American Journal of Molecular Biology
Vol.1 No.3(2011), Article ID:7778,9 pages DOI:10.4236/ajmb.2011.13014
EGR1 is essential for transcriptional regulation of BMPR2
![]()
Division of Allergy, Pulmonary and Critical Care Medicine, Department of Medicine, Vanderbilt University School of Medicine, Vanderbilt University, Nashville, USA.
Email: kirk.lane@vanderbilt.edu
Received 23 July 2011; revised 23 August 2011; accepted 9 September 2011.
Keywords: Transcription; BMPR2 Regulation; EGR1; NAB1; Pulmonary Hypertension
ABSTRACT
In this study, RLM-RACE was used to identify the transcriptional start site 387 bp upstream of the translational start. Evolutionarily conserved transcription factor binding sites were identified, and a series of luciferase reporter constructs driven by BMPR2 promoter elements used to determine their functional relevance. We found the promoter area from 983 bp to 90 bp upstream of the transcriptional start gave maximal activity, greater than longer constructs, with an area between 570 bp and 290 bp upstream of the transcriptional start containing an important repressor element. To characterize this repressor, we used a combination of EMSA, mutation of the EGR1 binding site, transfection with EGR1 and NAB1 constructs, and mutation of the NAB1 binding site within the EGR1 protein. From this we conclude that EGR1 is essential to BMPR2 transcription, but that NAB1 binding to EGR1 causes it to act as a repressor.
1. INTRODUCTION
Bone Morphogenic Proteins (BMP) belong to the TGF Beta superfamily of growth factors [1,2]. The BMPs are regulators of animal development [3-13]. BMPs were first identified in bone matrix [2], they were shown to be important in embryonic development [11,14]. The signaling of the BMP pathway involves six receptors BMPR1A, BMPR1B, BMPR2, ACTR1a, ACTR2 and ACTR2b [10-13]. BMPR2 is a transmembrane serine/threonine kinase receptor comprised of four domains; extracellular ligand binding, transmembrane, kinase and a cytoplasmic tail unique among TGF Beta superfamily receptors [15,16]. It forms heteromeric complexes with type 1 receptors and transmits the signal through the Similar to Mothers Against Decapentalegia (SMAD) pathway by activating SMAD 1, 5 and 8 by phosphorylating specific serines when the receptor complex is bound to its ligands, the BMPs [1]. It has also been reported to signal through multiple alternative pathways [13].
BMPR2 is highly conserved and the absence of BMPR2 was shown to be lethal in Bmpr2 null mice [14]. Mutations in BMPR2 are associated with more than 80% of Familial and 25% of sporadic cases of pulmonary arterial hypertension (PAH) tested [17-20]. BMPR2 expression is reported to be reduced in most forms of PAH whether experimental or pathological. To gain insight into this feature of PAH at the level of genetic regulation we have cloned and analyzed the proximal promoter of the human BMPR2 gene. Approximately 1800 base pairs of the region 5’ of the BMPR2 transcription start site was cloned into a luciferase reporter vector to determine the expression pattern of BMPR2.
The BMPR2 Promoter is a TATA-less promoter like TGF Beta receptor2 [21] and is very GC rich. Analysis of the sequence predicts many possible transcription factor binding regions including EGR1, AP1, ER half sites, SP1 and ETS1 sites. Of particular interest is Early Growth Response 1 (EGR1) gene (KROX24, Nerve growth factor–induced clone A (NGFI-A1)). EGR1 is expressed following mitogenic stimulation [22-26]. EGR1 is localized to 5q23-24 in humans [26] and contains three zinc finger DNA binding regions [27]. It acts as a transcription factor by binding to promoter regions defined by a GC rich binding site and activating transcription [28,29]. EGR1 has been demonstrated as an important modulator of TGF b [30] and leutenizing hormone [31-33] expression. EGR1 upregulates TGF b production and causes suppression of growth and transformation and induces apoptosis. It was shown that EGR1 controls the expression of LH by binding to its promoter at the GC rich binding domains. Mutating the EGR1 binding site is associated with decreased LH expression in response to GnRH.
NAB (NGFI-1A Binding) proteins are an evolutionarily conserved family of co-repressors that specifically interact with and repress the transcription mediated by NGFI-A family of immediate early gene transcription factors, NGFI-C, Krox20, and EGR3 [32,34-36]. The NAB1 gene is found on human chromosome 2 at 2q32.3-33 and its product localizes to the nucleus [34,36]. The mechanism of action of NAB1 does not involve prevention of the nuclear localization or DNA binding of its targeted transcription factors [34]. NAB1 functions in an active protein-protein co-repression manner [34]. NAB1 acts by binding to R1 binding domain of transcription factors [36]. Tethered NAB1 represses promoters even when distal to enhancer elements [34]. It represses the transcription from a TATA box or Inr element driven promoters. Recruitment of NAB1 does not merely negate NGFI-A mediated activation but converts NGFI-a binding sites to silencer elements that can repress multiple activators bound to promoters and enhancers [34].
In this report we determined that the transcriptional start site of BMPR2 mRNA is 387 base pairs upstream of the start of translation. A piece of DNA consisting of 1673 base pairs upstream of this start site was cloned into a luciferase reporter vector. Different truncations of reporter vector were assessed. The minimal promoter is shown to be –201 base pairs.
The expression of–894 was higher than all other constructs tested. The expression of –481 was very low; lower than the promoter less luciferase vector expression. We show here that if we mutate the EGR1 site on BMPR2 promoter at –298 the expression of promoter is decreased by half and loses its response to both EGR1 and NAB1.
2. MATERIALS & METHODS
2.1. RLM-RACE
To determine the transcriptional start site for BMPR2, mRNA Firstchoice RLM-RACE kit (Ambion, Austin, TX ) was used. Total RNA was derived either from RACE-READY lung cDNA (Ambion, Austin, TX) or from U937 cells (a human monocyte macrophage cell line, ATCC CRL-1593.2) extracted using an RNEasy kit (Qiagen, Valencia,CA). Ambion’s Firstchoice RLM-RACE Kit was used to capture the 5’ prime end of BMPR2 mRNA. The RLM-RACE kit is dependent on dephosphorylation of free phosphate bonds on the ends of all non-capped RNAs by calf intestinal phosphatase followed by digestion with Tobacco Alkaline Pyrophophatase to remove phosphate-phosphate bonds at the 5’ prime ends of full length mRNAs. A synthetic RNA Adapter oligonucliotide is ligated 5’ prime end of decapped mRNAs using T4 ligase. Using an outer linker specific 5’ Primer (supplied by RLM-RACE Kit) and an outer 3’ prime gene specific primer for BMPR2 (GS1: GCAAAGAAAAGAAATATGGGAAGT) an outer PCR was done at 55˚C annealing temp. A nested PCR was done using 5’ linker specific and 3’ gene specific inner primers (GS2: TCACAATACAGTTTGAAAGGACAA) and the product was cloned into pGEMT Easy. The clones were sequenced to determine the absolute transcriptional start site for BMPR2 mRNA. Another nested PCR was done using BMPR2 gene specific 5’ prime primer (GS3: TCGTGAAACTACGAGGGAAATAAT) and 3’ prime inner primer to confirm the DNA segment obtained from the earlier PCR is BMPR2.
2.2. Promoter Analysis
Overall alignment of promoters was performed using BLAST (http://blast.ncbi.nlm.nih.gov/). Determination of highly conserved regions was performed using MULAN [37] (http://mulan.dcode.org/). Determination of conserved transcription factor binding sites was performed using consensus between ConTra and multiTF [37] ( http://multiTF.dcode.org/).
2.3. Reporter Plasmids and Expression Vectors
The human BMPR2 promoter was cloned by PCR amplification of a genomic fragment corresponding to –1834 through +103, where the transcriptional start is +0. The resulting fragment was cloned into pGEM-T EASYÒ (Promega, Madison, WI). The clone was confirmed by sequencing and comparison to the 5’ region associated with the human BMPRII locus (Entrez GeneID: 659).
This promoter was sub cloned by utilizing primers corresponding to the desired 5’ and 3’ ends. These primers were further modified to include a Kpn I site on the 5’ primers and a Sac I site to facilitate insertion into the luciferase reporter plasmid pGL3E (Promega, Madison, WI). Following PCR amplification promoter amplicons were digested with Kpn I and Sac I and the resulting fragment was cloned into similarly digested pGL3E. The promoter-reporter constructs were amplified by passage through DH5 E. coli and the confirmed by DNA sequencing. Construct start and endpoint are as follows, with the transcriptional start site as +0: p1777 (–1673 to +103); p1583 (–1673 to –90); p894 (–983 to –90); p481 (–570 to –90); p201 (–290 to –90).
2.4. Cell Culture and Transfection
Normal Mouse mammary gland epithelial (NMuMG) cells from ATCC were used to do most of the cell culture experiments. Hela cells (ATCC), primary Human Umblical Vein endothelial cells (HUVECs), Human Microvascular endothelial cells (HMECs) were also used in some transfection reactions. The cells were plated and grown in 12 well plates in DMEM with 10% FBS and 1% pen/strep. When the cells were 60 to 70% confluent they were transfected with 0.4 ug of BMPR2 promoter reporter vector and 0.04 ug of Renella (for internal control) using FUGENE 6 (ROCHE, Indianapolis, IN) as transfecting medium. EGR1 and NAB1 vectors were obtained from Dr. Milbrant at Washington University and used at 20 to 80 ng concentrations in identified transfections following the same protocol.
2.5. Dual Luciferase Assay
The transfected cells were lysed directly in the plates using 200ul of 1X lysis buffer from Dual Luciferase Assay Kit (Promega). Twenty microliters of the lysate and 100ul each of Luciferase Assay Reagent II and Stop & Glo Reagent were used for each assay. Monolight 3010 (Analytical luminescence laboratory, Ann Arbor, MI) was used to read the assay.
2.6. Site Directed Mutagenesis
QuickChange Site-Directed Mutagenesis Kit (Stratagene cloning Systems, La Jolla,CA) was used to change the EGR1 binding site configuration in BMPR2 promoter reporter vectors and to alter the NAB binding domain from isoleucine to phenylalanine [38] of the Egr1 expression vector following the manufacturer’s instructions. On brief, plasmid vectors were subjected to 10-15 rounds of PCR amplification. The reaction was primed with sense and anti-sense primers carrying the desired base pair change. Dpn1 restriction endonuclease was added following PCR to digest the parental plasmids. The resulting solution was transfected into supercompetent XL-1 Blue cells and plated on selection media. Twenty-four hours later colonies were picked amplified and sequenced to confirm the desired changes.
The EGR1 binding sequence in the BMPR2 promoter GGCG was changed to AATT. The NAB binding domain in the Egr1 expression plasmid was changed from CTGTCTACTATTAAGGCC to CTGTCTACTTTTAAGGCC.
2.7. Gel-Shift Assays
Gel shifts assays were performed using a Lightshift Chemiluminescent EMSA Kit (Pierce,) using NMuMG cellular protein extracts according to the manufacturer’s directions. Biotin labeled primers were ordered from IDT containing the BMPR2 promoter section containing the EGR binding site, with the following sequence (sense primer 5’-/5Bio/GCTCACAGTCCCCAGGCGTTCGCAGAACAACCGTGA). Protein extracts were pre-incubated for 15 minutes with EGR1 or NAB1 antibodies (EGR1, #4152 Cell Signaling Technology, Inc. NAB1, H-106 sc-22813, Santa Cruz biotechnology, Inc) or unlabelled primers (cold competition) as indicated.
2.8. Statistical Methods
One or two-way ANOVA, with post-hoc t-tests, or correlation z-tests as appropriate were preformed using statistical software JMP (SAS Institute, Cary, NC).
3. RESULTS
3.1. The BMPR2 Transcriptional Start Site is 387 bp Upstream of the Translational Start
We identified the transcriptional start site (TSS) by using RACE ready human lung cDNA and cDNA prepared from total RNA of U937 cells. The methodology used was specific to 5’ capped RNA, increasing confidence that this represents a functional TSS, at least in RNA from these sources. A single PCR product was observed in PCR amplification using cDNAs from lung and U937 cells as templates (Figure 1(b)). Sequencing of the PCR products using two different templates indicated that both started at the same nucleotide, a G located 387 bases upstream of translation start site. Figure 1(a) is a schematic indicating location of primers used for identification and verification of the TSS.
 (a)
(a) (b)
(b)
Figure 1. RLM-RACE analysis of the transcription start site of BMPR2. (a) Diagrammatic presentation of primers used in RLM-RACE assays. Two adaptor specific primers (RLM1, RLM2) and two BMPR2 gene specific primers (GS1, GS2) were used to capture the 5’ end of BMRP2 mRNA using 5’RACE Ready lung cDNA from Ambion and cDNA from the U937 (human monocyte) cell line; (b) Gel electrophoresis of PCR products demonstrating size of 5’ UTR. GS2-GS3; positive control. GS1-GS2 and RLM1-RLM2 are negative controls. RLM1-GS1 and RLM2-GS2 demonstrate product sizes setting the transcriptional start site as shown in part A, 387 bp upstream of the translational start.
3.2. The BMPR2 Promoter is Highly Conserved within Mammals and Contains Multiple Conserved Transcription Factor Binding sites
The 1673bp of promoter sequence upstream of the human transcription start site, included in our longest promoter construct (discussed below), was compared to mouse, rat, and dog BMPR2 promoters. We found that there was overall identity of 63%, 64%, and 69% respecttively between mouse, rat, and dog promoter sequences and the human promoter. The alignment was divided into islands of very highly conserved regions (indicated by bars in Figure 2), of greater than 75% identity, separated by regions of lower conservation. This level of identity is very high for non-coding sequence (and is comparable to identity in coding sequences between rodents and humans). The sequence has 63% overall GC content, and does not appear to have a TATA box.
We found that the evolutionarily conserved regions (ECR) contained numerous conserved transcription factor binding sites, with the highest concentration in the ECR bracketed by p481 and p894 (discussed below). These included multiple binding sites for both EGR1 (triangles in Figure 2) and related transcription factors ETF and EGR2-4. Other transcription factors with multiple binding sites include SP1, AP2, ZF5, and KROX. Transcription factors with single sites conserved across species include NFB, ER, p53, CREB, PAX3-5 and 9, MyoD, PPARa, HEN1, and SMAD3. The only conserved site found in the distal ECR around bp-1400 was the sex-determination fac tor SRY.
3.3. Deletional Analysis of the BMPR2 Promoter
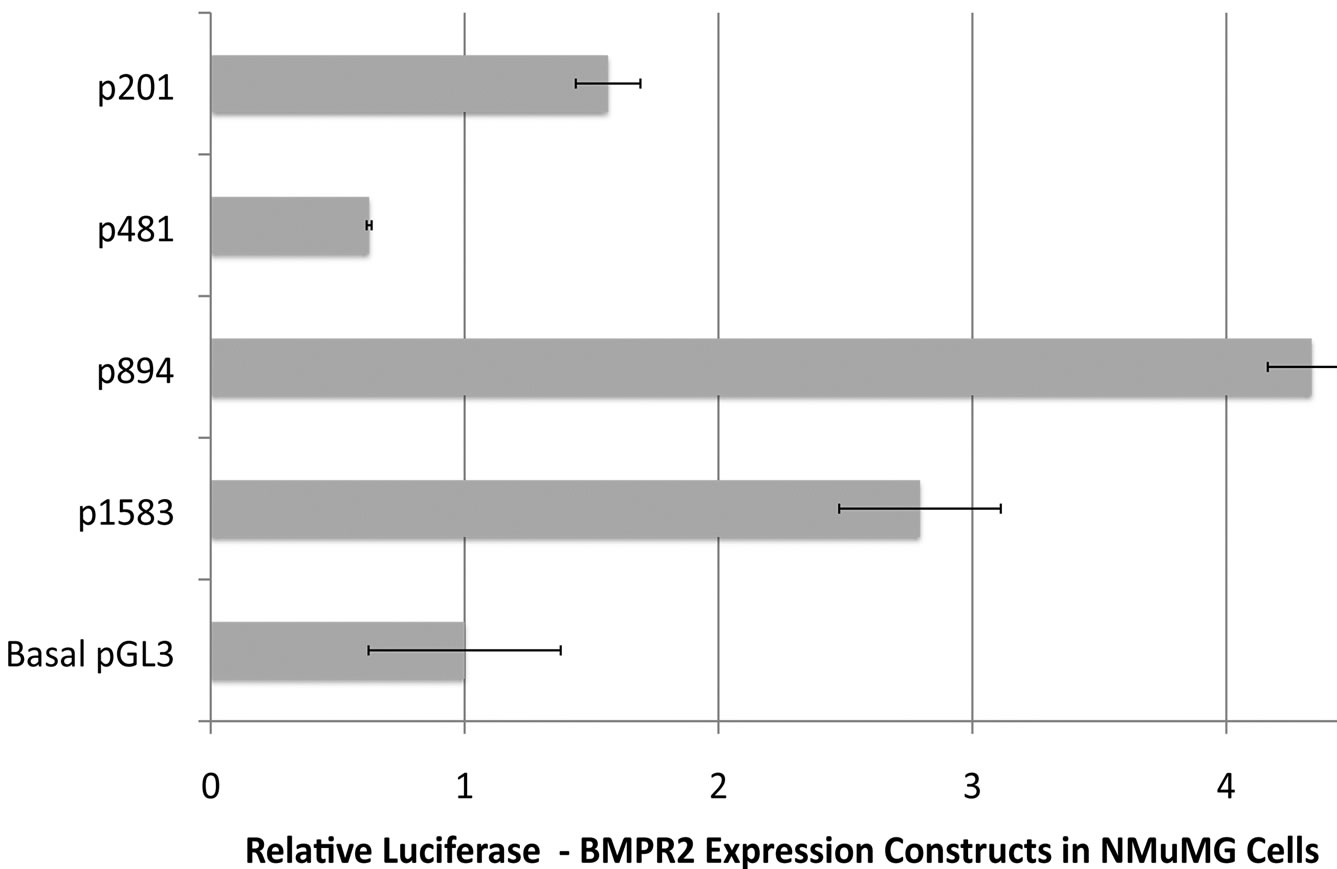
To determine the sequences essential for efficient transcription of human BMPR2 promoter in NMuMG cells, promoter constructs were created containing various sections of the BMPR2 promoter (depicted in Figure 2,). NMuMG cells have intact BMP signaling and therefore make a suitable platform to study the general transcripttional regulation of BMPR2. These constructs are referred to as pX, where X represents the size of insert in base pairs. Upon transient transfection into NMuMG cells, all drove higher activity than the basal pGL3 enhancer, aside from p481 (Figure 3(a)). Further deletion from p481 to p201 increased promoter activity (Figure 3(a)). Similar observations were made when the p481 and p1583 were transfected in different cell types including HUVEC, RSMC and HMECs (Figure 3(b)). The consistency of response across mouse, rat, and human as well as vascular and epithelial cells supports the general applicability of the findings reported here. This finding suggests that there is a strong repressive element in the region between bp –292 and –571 of the BMPR2 promoter.
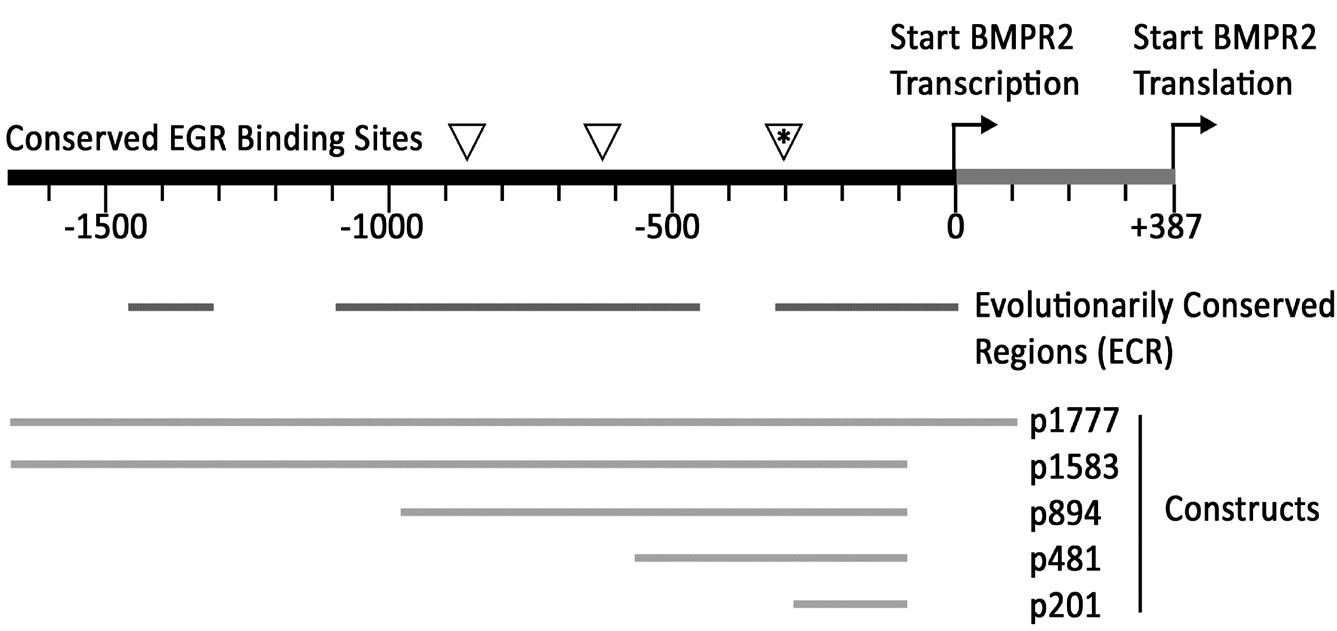
Figure 2. Schematic of BMPR2 promoter, showing EGR binding sites conserved across species (triangles, top line); Evolutionarily Conserved Regions (ECR) with greater than 75% sequence identity across mice, rats, humans, and dogs; and the positions of promoter constructs created to examine expression (bottom section).
 (a)
(a)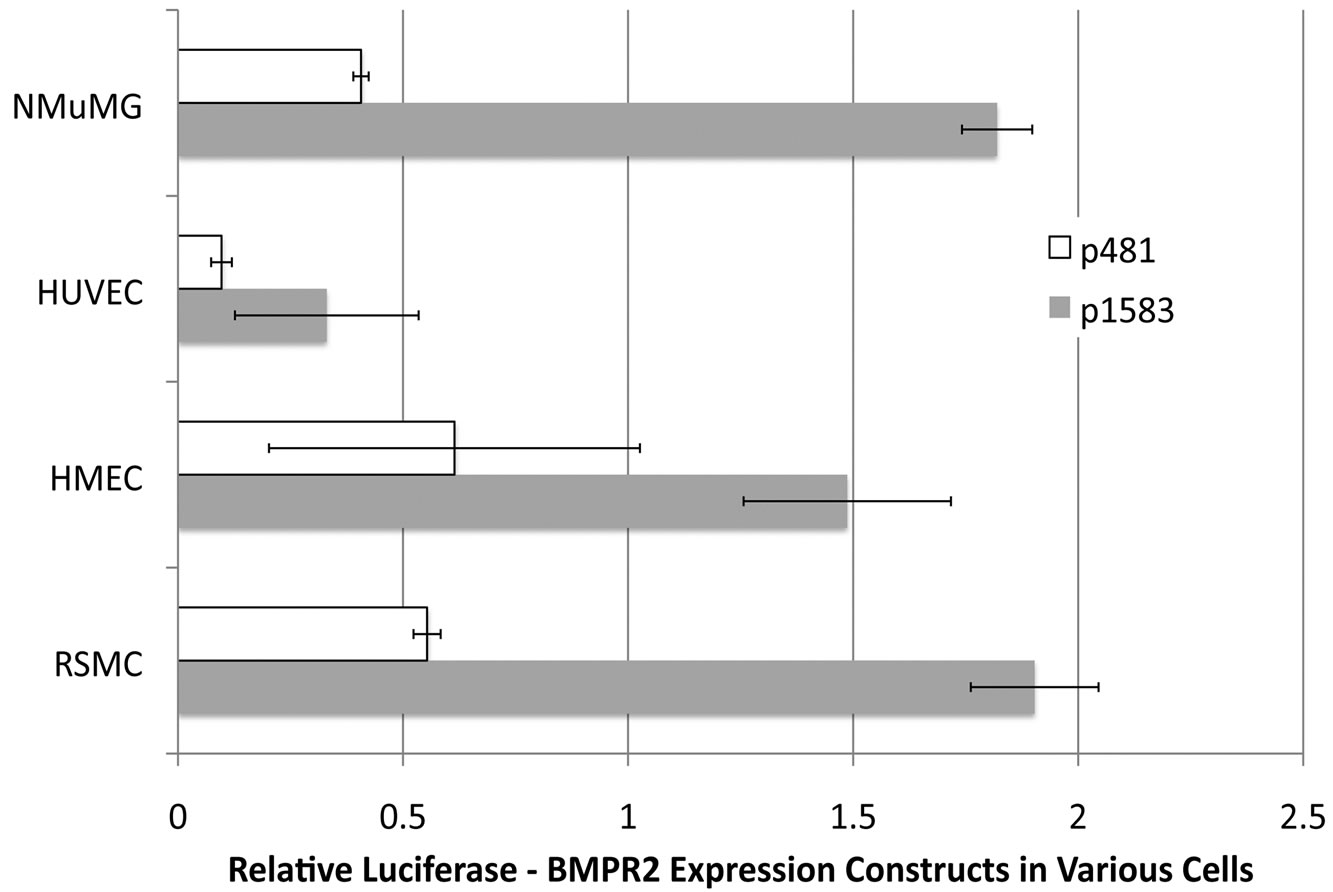 (b)
(b)
Figure 3. (a) Sections of the BMPR2 promoter were used to drive expression from the pGL3 luciferase reporter construct. Data was normalized to Renilla, and then to basal pGL3 expression. All constructs’ activities are significantly different than all others by ANOVA with post-hoc t-test. In particular note that expression from p201 is almost three times that of p481, implying the existence of a repressor element in p481 not present in p201; (b) Loss of BMPR2 promoter activity with truncation to p481 is similar across cell types. By two-way ANOVA, p < 0001 for cell type effect and p < 0001 for construct effect.
We attribute the increase in BMPR2 expression with the longer promoter constructs (p894 and p1583, Figure 3(a)) to be due to the presence and context of gene-appropriate additional transcription factors. The presence of ECRs in the region between –481 and –894 (Figure 2) would support this contention.
3.4. Unbound EGR Drives and NAB1 Bound EGR Represses BMPR2 Promoter Activity
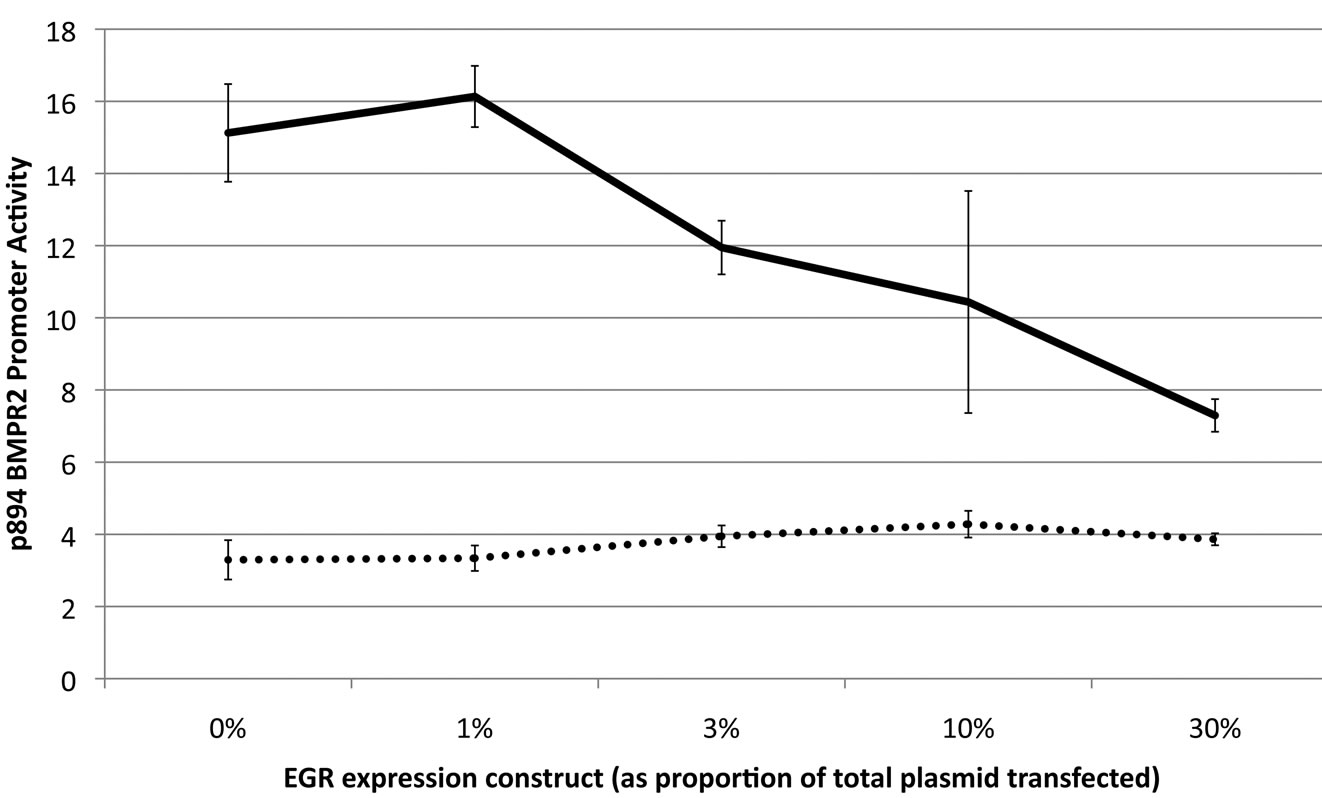
In our search for the repressive element between bp –292 and –571, we focused on an evolutionarily conserved EGR binding site at bp –292 to –303. Using site-directed mutagenesis, we created a promoter construct in which this EGR site had a 4-base mutation. To examine the impact of EGR, NMuMG cells were transfected with increasing quantities of EGR expression plasmid and either wild-type or EGR-mutant luciferase-reporter construct. We found that mutating the EGR site in p894 dramatically inhibited luciferase activity, but paradoxically, increasing quantities of EGR also inhibited luciferase activity (Figure 4(a)).
Because of literature reviewed in the introduction, we hypothesized that this effect was driven by EGR interaction with NAB1. To test this hypothesis, first we contransfected NMuMG cells with increasing quantities of a NAB1 expression construct and three of our BMPR2 promoter constructs. Those which contained the EGR binding site (p481, p1777) were roughly twofold inhibited by increasing quantities of NAB1; p201, which does not contain the EGR binding site, was not inhibited by increasing NAB1 and in fact showed an enhanced expression with increasing NAB1 levels (Figure 4(b)). We believe this increased expression in p201 is a secondary effect due to NAB1 reducing transcription of other factors that may be important in driving the shorter constrct.
To demonstrate that the NAB1 effect was through EGR binding, we created a plasmid encoding an EGR protein with a mutated NAB1 binding site. Cotransfecting EGR with NAB1 binding mutation into NMuMG cells with p894 BMPR2 luciferase reporter with intact or mutated EGR binding site shows abolition of the inhibitory effect of increasing EGR (Figure 4(c)). As before, a mutated EGR binding site abolishes transcriptional activity from this promoter. These data strongly suggest that EGR not bound by NAB1 is required to drive BMPR2 expression but that EGR bound by NAB1 becomes an inhibitor of BMPR2 expression.
To confirm that this is a direct effect, we conducted a gel-shift assay, using the EGR binding site from the BMPR2 promoter as a target and either EGR or NAB1 antibodies (Figure 5). We used over expression of EGR to identify the correct ‘shift’ band (lane 10), and found that both EGR and NAB1 antibodies created a ‘supershift’ band, indicating binding to the EGR site in the
 (a)
(a)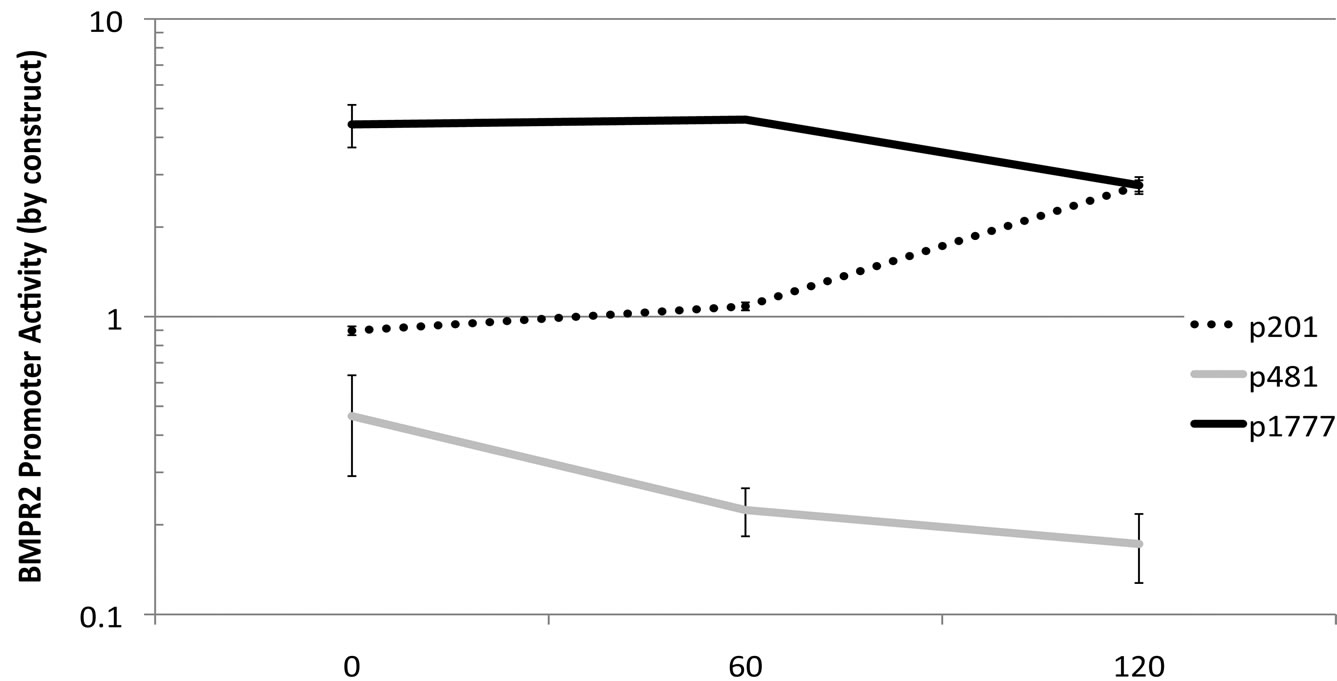 (b)
(b) (c)
(c)
Figure 4. Expression driven by the BMPR2 p894 construct is regulated by an interaction between EGR and NAB1. (a) Increasing quantities of EGR expression construct causes decreasing expression from a native p894 BMPR2 promoter, solid line (p = 0017 by correlation z-test), however mutating the EGR binding site in the promoter strongly suppresses transcription, broken line, (p < .0001 by 2-way ANOVA); (b) Increased NAB1 expression construct transfection results in decreased transcriptional activity in p481 and p1777 expression constructs, but not p201 (which is lacking the EGR binding site) (p <. 0001 for differential NAB1 effect between constructs by 2-way ANOVA); (c) Mutating the NAB1 binding site in the EGR protein removes the inhibitory effect of increasing EGR, solid line, and almost eliminates transcription from this promoter in the context of a mutated EGR binding site, broken line.
BMPR2 promoter. In the case of the EGR supershift, it almost entirely eliminated the ‘shift’ band, indicating that this band was almost entirely made up of EGR.

Figure 5. Gel shift assay demonstrating EGR and NAB1 binding to the EGR binding site in the BMPR2 promoter. Proteins were extracted from NMuMG cells and incubated with a biotin-labeled BMPR2 promoter fragment containing the putative EGR binding site. Lane 1: no antibody; Lanes 2, 5, 8: preincubation of extract with 5 μg, 1 μg, or 0.5 μg NAB1 antibody (respectively) causes a supershift band to appear; Lanes 3, 6, 9: Preincubation of extract with with 5 μg, 1 μg, or 0.5 μg EGR antibody (respectively) causes a supershift band to appear and the EGR band to disappear; Lanes 4,7: cold competition removes both EGR (“shift”) band, and supershift bands; Lane 10: Proteins extracted from NMuMG cells transfected with an EGR-expression construct show increased strength of the EGR shift band.
4. DISCUSSION
BMPR2 has come under increased scrutiny outside of developmental biology due to the role it plays in the familial form of pulmonary arterial hypertension (PAH). The region 5’ of BMPR2 has been investigated in the context of PAH [39]. We chose to approach BMPR2 transcriptional regulation from a classic promoter identification strategy. Initially we identified the primary transcription start site from mature capped mRNA from the human monocytic cell line U937 (ATCC# CRL- 1593.2). We confirmed the detected start site (positioned at –387 from the translation start) in commercially obtained whole lung cDNA. These 2 sources produce the same transcription start site that differed from the report of Aldred et al. [39]. This discrepancy may represent alternative start sites utilized or the detection of immature mRNA. Our transcriptional start site is supported by the work of Wang et al. [40] who reported a promoter polymorphism at –669 that altered a proposed SP3 site that inhibited BMPR2 expression. This change would be found in the promoter we predict while contained in the transcribed region proposed by Aldred et al. Our promoter findings do not alter the principle finding reported by Aldred et al.
Once the transcription start site was determined approximately 1600 base pairs 5’ of the transcription start site was cloned. This promoter was found to be active in many cell types although the magnitude of expression varied depending on cell type. The literature suggest a similar pattern of expression for BMPR2 [2,10,12]. Through truncation analysis the minimal promoter was determined to be the proximal 200bp.
Interestingly a promoter containing nearly 300bp more than the minimal promoter had lower expression levels than even the promoter-less backbone the promoter fragments were cloned into. Comparative genomics uncovered a conserved region that contained an EGR1 (KROX24, NGFIA, ZIF268) binding site in this fragment. EGR1 may act as both a positive and negative transcriptional modulator though interactions with cognate binding sites and co-repressors of the NAB family. Protein-protein interactions between EGR1 and NAB do not keep EGR1 from binding to DNA but do create a transcriptional repressor complex.
To confirm this site as essential in transcriptional regulation of BMPR2 we both altered the binding site, and altered the EGR1 or NAB1 protein levels by transfection of expression vectors. Paradoxically, when EGR1 plasmids were added expression from the BMPR2 promoter decreased. We interpret this finding to an excess of NAB proteins available in NMuMG cells. By adding additional EGR1 we allow these excess Nab proteins to act as repressors in conjunction with the added Egr1. This interpretation is supported by the diminished transcription seen when a similar BMPR2 promoter-reporter plasmid in which the EGR1 binding site has been eliminated is delivered to these cells. Expression for this construct was much reduced but was not influenced by addition of EGR1. These data point to the importance of the EGR1 site and the potential of both positive and negative regulation associated with this site.
We confirmed the role of Nab by adding exogenous NAB1 via expression construct. In BMPR2 promoter reporter constructs those that contain an EGR1 binding site (p481 and p1777) exogenous NAB1 expression resulted in similar diminished reporter expression. With the minimal BMPR2 promoter-reporter construct, which does not include the EGR1 site, there was no fall in reporter expression.
To provide evidence that the EGR1 and NAB1 protein-protein interaction is important in BMPR2 expression we altered the NAB binding site of EGR1. This altered EGR1 was transfected into cells resulting in relatively unchanged reporter expression in place of falling expression if the NAB binding site were intact. This finding confirmed the interaction between EGR1 and NAB to modulate BMPR2 expression.
Finally to confirm our interpretation of the interaction of EGR1 and NAB1 with the putative binding site associated with the BMPR2 promoter we conducted Electophoretic Mobility Shift Assays employing the EGR1 binding site of the human BMPR2 promoter. In these assays the mobility of the labeled EGR1 oligos was retarded and coincided with a positive control sample. Further more this band was supershifted by both the presence of antibodies to EGR1 or NAB1. These findings confirm our interpretation that the EGR1 binding site is functional and critical to the expression of BMPR2. We were led to this site by the conserved nature of this region suggesting that this regulation has been important through out evolution of vertebrates.
The role of EGR1 in the regulation of BMPR2 may be linked in development. EGR1 is expressed early in development and regulates brain maturation [36]. BMP signaling is also important early in development [41]. Along with the early developmental role EGR1 is involved in angiogenesis [42-45]. Lack of EGR1 results in failure of effective angiogenesis and has been proposed to be linked to NSAIDs potential anti tumor effect [46]. Additionally, EGR1 effects late terminal differentiation modulated by environmental stimulation. EGR1 is essential for macrophage response to endotoxin stimulation[47].
5. CONCLUSIONS
In this report we have demonstrated that BMPR2 is differentially expressed in various cell types. Furthermore, we have shown that BMPR2 expression is strongly controlled by EGR1 and its co-repressor NAB1. These findings are particularly interesting in that heterogenous mutations in BMPR2 are responsible for the familial form of pulmonary arterial hypertension (PAH). Additionally, in non-familial PAH BMPR2 expression is reported to be decreased [48,49]. To our knowledge there has not been a systematic evaluation of the role of BMPR2 transcriptional regulation in the context of PAH.
EGR1 is important in regulating transcription of many immediate early response genes following a variety of stresses [50-54]. The role of NAB1 has yet to be assessed in the same detail. It is likely that pulmonary vascular stress results in EGR1 stimulation. This may result in down regulation of BMPR2 depending on the relative concentrations of NAB1. This scenario would fit well with the finding of BMPR2 suppression in the non-familial form of PAH. The data presented here should allow the initiation of such a study to assess if the decreases in BMPR2 expression associated with PAH are transcriptional in origin. This data my also suggest targets for intervention if decreased BMPR2 expression is determined to be essential in PAH progression.
REFERENCES
- Heldin, C.H., Miyazono, K. and ten Dijke, P. (1997) TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature, 390, 465-471. doi:10.1038/37284
- Ducy, P. and Karsenty, G. (2000) The family of bone morphogenetic proteins. Kidney International, 57, 22- 07-2214. doi:10.1046/j.1523-1755.2000.00081.x
- Bellusci, S., et al. (1996) Evidence from normal expression and targeted misexpression that bone morphogenetic protein (Bmp-4) plays a role in mouse embryonic lung morphogenesis. Development, 122, 1693-1702.
- Frisch, A. and Wright, C.V. (1998) XBMPRII, a novel Xenopus type II receptor mediating BMP signaling in embryonic tissues. Development, 125, 431-442.
- Hemmati-Brivanlou, A. and Thomsen, G.H. (1995) Ventral mesodermal patterning in Xenopus embryos: Expression patterns and activities of BMP-2 and BMP-4. Devepment and Genetics, 17, 78-89.
- Hild, M., et al. (2000) The roles of BMPs, BMP antagonists, and the BMP signaling transducers Smad1 and Smad5 during dorsoventral patterning of the zebrafish embryo. Ernst Schering Research Foundation Workshop, 29, 81-106.
- Smith, W.C. (1999) TGF beta inhibitors. New and unexpected requirements in vertebrate development. Trends in Genetics, 15, 3-5. doi:10.1016/S0168-9525(98)01641-2
- Thomsen, G.H. (1997) Antagonism within and around the organizer: BMP inhibitors in vertebrate body patterning. Trends in Genetics, 13, 209-211. doi:10.1016/S0168-9525(97)01117-7
- Wozney, J.M. (1998) The bone morphogenetic protein family: Multifunctional cellular regulators in the embryo and adult. European Journal of Oral Sciences, 1, 1- 60-166.
- Iwasaki, S., et al. (1995) Distribution and characterization of specific cellular binding proteins for bone morphogenetic protein-2. Journal of Biological Chemistry, 270, 5476-82. doi:10.1074/jbc.270.10.5476
- Josso, N. and di Clemente, N. (1997) Serine/threonine kinase receptors and ligands. Current Opinion in Genetics and Development, 7, 371-377. doi:10.1016/S0959-437X(97)80151-7
- Kawabata, M., Imamura, T. and Miyazono, K. (1998) Signal transduction by bone morphogenetic proteins. Cytokine and Growth Factor Reviews, 9, 49-61. doi:10.1016/S1359-6101(97)00036-1
- Nohe, A., et al. (2002) The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. Journal of Biological Chemistry, 277, 5330-5338. doi:10.1074/jbc.M102750200
- Beppu, H., et al. (2000) BMP type II receptor is required for gastrulation and early development of mouse embryos. Developmental Biology, 221, 249-258. doi:10.1006/dbio.2000.9670
- Kawabata, M., Chytil, A. and Moses, H.L. (1995) Cloning of a novel type II serine/threonine kinase receptor through interaction with the type I transforming growth factor-beta receptor. Journal of Biological Chemistry, 270, 5625-5630. doi:10.1074/jbc.270.10.5625
- Nohno, T., et al. (1995) Identification of a human type II receptor for bone morphogenetic protein-4 that forms differential heteromeric complexes with bone morphogenetic protein type I receptors. Journal of Biological Chemistry, 270, 22522-22526. doi:10.1074/jbc.270.38.22522
- Cogan, J.D., et al. (2006) High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine, 174, 590-598. doi:10.1164/rccm.200602-165OC
- Deng, Z., et al. (2000) Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. American Journal of Human Genetics, 67, 737-744. doi:10.1086/303059
- Lane, K.B., et al. (2000) Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nature Genetics, 26, 81-84. doi:10.1038/79226
- Thomson, J.R., et al. (2000) Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. Journal of Medicine Genetics, 37, 741-745. doi:10.1136/jmg.37.10.741
- Humphries, D.E., et al. (1994) Structure and expression of the promoter for the human type II transforming growth factor-beta receptor. Biochemical and Biophysical Research Communications, 203, 1020-1027. doi:10.1006/bbrc.1994.2284
- Ahn, B.H., et al. (2007) Phorbol myristate acetate-induced EGR-1 expression is suppressed by phospholipase D isozymes in human glioma cells. FEBS Letters, 581, 5940-5944. doi:10.1016/j.febslet.2007.11.077
- Dunnmon, P.M., et al. (1990) Phorbol esters induce immediate-early genes and activate cardiac gene transcription in neonatal rat myocardial cells. Journal of Molecular and Cellular Cardiology, 22, 901-910. doi:10.1016/0022-2828(90)90121-H
- Maass, A., et al. (1994) Mitogenic signals control translation of the early growth response gene-1 in myogenic cells. Biochemical and Biophysical Research Communications, 202, 1337-1346. doi:10.1006/bbrc.1994.2077
- You, L. and Jakowlew, S.B. (1997) Identification of early growth response gene-1 (EGR-1) as a phorbol myristate acetate-induced gene in lung cancer cells by differential mRNA display. American Journal of Respiratory and Critical Care Medicine, 17, 617-624.
- Sukhatme, V.P., et al. (1988) A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell, 53, 37-43. doi:10.1016/0092-8674(88)90485-0
- Matheny, C., Day, M.L. and Milbrandt, J. (1994) The nuclear localization signal of NGFI-A is located within the zinc finger DNA binding domain. Journal of Biological Chemistry, 269, 8176-8181.
- Kubosaki, A., et al. (2009) Genome-wide investigation of in vivo EGR-1 binding sites in monocytic differentiation. Genome Biology, 10, 41. doi:10.1186/gb-2009-10-4-r41
- Seyfert, V.L., et al. (1990) EGR-1 expression in surface Ig-mediated B cell activation. Kinetics and association with protein kinase C activation. Journal of Immunology, 145, 3647-3653.
- McCaffrey, T.A., et al. (2000)High-level expression of EGR-1 and EGR-1-inducible genes in mouse and human atherosclerosis. Journal of Clinical Investigation, 105, 653-662. doi:10.1172/JCI8592
- Lee, S.L., et al. (1996) Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (EGR-1). Science, 273, 1219-1221. doi:10.1126/science.273.5279.1219
- Sevetson, B.R., Svaren, J. and Milbrandt, J. (2000) A novel activation function for NAB proteins in EGR-dependent transcription of the luteinizing hormone beta gene. Journal of Biological Chemistry, 275, 97- 49-9757. doi:10.1074/jbc.275.13.9749
- Tourtellotte, W.G., et al. (2000) Functional compensation by EGR4 in EGR1-dependent luteinizing hormone regulation and Leydig cell steroidogenesis. Molecular and Cellular Biology, 20, 5261-5268. doi:10.1128/MCB.20.14.5261-5268.2000
- Swirnoff, A.H., et al. (1998) NAB1, a corepressor of NGFI-A (EGR-1), contains an active transcriptional repression domain. Molecular and Cellular Biology, 18, 512-524.
- Svaren, J., et al. (1996) NAB2, a corepressor of NGFI-A (EGR-1) and Krox20, is induced by proliferative and differentiative stimuli. Molecular and Cellular Biology, 16, 3545-3553.
- Russo, M.W., Sevetson, B.R. and Milbrandt, J. (1995) Identification of NAB1, a repressor of NGFI-Aand Krox20-mediated transcription. Proceedings of the National Academy of Sciences, USA, 92, 6873-6877.
- Loots, G.G. and Ovcharenko, I. (2005) Dcode.org anthology of comparative genomic tools. Nucleic Acids Research, 33, 56-64. doi:10.1093/nar/gki355
- Wang, C., et al. (2005) EGR-1 negatively regulates expression of the sodium-calcium exchanger-1 in cardiomyocytes in vitro and in vivo. Cardiovascular Research, 65, 187-194. doi:10.1016/j.cardiores.2004.09.026
- Aldred, M.A., et al. (2007) Characterization of the BMPR2 5'-untranslated region and a novel mutation in pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine, 176, 819-824. doi:10.1164/rccm.200701-164OC
- Wang, H., et al. (2009) Novel promoter and exon mutations of the BMPR2 gene in Chinese patients with pulmonary arterial hypertension. European Journal of Human Genetics, 17, 1063-1069. doi:10.1038/ejhg.2009.3
- Beppu, H., et al. (2000) BMP type II receptor is required for gastrulation and early development of mouse embryos. Developmental Biology, 221, 249-258. doi:10.1006/dbio.2000.9670
- Svaren, J., et al. (2000) EGR1 target genes in prostate carcinoma cells identified by microarray analysis. Journal of Biological Chemistry, 275, 38524-38531. doi:10.1074/jbc.M005220200
- Abdel-Malak, N.A., et al. (2009) Early growth response-1 regulates angiopoietin-1-induced endothelial cell proliferation, migration, and differentiation. Arteriosclerosis, Thrombosis, and Vascular Biology, 29, 209-216. doi:10.1161/ATVBAHA.108.181073
- Lee, Y.S., et al. (2005) Adenoviral-mediated delivery of early growth response factor-1 gene increases tissue perfusion in a murine model of hindlimb ischemia. Molecular Therapy, 12, 328-336. doi:10.1016/j.ymthe.2005.03.027
- Fahmy, R.G., et al. (2003) Transcription factor EGR-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nature Medicine, 9, 1- 026-1032. doi:10.1038/nm905
- Szabo, I.L., et al. (2001) NSAIDs inhibit the activation of Egr-1 gene in microvascular endothelial cells: A key to inhibition of angiogenesis? Journal of Physiology, Paris, 95, 379-383. doi:10.1016/S0928-4257(01)00051-1
- Sweet, M.J. and Hume, D.A. (1996) Endotoxin signal transduction in macrophages. Journal of Leukocyte Biology, 60, 8-26.
- Dewachter, L., et al. (2009) Bone morphogenetic protein signalling in heritable versus idiopathic pulmonary hypertension. European Respiratory Journal, 34, 110- 0-1110. doi:10.1183/09031936.00183008
- Rondelet, B., et al. (2005) Prevention of pulmonary vascular remodeling and of decreased BMPR-2 expression by losartan therapy in shunt-induced pulmonary hypertension. American Journal of Physiology-Heart and Circulatory Physiology, 289, 2319-2324. doi:10.1152/ajpheart.00518.2005
- Pritchard, M.T. and Nagy, L.E. (2005) Ethanol-induced liver injury: Potential roles for EGR-1. Alcohol Clinical Experiment Research, 29, 146S-150S. doi:10.1097/01.alc.0000189286.81943.51
- Revest, J.M., et al. (2005) The MAPK pathway and EGR-1 mediate stress-related behavioral effects of glucocorticoids. Nature Neuroscience, 8, 664-672. doi:10.1038/nn1441
- Quinones, A., Dobberstein, K.U. and Rainov, N.G. (2003) The EGR-1 gene is induced by DNA-damaging agents and non-genotoxic drugs in both normal and neoplastic human cells. Life Science, 72, 2975-2992. doi:10.1016/S0024-3205(03)00230-3
- Yan, S.F., et al. (2000) EGR-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nature Medicine, 6, 1355-1361. doi:10.1038/82168
- Stula, M., et al. (2000) Influence of sustained mechanical stress on EGR-1 mRNA expression in cultured human endothelial cells. Molecular and Cellular Biochemistry, 210, 101-108. doi:10.1023/A:1007126218740