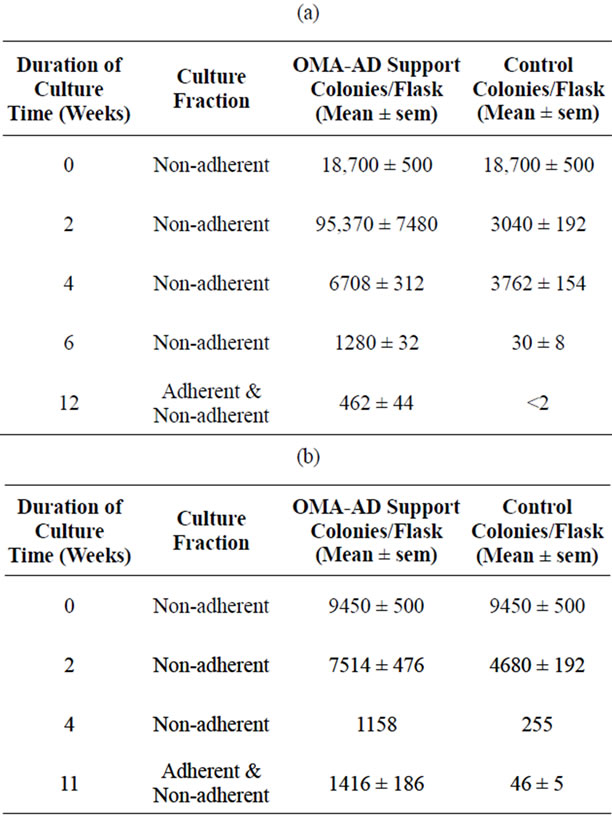
Table 2. OMA-AD cells supported human long-term hematopoiesis in vitro. (a) OMA-AD supported human CFU-GM from monocyte depleted cord blood. (b) OMA-AD supported CFUGM from T-Cell depleted cord blood. CFU-GM were maintained for a longer time in the presence of an adherent layer formed by OMA-AD cells compared to controls (p < 0.05) OMAAD long-term support of human granulopoiesis did not require the presence of human T lymphocytes or monocytes. Controls were grown in the absence of OMA-AD as adherent layer.
not injected with OMA-AD cells. Despite cell depletion, all mice injected with OMA-AD cells maintained 80% - 90% circulating cells expressing GFP indicating that OMA-AD did not contribute significantly to the circulating cell pool. All lethally irradiated mice died 11 days after irradiation demonstrating that OMA-AD cells were unable to rescue hematopoiesis (Figure 4). However, mice receiving sub lethal irradiation and OMA-AD injection, survived several months post irradiation (Figure 4) up to 125 days post irradiation but did not have evidence of progeny derived from OMA-AD cells (data not shown).
3.6. Tumorigenicity
No tumors were formed by OMA-AD cells injected subcutaneously or intraperitoneally into 6 syngeneic mice after monitoring them for over 120 days (data not shown). The non-tumorigenic properties of OMA-AD are vital for their use in homing studies as well as regenerative medicine.
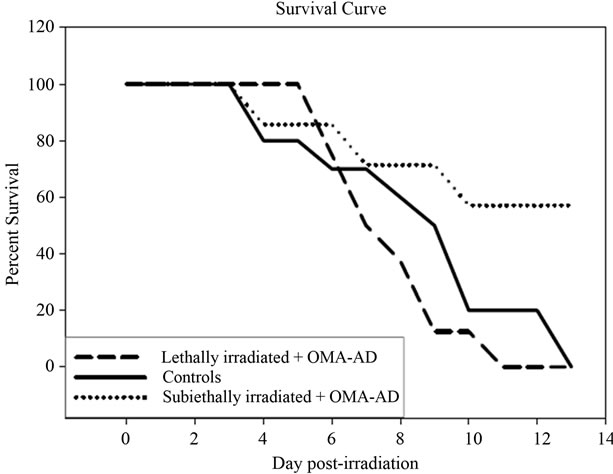
Figure 4. Injection of OMA-AD cells into lethally irradiated recipients for radioprotection. C57BL/6J mice were irradiated with lethal dose (10 Gy) or sub lethal dose (7.5 Gy) and subsequently injected with 1 × 106 OMA-AD cells. Control group received sub lethal irradiation but no OMA-AD injection. All mice receiving lethal dose died by 11 days after irradiation demonstrating that OMA-AD did not rescue hematopoiesis. Mice receiving sub lethal dose had about 50% survival by 13 days, with all mice dead by 125 days post irradiation (not shown).
3.7. Immunosuppression
Irradiated DBA spleen cells stimulated an immune response from non-irradiated B6 cells (experimental group) greater than that in the control group, p < 0.05 (irradiated B6 cells with non-irradiated B6 cells) (Figure 5). When OMA-AD cells were added to the experimental group (irradiated DBA with non-irradiated B6); termed “Experimental + OMA-AD”, there was inhibition of immune response compared to the experimental group alone (p < 0.05). Similarly, control + OMA-AD (irradiated B6 + non-irradiated B6 + OMA-AD) suppressed the immune response even further (p < 0.05). These data clearly show that OMA-AD cells were immunosuppressive in a mixed lymphocytic reaction (Figure 5) making them ideal for in vivo studies.
4. DISCUSSION
OMA-AD cells represent a spontaneously immortalized mouse MSC line. OMA-AD cells are capable, under appropriate inductive conditions, of generating all of the primary differentiated cell lineages of mesenchymal (connective) tissue lineages: chondroblasts, osteoblasts and adipocytes. Further, OMA-AD cells not only maintained mouse quiescent pluripotent hematopoietic stem cells in vitro [21] but also supported human normal and leukemic cells in culture [24]. In addition to mimicking the properties of primary mouse MSC, this cell population is non-tumorigenic, even though it is immunosup pressive. This is an important component of their function

Figure 5. Suppression of Mixed Lymphocyte Reactions (MLR). Spleen cells were obtained from DBA and B6 mice where DBA spleen cells were stimulators and B6 spleen cells were responders. Cell proliferation was measured by thymidine uptake. The presence of OMA-AD cells significantly suppressed the MLR as shown by decreased cell proliferation (p < 0.05).
since they are able to ameliorate the consequences of graft versus host disease (GVHD) (data not shown). Although cloned and subsequently re-cloned, OMA-AD cells continued to generate heterogeneous progeny in culture with a doubling time of approximately 35 hours. This is characteristic of MSC and is comparable to primary rat MSC described by Ho et al. 2008 [25]. Since OMA-AD cells are clonally derived, generation of such progeny indicates heterogeneity of their differentiation abilities, rather than origins from a heterogeneous collection of progenitor cells. Under appropriate culture conditions, OMA-AD cells were able to differentiate into adipocytes, osteoblasts and chondrocytes. However, when cultured, OMA-AD cells piled up and they differentiated spontaneously to osteoblasts, suggesting the possible involvement of locally acting autocrine or paracrine factors, although a role for cell-cell contact cannot be excluded. OMA-AD cells did not express markers typically associated with primary MSC. The reasons for this are unclear because they differentiated in a manner identical to primary MSC. They did have a low expression of several markers of hematopoietic cells, although they showed no evidence of hematopoietic function (Table 1). Potentially, they represent an undifferentiated multipotential stem/progenitor population with either little or no expression of differentiated cell markers.
The primary role of MSC in the hematopoietic microenvironment is support of hematopoietic stem cell survival as well as maintenance of quiescence and differentiation. OMA-AD cells supported hematopoiesis in vitro when cultured with BM cells (from green fluorescent mice) for 11 weeks and also supported human hematopoietic cells long term in vitro (Tables 2(a) and (b)). This demonstrated that OMA-AD mimic primary MSC in regulation of hematopoietic progenitors [25] and provision of stem cell niches. MSC can protect the ovaries from chemotherapy induced damage and potentially protect against radiation damage [26] therefore, an approach was devised to test the effects of OMA-AD cells in irradiated mice. Two doses of radiation were employed, one sufficient to eliminate most endogenous hematopoietic stem cells (10 Gy) and a second (7.5 Gy), a sub lethal dose that permits survival of some, albeit low numbers, of endogenous stem cells. In addition to survival, the repopulation of blood cells in these mice, if any, was tracked, as well as the expression of GFP by the nucleated cells to determine the origins of the repopulating cells. No production of blood cells by OMA-AD cells was observed. OMA-AD cell injections into lethally irradiated recipients did not rescue hematopoiesis. This further confirmed that OMA-AD cells represented the nonhematopoietic component of BM and did not possess hematopoietic progenitors, or at least in sufficient numbers, to effect hematopoietic repopulation. However, the observation that a majority of the mice receiving a high sub lethal dose of irradiation and 1 × 106 OMA-AD cells survived for up to 125 days post irradiation, hints that OMA-AD cells might help support endogenous HSC.
This study also demonstrated an approach that can be employed to isolate additional MSC cell lines which can be engineered to express fluorescent cell markers (data not shown).
The MSC component of BM can be tracked and has the ability to give rise to rare types that integrate into injured target tissues [27]. Given that the frequency of cells with such ability is low and because intravenously injected MSC are found for only a short time in lung, liver and bone marrow (in that order) and are mostly undetectable after 72 hours (data not shown), this direct approach which depends on integration and plasticity is unlikely to result in significant tissue repair. More information on the regulation of self renewal and differentiation of MSC is needed as well as the mechanisms that target these cells to sites of tissue injury. Additionally, the relative contribution of these cells to target tissues by “plasticity”, as opposed to their ability to promote repair by stimulating endogenous cells and the mechanisms of these effects needs to be probed. Currently, it appears that their ability to immunosuppress and promote repair by tropic factors dominates [18]. Despite these uncertainties, it is clear that MSC can support hematopoietic stem cells and can contribute to repair of tissue injury. OMA-AD cells provide a useful approach to improving our understanding of the mechanisms of these effects in mouse models [18].
5. CONCLUSION
OMA-AD cells are easier to grow and exhibit more consistent properties than primary mouse MSC. Consequently, their use is more efficient and economical than that of primary cells. Most importantly, this consistency presents an opportunity to further investigate the mechanisms of MSC differentiation to bone, support of hematopoietic stem cells, amelioration of GVHD and promotion of tissue repair and regeneration which have previously been challengingntly, OMA-AD mouse MSC line exhibits all of the properties of given the heterogeneity of primary MSC populations. Conseque primary MSC, is easier to grow, exhibits consistent properties, is non-tumorigenic and immunosuppressive and has significant promise to advance our mechanistic understanding of the role of MSC in tissue repair and regenerative medicine.
6. ACKNOWLEDGEMENTS
The authors would like to thank the following individuals who assisted with or advised these studies: Shantaram Joshi, PhD, Molly Lang, MS, Greg Perry, PhD, Sam Pirruccello, MD, Lisbeth Welniak, PhD and Tami Houdesheldt who typed the manuscript. These studies were supported as a part of assay development by funds from the National Institute on Aging and the State of Nebraska Department of Health and Human Services. This support is gratefully acknowledged.
REFERENCES
- Zhang, J., Niu, C., Ye, L., Huang, H., He, X., Tong, W.G., Ross, J., Haug, J., Johnson, T., Feng, J.Q., Harris, S., Wiedemann, L.M., Mishina, Y. and Li, L. (2003) Identification of the haematopoietic stem cell niche and control of the niche size. Nature, 425, 836-841. doi:10.1038/nature02041
- Bruder, S.P., Fink, D.J. and Caplan, A.I. (1994) Mesenchymal stem cells in bone development, bone repair, and skeletal regeneration therapy. Journal of Cellular Biochemistry, 56, 283-294. doi:10.1002/jcb.240560303
- Pereira, R.F., Halford, K.W., O’Hara, M.D., Leeper, D.B., Sokolov, B.P., Pollard, M.D., Bagasra, O. and Prockop, D.J. (1995) Cultured adherent cells from marrow can serve as long-lasting precursor cells for bone, cartilage, and lung in irradiated mice. Proceedings of the National Academy of Sciences of the USA, 92, 4857-4861. doi:10.1073/pnas.92.11.4857
- Horwitz, E.M., Prockop, D.J., Fitzpatrick, L.A., Koo, W.W., Gordon, P.L., Neel, M., Sussman, M., Orchard, P., Marx, J.C., Pyeritz, R.E. and Brenner, M.K. (1999) Transplantability and therapeutic effects of bone marrowderived mesenchymal cells in children with osteogenesis imperfecta. Nature Medicine, 5, 309-313. doi:10.1038/6529
- Koc, O.N., Gerson, S.L., Cooper, B.W., Dyhouse, S.M., Haynesworth, S.E., Caplan, A.I. and Lazarus, H.M. (2000) Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. Journal of Clinical Oncology, 18, 307-316.
- Le Blanc, K. and Ringden, O. (2005) Immunobiology of human mesenchymal stem cells and future use in hematopoietic stem cell transplantation. Biology of Blood and Marrow Transplantation, 11, 321-334. doi:10.1016/j.bbmt.2005.01.005
- Le Blanc, K., Frassoni, F., Ball, L., Locatelli, F., Roelofs, H., Lewis, I., Lanino, E., Sundberg, B., Bernardo, M.E., Remberger, M., Dini, G., Egeler, R.M., Bacigalupo, A., Fibbe, W., Ringden, O. and Developmental Committee of the European Group for Blood and Marrow Transplantation (2008) Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: A phase II study. Lancet, 371, 1579-1586. doi:10.1016/S0140-6736(08)60690-X
- Tyndall, A. and Uccelli, A. (2009) Multipotent mesenchymal stromal cells for autoimmune diseases: Teaching new dogs old tricks. Bone Marrow Transplant, 43, 821-828. doi:10.1038/bmt.2009.63
- Kobayashi, N., Yasu, T., Ueba, H., Sata, M., Hashimoto, S., Kuroki, M., Saito, M. and Kawakami, M. (2004) Mechanical stress promotes the expression of smooth muscle-like properties in marrow stromal cells. Experimental Hematology, 32, 1238-1245. doi:10.1016/j.exphem.2004.08.011
- Falanga, V., Iwamoto, S., Chartier, M., Yufit, T., Butmarc, J., Kouttab, N., Shrayer, D. and Carson, P. (2007) Autologous bone marrow-derived cultured mesenchymal stem cells delivered in a fibrin spray accelerate healing in murine and human cutaneous wounds. Tissue Engineering, 13, 1299-1312. doi:10.1089/ten.2006.0278
- Semont, A., Francois, S., Mouiseddine, M., Francois, A., Sache, A., Frick, J., Thierry, D. and Chapel, A. (2006) Mesenchymal stem cells increase self-renewal of small intestinal epithelium and accelerate structural recovery after radiation injury. Advances in Experimental Medicine and Biology, 585, 19-30. doi:10.1007/978-0-387-34133-0_2
- Li, Y., Chen, J., Chen, X.G., Wang, L., Gautam, S.C., Xu, Y.X., Katakowski, M., Zhang, L.J., Lu, M., Janakiraman, N. and Chopp, M. (2002) Human marrow stromal cell therapy for stroke in rat: Neurotrophins and functional recovery. Neurology, 59, 514-523.
- Laflamme, M.A. and Murry, C.E. (2005) Regenerating the heart. Nature Biotechnology, 23, 845-856. doi:10.1038/nbt1117
- Tang, Y.L. (2005) Autologous mesenchymal stem cells for post-ischemic myocardial repair. Methods in Molecular Medicine, 112, 183-192.
- Da Silva Meirelles, L., Chagastelles, P.C. and Nardi, N.B. (2006) Mesenchymal stem cells reside in virtually all post-natal organs and tissues. Journal of Cell Science, 119, 2204-2213. doi:10.1242/jcs.02932
- Sudo, K., Kanno, M., Miharada, K., Ogawa, S., Hiroyama, T., Saijo, K. and Nakamura, Y. (2007) Mesenchymal progenitors able to differentiate into osteogenic, chondrogenic, and/or adipogenic cells in vitro are present in most primary fibroblast-like cell populations. Stem Cells, 25, 1610-1617. doi:10.1634/stemcells.2006-0504
- Pilgaard, L., Lund, P., Rasmussen, J.G., Fink, T. and Zachar, V. (2008) Comparative analysis of highly defined proteases for the isolation of adipose tissue-derived stem cells. Regenerative Medicine, 3, 705-715. doi:10.2217/17460751.3.5.705
- Phinney, D.G. and Prockop, D.J. (2007) Concise review: Mesenchymal stem/multipotent stromal cells: The state of transdifferentiation and modes of tissue repair—Current views. Stem Cells, 25, 2896-2902. doi:10.1634/stemcells.2007-0637
- Phinney, D.G., Kopen, G., Isaacson, R.L. and Prockop, D.J. (1999) Plastic adherent stromal cells from the bone marrow of commonly used strains of inbred mice: Variations in yield, growth, and differentiation. Journal of Cellular Biochemistry, 72, 570-585. doi:10.1002/(SICI)1097-4644(19990315)72:4<570::AID-JCB12>3.0.CO;2-W
- Shen, F.H., Visger, J.M., Balian, G., Hurwitz, S.R. and Diduch, D.R. (2002) Systemically administered mesenchymal stromal cells transduced with insulin-like growth factor-I localize to a fracture site and potentiate healing. Journal of Orthopaedic Trauma, 16, 651-659. doi:10.1097/00005131-200210000-00007
- Klarmann, K., Ortiz, M., Davies, M. and Keller, J.R. (2003) Identification of in vitro growth conditions for c-Kit-negative hematopoietic stem cells. Blood, 102, 3120-3128. doi:10.1182/blood-2003-04-1249
- Crouse, D.A., Mann, S.L. and Sharp, J.G. (1984) Segregation and characterization of lymphohematopoietic stromal elements. Kroc Found Ser Journal, 18, 211-231.
- Kessinger, A., O’Kane Murphy, B., Jackson, J.D. and Sharp, J.G. (2005) An ex vivo model of hematopoietic stem cell mobilization. Cytotherapy, 7, 463-469. doi:10.1080/14653240500361418
- Pirruccello, S.J., Jackson, J.D., Lang, M.S., DeBoer, J., Mann, S., Crouse, D., Vaughan, W.P., Dicke, K.A. and Sharp, J.G. (1992) OMA-AML-1: A leukemic myeloid cell line with CD34+ progenitor and CD15+ spontaneously differentiating cell compartments. Blood, 80, 1026-1032.
- Ho, A.D., Wagner, W. and Franke, W. (2008) Heterogeneity of mesenchymal stromal cell preparations. Cytotherapy, 10, 320-330. doi:10.1080/14653240802217011
- Fu, X., He, Y., Xie, C. and Liu, W. (2008) Bone marrow mesenchymal stem cell transplantation improves ovarian function and structure in rats with chemotherapy-induced ovarian damage. Cytotherapy, 10, 353-363. doi:10.1080/14653240802035926
- Abe, S., Lauby, G., Boyer, C., Rennard, S.I. and Sharp, J.G. (2003) Transplanted BM and BM side population cells contribute progeny to the lung and liver in irradiated mice. Cytotherapy, 5, 523-533. doi:10.1080/14653240310003576