New Journal of Glass and Ceramics
Vol.04 No.02(2014), Article ID:45415,8 pages
10.4236/njgc.2014.42004
The Behavior of Zeta Potential of Silica Suspensions
José Antonio Alves Júnior, João Baptista Baldo
Departamento de Materiais, Universidade Federal de São Carlos, São Carlos, Brazil
Email: baldo@ufscar.br, jjunior84@live.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 29 January 2014; revised 15 March 2014; accepted 22 March 2014
ABSTRACT
Zeta potential is one of the most relevant parameters controlling the rheological behavior of ceramic suspensions. In this work, it was observed that for pH values below the isoelectric point (IEP), the positive value of zeta potential of water suspensions of α-quartz and α-cristobalite, experiences a sudden steep increase with the increase in specific surface area of the powders. For pH values above the IEP, the zeta potential values of crystalline forms of silica (α-quartz and α-cris- tobalite), get gradually more negative with the increase in pH. Conversely in the case of vitreous silica, for pH values above 6, there occurs a steep change towards more negative values of zeta potential than those presented by quartz and cristobalite. These findings have not yet been accounted for in the DLVO theory but may provide subsidies for better understanding of how to stabilize and destabilize crystalline and vitreous silica water suspensions.
Keywords:
Silica Slurries, Zeta Potential, Stabilization, Iso-Electronic Point, Silica

1. Introduction
Zeta Potential is an electro-kinetic parameter indirectly determined by the surface charge of particles when suspended in a polar media. The magnitude of zeta potential by itself can not guaranty the stability of suspensions [1] . However, through its manipulation, guidelines can be taken towards stabilization or destabilization of ceramic slurries. The charged particles in suspensions can be set into motion under the action of an electric field [1] [2] . The movement is in the direction of the oppositely charged electrode and can be dragged of by the viscous hydrodynamic forces of the solution. At equilibrium the particles can move at constant speed. The constant velocity acquired under unitary electric field strength is called unitary electrophoretic mobility (µe) [3] .
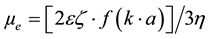 [3] (1)
[3] (1)
where:
ɛ = dielectric constant of the solution;
ζ = zeta potential;
η = viscosity of the suspension fluid phase;
 = Henry’s function;
= Henry’s function;
k = Debye length (1/k = electric double layer thickness);
a = particle equivalent spherical diameter.
For conditions of low electrolyte concentration and particle size bigger than 0.2 µm the Henry’s function turns out to be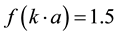 .
.
The zeta potential indicated in Equation (1), is an indirect measure of the net charge created in the particles of the suspension, either by adsorption of contra ions or dissociation of particles surface groups. Following the DLVO theory the larger the zeta potential absolute value is, more stable the suspension can be. The adjustment of this parameter makes it possible to control the stability of colloidal ceramic suspensions. It is accepted that zeta potential values of ±30 mV are enough to promote stable water suspensions. This occurs because particles like charge repel each other, overcoming the tendency to aggregation caused by the action of the Van der Waals forces [1] - [3] .
The measurement of zeta potential is an information, frequently used in coagulation (aggregation) and dispersion processes in several applications, such as colloidal processing of ceramic pastes and slurries, water purification, paints and cosmetics [4] - [7] .
On the other hand, the stability of suspensions is also affected by the sedimentation tendency of particles suspended in a liquid. Under conditions of low solid concentration, the sedimentation velocity is expressed by Stokes law as in Equation (2).
 (2)
(2)
where:
Vs = sedimentation velocity;
∆ρ = Difference between the suspended solid density and the fluid phase density;
r = equivalent spherical diameter of particle in suspension;
η = viscosity of the fluid phase.
It results in that for low concentration suspensions, sedimentation may compete with the electrophoretic mobility (Equation (1)), in this context they are perpendicular to each other.
Comparing Equations (1) and (2), we may write:
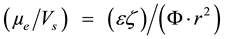 (3)
(3)
where .
.
From Equation (3) we can see that by decreasing particle size there is a relative increase in eletrophoretic mobility and indirectly also an increase in the apparent zeta potential.
In milled silica particles the  coordination tetrahedron is imperfect leaving silicon and oxygen unsatisfied bonds at each particle surface. In aqueous suspensions of silica particles, these free bonds are neutralized by OH− and H+ species. In this sense partial or total particle surface hydroxylation can result in the formation of silanol groups [Si(OH)n]. The silanol groups in pure water dissociate through the following reactions:
coordination tetrahedron is imperfect leaving silicon and oxygen unsatisfied bonds at each particle surface. In aqueous suspensions of silica particles, these free bonds are neutralized by OH− and H+ species. In this sense partial or total particle surface hydroxylation can result in the formation of silanol groups [Si(OH)n]. The silanol groups in pure water dissociate through the following reactions:
 (4)
(4)
 (5)
(5)
That is why silica particles may end negatively charged in water suspension for basic pH and positively charged under acidic pH [8] [9] . These phenomena impact directly the zeta potential behavior of silica suspensions. Considering that SiO2 is inherently an acidic oxide, the pH of an aqueous suspension of silica is slightly acidic. However, it also may depend on the crystal structure, that planes are exposed on the fractured surface of particles, their atomic density and presence of contaminants of the SiO2 source [10] - [12] .
The aim of this paper is to discuss the behavior of zeta potential versus pH, in the vicinity and away from the isoelectric point (IEP), presented by suspensions of several silica raw materials of different crystal structures and specific surface area. The chosen silica powdered raw materials for this study were two crystalline polymorphs of silica (α-quartz and α-cristobalite), as well as vitreous silica.
2. Materials and Methods
The silica raw materials used were the following:
· α-Quartz in the following granulometries (mesh or micrometers undersized): #200 (d50 = 13 µm ), #325 (d50 = 7 µm) and #500 (d50 = 4 µm) mesh sizes.
· α-Cristobalite #500 (d50 = 10 µm).
· α-Cristobalite #500 additionally wet bal milled for 24 hours in plastic jars containing alumina balls grinding media in a 4:1 balls: powder mass ratio (d50 = 4 µm).
· Silica mix a blend containing 52% of quartz #200, 32% of quartz #325 and 16% of quartz #500 (d50 = 7 µm).
· Vitreous silica (d50 = 30 µm).
In Table 1 below the values of the respective densities for samples: α-quartz, α-cristobalite and vitreous silica are shown.
Obs: All the quartz samples came from the same quarry and same producer Minas Minérios (Brazil).
The cristobalite was from from Hoben International (England).
The vitreous silica came from Treibacher Company (Brazil).
The measurement of the respective zeta potential was performed as a function of pH change, using an ELS Analysis Zeta Meter®. Initially 0.05 g of each sample was added to 50 mL of KCl (supporting electrolyte), at a concentration 0.02 mol/L. The six samples were submitted to an ultrasound probe in order to eliminate any agglomerates present. The tests were made by diluting the samples in one-liter balloon in distilled water. The reagents used for varying the pH were HCl 0.1 mol/L and KOH 0.1 mol/L.
The equipment used for determining the specific surface was a Surface Area and Porosity Analyzer Micromeritics ASAP® 2020. The specific surface area was determined for each powdered sample, using the B.E.T. technique (Brunauer, Emmett and Teller), using nitrogen gas. The particle size distribution of each sample was made by means of Sedigraph Micromeritics®-Particle Analyser 5000-D.
3. Results and Discussions
In Figure 1 it is presented the grain size distribution of all the eight silica raw materials investigated in this work. It can be seen that the vitreous silica #100 was the coarsest among all the samples.
In Table 2 the pH of distilled aqueous suspensions of the eight raw materials investigated are presented.
It can be seen that the pH in distilled water of all the quartz samples are practically the same. However, the cristobalite samples show higher values of pH in distilled water than all the quartz samples and vitreous silica #100. However, vitreous silica #325 sample displayed the highest pH in distilled water suspension. Dove [9] , pointed out that the SiO2 activity (ac) differs for the different crystalline species of silica in the following order: acquartz < accristobalite < acvitreous silica.
This could explain the higher pH presented in aqueous solution by cristobalite when compared to quartz. This behavior, is apparently in a slight contradiction to the set ranking, considering it would be expected a bigger pH for both of the vitreous silica samples in aqueous solution, when compared to all the other silica samples investigated. However, we should recall that the particles of the vitreous silica #100 are bigger (smallest specific surface area), than the other forms of silica investigated, such that its dissolution in water may not be as thorough as expected.
In Table 3 the respective specific surface areas of all samples are presented. The values follow approximately the relation for specific surface area and average particle size.
The specific surface area of the #500 quartz is respectively 58% bigger than that of #325 quartz sample and

Table 1. The values of the respective densities.

Figure 1. Particle size distribution for each of the seven silica samples.
Table 2. pH values for the different silica raw materials in distilled water.
Table 3. The values of the respective densities.
84% bigger than that of #200 quartz sample. From Figure 1 it can be seen that the #325 quartz sample, displays a more open particle size distribution than the #200 quartz sample.
From Table 3 it can be concluded that after ball milling for 24 hours, the cristobalite sample presented a comparable specific surface area to that of #500 quartz. The vireous silica #325 presented a different specific surface area than that quartz #325 sample which nominally should be similar. On the other hand the vitreous silica #100 sample obviously presented the smallest specific surface area among all the eight silica raw materials.
In Figure 2 it can be seen that the IEP values of the three quartz samples and the silica mix, are practically the same (≈2). These are compatible to the IEP values reported in the literature for pure silica raw materials, inde-

Figure 2. Variation of zeta potential for samples of quartz.
pendent of the type of the polymorph being it crystalline or vitreous in character. Based on what was treated in the introduction of this paper, the zeta potential versus pH behavior is consistent with the expected trend. However, interestingly, for pH values just below the isoelectric point (IEP), the #500 quartz sample displayed an unexpected sudden increase (in positive mV) for its zeta potential. The same sudden increase was not observed for the #325 and #200 quartz and silica mix samples. This is remarkable, considering that all the quartz samples investigated presented the same chemical and mineralogical composition and differ basically only in the specific surface area.
It can be concluded from Figure 2 and Table 2, that for the quartz samples the bigger the specific surface area, the bigger the increase in zeta potential for pH values just below the IEP. There is a small inversion observed between the #200 and #325 quartz samples which can be accounted by the less than 20% difference in the respective specific surface areas.
The relationship of surface area effect on the zeta potential versus pH behavior followed the same trend by cristobalite samples. This can be seen in Figure 3. Note that cristobalite #500 and cristobalite #500 re-milled for 24 hours, displayed a similar trend of specific surface area effect in zeta potential versus pH behavior, presented by quartz #325 and #500 species. This behavior extends to high pH values. The bigger surface area of re-milled cristobalite #500 lead to a more negative zeta potential than the cristobalite #500, in the full range of pH = 6 and over. In addition we may conclude that the behavior of positive zeta potential versus pH, below IEP for quartz #500 and re-milled cristobalite #500 are comparable because of their similar specific surface areas which differ by only 15%. It can be noticed that #500 quartz sample, shows a variation of zeta potential as a function of pH in the range from about −40 mV to +80 mV. On the other hand for the #500 cristobalite this variation is in the range from +25 mV to −55 mV and the re-milled cristobalite #500 shows a variation from −70 to +75 mv. We also see a similar deviation of quartz #500, cristobalite #500 and re-milled cristobalite #500 for pH values below the IEP (high acidity). The differences between the respective zeta potential variation range of the three samples, apparently rest more in their specific surface area and less on their crystalline structures. The α-quartz crystal structure is trigonal, while α-cristobalite belongs to the tetragonal crystal system [9] . Quartz structure presents smaller number of structural sites able to be hydroxylated. However, the surfaces of cristobalite particles, may acquire higher charge density at high pH than quartz, probably because of its greater number of structural holes and higher activity.
With respect to the silica mix sample (Figure 4), it can be observed that its zeta potential versus pH behavior is dominated by the #200 quartz which is its constituent in largest quantity. This indicates a dependence on the predominating mix component for zeta potential versus pH behavior for quartz samples. For pH values above the IEP the zeta potential versus pH behavior, is almost identical for all the four different quartz samples reaching a minimum around minus −40 mV, exception made to the silica mix, which presented a deviation for more negative values for pH ≥ 12.
The representative dissolution of silica species in water is given by [9] :
 (6)
(6)

Figure 3. Zeta Potential versus pH for suspensions of quartz #500, cristobalite #500 and re-milled cristoballite #500.

Figure 4. Variation of Zeta potential for samples of quartz #200 and “silica mix”.
There are two types of hydration water in equilibrium with solid quartz:
· One bonded directly to SiO2 surfaces forming OH− groups.
· The other type is the water bonded through hydrogen bonding.
This fact implies that the SiO2:H2O ratio is less than 1:2.
In addition the H4SiO4(aq) species is a weak acid that dissociates appreciably for pH values over two pH points above IEP. This is shown schematically by Reaction (7):
 (7)
(7)
There is also the presence of dimmers 2H4SiO4(aq), which exist in greater numbers in vitreous silica water suspensions. These species are in equilibrium in water as shown by Reaction (8):
 (8)
(8)
From Reaction (7), a buffering like behavior can be expected considering that for each mole of SiO2 dissolved there is a counterpart of one mole of H+ formation. This fact comes to explain why the zeta potential of silica suspensions does not change appreciably for pH values greater than two pH points above the IEP. This is more evident for quartz samples. As pointed out cristobalite is expected to display a lesser buffering capacity than quartz because of its lower activity and this may be the cause of its somewhat higher increase in negative zeta potential above pH = 6.
Vitreous silica seems to violate strongly the regularities found with the crystalline silica species, considering that the zeta potential versus pH behavior of this material does not show the same relationship with specific surface area for pH values smaller than IEP, neither follows the buffering action for pH values above two pH points of its IEP as shown in Figures 5-7.
From Figures 5-7 it can be noticed that for values below the IEP, the two samples of vitreous silica display almost the same behavior, reaching the maximum common value of about +33 mV. This value is bigger than those of quartz samples of medium to low specific surface areas. On the other hand for pH values above IEP, the specific surface vitreous silica (#325) is 30% higher than that of vitreous silica #100. It seems that this difference contributed to the higher rate of change of its negative zeta potential, reaching a considerably more negative value at very high pH. Based on this fact we may infer that higher specific surface areas for vitreous silica results in a higher concentration of dimmer species which leads to higher adsorption of OH- groups, increasing the negative charge on vitreous silica particles in water suspension, turning its zeta potential more negative.
Finally in Figure 8 it can be observed that when the sample of cristobalite #500 was re-milled, its specific surface area increased about 31%, causing equally a steep increase in the variation of zeta potential with pH, similar to what happened to the quartz sample. These facts demonstrate a visible relation between the values of zeta potential and the specific surface area for silica samples, even in the case of samples of different crystalline structures or being vitreous in nature.

Figure 5. Variation of Zeta potential for samples of quartz #325 and vitreous silica #100.
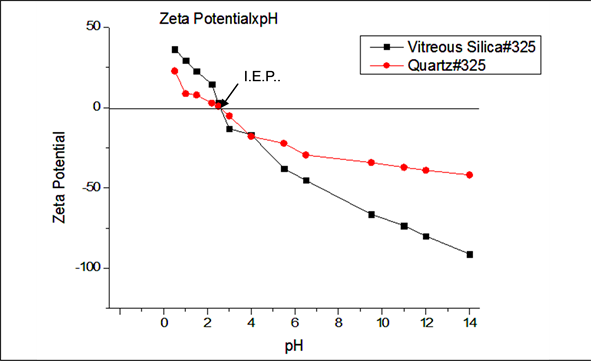
Figure 6. Variation of Zeta potential for samples of quartz #325 and vitreous silica #325.

Figure 7. Variation of Zeta potential for samples of vitreous silica #100 and vitreous silica #325.

Figure 8. Variation of Zeta potential for samples of quartz #500, cristobalite #500 and re-milled cristobalite #500.
4. Conclusions
Through this work it was possible to identify different behaviors of the zeta potential versus pH for silica samples, crystalline or vitreous, which was found remarkably different from pH values below and above the IEP.
The pH at IEP was practically the same for all the different silica samples studied. However, it was found that for the crystalline samples of silica (quartz and cristobalite) the higher the specific surface area of the used powders, the greater the rate of increase in positive mV of the zeta potential, for pH values just below the respective IEP. For pH values in a wide range above the IEP, all the crystalline samples displayed practically the same behavior of zeta potential versus pH.
Conversely, for vitreous samples the opposite was found. That is, the surface area the rate of change in zeta potential was higher towards more negative values, while for pH values below IEP the variation was small and identical for samples of vitreous silica with smaller specific surface areas.
Understanding how the zeta potential varies with crystalline and vitreous character and its dependence on powder surface area of silica raw materials will help to predict and control the rheological behavior of the silica slips. In this manner the stabilization and destabilization of these ceramic materials under colloidal processing, can be considerably facilitated.
Acknowledgements
The authors thank FAPESP process number 98/14324-0 Capes Agency, process number 33001014004P9.
References
- Hunter, J. (1988) Zeta Potential in Colloid Science: Principles and Applications. Academic Press, UK, 187-203.
- Hunter, J. (1993) Everything You Want to Know about Coagulation & Floculation and Zeta Meter. Academic Press, UK, 26-39.
- Reed, J.S. (1988) Introduction to the Principles of Ceramic Processing. EBS, New York, 265-274.
- Zhang, Y. and Binner, J. (2008) Effect of Dispersants on the Rheology of Aqueous Silicon Carbide Suspensions. Ceramics International, 34, 1381-1386. http://dx.doi.org/10.1016/j.ceramint.2007.03.030
- Pugh, R.J. and Bergström, L. (1994) Surface and Colloidal Chemistry in Advanced Ceramics Processing. Journal of the American Ceramic Society, 22, 94-104.
- Gauckler, L.J., Graule, T.H. and Baader, F. (1999) Ceramic Forming Using enzime Catalyzed Reactions. Materials Chemistry and Physics, 68, 78-102. http://dx.doi.org/10.1016/S0254-0584(99)00117-0
- Bisesier, J.G.P., Mcdermott, A.M., Yin, Y., Sambrook, R.M. and Vaidhyamatham, B. (2006) In Situ Coagulation Moulding—A New Route for High Quality Net Shape Ceramics. Ceramics International, 32, 29-35. http://dx.doi.org/10.1016/j.ceramint.2004.12.006
- Frenkel, D. (2002) Colloidal Systems: Playing Tricks with Designers Atoms. Ceramics International, 296, 65.
- Dove, P.M. and Rimstidt, J.D. (1994) Silica Water Interactions—In Silica Reviews in Mineralogy. Mineral Society of America, 29, 259-307.
- Ning, S., Li, H., Chen, W., Liu, B. and Chen, S. (2005) Effects of Surface Oxide Species and Contents on SiC Slurry Viscosity—Rare Metals. Mineral Society of America, 24, 240-245.
- Prezzi, M., Monteiro, P.J.M. and Sposito, G. (1997) Use of Double Layer Theory to Explain the Behavior of Reaction Products Gels. ACI Materials Journal, 11, 10-11.
- Alexander, S., Chaikin, P.M., Grant, P., Morales, G.P., Pincus, P. and Hone, D. (1984) Charge Renormalization, Osmotic Pressure, and Bulk Modulus of Colloidal Crystals Theory. Journal of Chemical Physics, 80, 5776-5781. http://dx.doi.org/10.1063/1.446600