International Journal of Organic Chemistry
Vol.06 No.03(2016), Article ID:70839,9 pages
10.4236/ijoc.2016.63018
Semi-Automated Synthesis of [F-18]FBAM, a Thiol Reactive Prosthetic Group, Using Continuous Flow Chemistry
Murthy R. Akula1, Srinivasa R. Marepally1, Lee T. Collier2, W. George Kabalka1
1Departments of Chemistry and Radiology, University of Tennessee, Knoxville, Tennessee, USA
2Advion Biosystems, Ithaca, NY, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: May 3, 2016; Accepted: September 23, 2016; Published: September 26, 2016
ABSTRACT
[F-18]FBAM, a thiol reactive bifunctional agent, was successfully synthesized using continuous flow chemistry in a micro reactor that is part of Advion NanoTek Microfluidic Synthesizer. As the radiofluorination was carried out microfluidically, a very small amount of precursor was used and over all radiochemical yield was 38% ± 4% (n = 8, decay corrected) and the radiochemical purity was ≥98% with specific activity of 430 mC/µmol. The total reaction time including HPLC purification was 55 min that is 14 min more than manual synthesis and 6 min less than fully automated synthesis.
Keywords:
Bifunctional Agent, Micro-Reactor, 6-Bromohexnol, Radiofluorination, Appel Reaction

1. Introduction
Bioactive peptides and proteins are important key regulators in cell growth and cellular functions in living organisms [1] - [3] . Various peptides, proteins, antibodies, antibody fragments and nucleotides have been radiolabeled and used to image tumors and inflammatory processes [4] . Among these tracers, [F-18] labeled molecular probes are increasingly popular because of the ease of production of 18F and its favorable properties. However, harsh reaction conditions required to directly radiofluorinate these sensitive biomolecules hampered the preparation of the requisite tracers in good yields and high specific activity. To address these problems, radiochemists have taken advantage of bifunctional agents, also known as prosthetic groups. These prosthetic groups are categorized into three classes amine, thiol and carboxylic reactive. Notable thiol reactive bifunctional agents include [18F] FBAM [5] [6] , [18F]FBOM [7] , [18F]FBABM [8] , [18F]FPyAM [9] and [18F]FPyMe [10] (Figure 1).
Microfluidics represents a useful approach to conduct the reactions with minimal quantities of expensive precursors; other advantages include reduced reaction times and increased radiolabeling yields. These effects are realized due to the high surface to volume ratio encountered while the reagents are flowing through the microchannel which is accompanied by rapid mixing of the reagents leading to increased heat transfer between the reactants. One of the commercially available microfluidic devices with micro-channel system (MCS) is the Advion NanoTek Microfluidic Synthesizer. This unit consists of three modules called the reagent, reactor and concentrator modules. The isotope is dried and dissolved in the concentrator module and transferred to a loop attached to the reactor module while the precursor is stored in a second loop attached to reagent module. The reagents are then meter delivered and passed through the micro- reactor consisting of a 100 μm channel made of quartz. We wish to report an improved synthesis of [18F] FBAM utilizing this microfluidic system involving continuous flow.
2. Results and Discussion
2.1. Chemistry
The reported procedure for preparing requisite precursor 16 required synthesizing [18F] FBAM via the Mitsunobu reaction of tert-butyl N-[(6-hydroxyhexyl)oxy] carbamate, 6 with maleimide, 8, in the presence of Ph3P, diisopropyl acetylene dicaboxylate, and DMF at −78˚C to obtain 7 (Scheme 1).

Figure 1. Notable thiol reactive bifunctional agents.
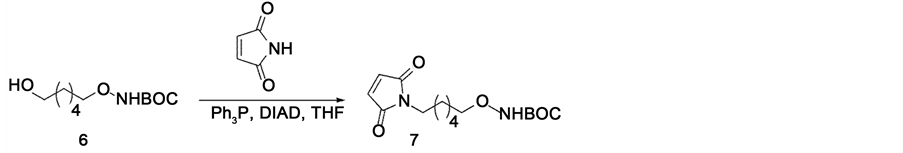
Scheme 1. Mitsunobu reaction.
Attempts to directly N-akylate maleimide 8 with various alkyl bromides failed or produced very poor yields (Scheme 2). We then treated maleimide 8 with either NaH or K2CO3 in THF or DMF at room temperature followed by the addition of 1,6-dibro- mohexane, 9; after, stirring the reaction mixture at reflux only trace amounts of the desired product 11 were produced. Adding sodium iodide to catalyze the reaction did not improve the yield. Further experiments using the bromide 10 as an alkylating agent also did not result in the desired product 7. It is possible that the N-Alkylation of maleimide does not proceed as expected because the maleimide anion underwent a 1,4-addition to another molecule of maleimide instead of reacting with alkyl bromide.
Precursor 16 was successfully prepared from 6-bromohexanol, 12, in six steps in an overall yield of 24.6% (Scheme 3). tert-Butyl-N-hydroxycarbamate, 13, was then O-al- kylated with 6-bromohexanol, 12, to obtain alcohol 6, using 1,8-diazabicyclo[5.4.0] undecene (DBU) as the base in DMF at room temperature. Appel reaction [11] of alcohol 6 to bromide 10 was achieved with carbon tetrabromide and triphenylphosphine in CH2Cl2 by stirring the reaction mixture at room temperature. Bromide 10 was allowed to react with sodium azide in DMF at 80˚C to obtain azide 14 in nearly quantitative yield. The crude azide 14 was then hydrogenated with 5% Pd-C in EtOAc to obtain amine 15 [12] . Reaction of amine 15 with N-methoxycarbonyl-maleimide in thepresence of NaHCO3 bin refluxing in dioxane afforded 7. The Boc-protecting group was readily removed with HCl/EtOAC at room temperature to obtain 16 as the HCl salt. The condensation of 16 with 4-fluorobenzaldehyde in DMF at room temperature gave the 17 as a white solid.
The two step radiosynthesis of [18F]FBAM, 1, was performed in the Advion NanoTek Microfludic Synthesizer (Scheme 4). Using the drying macros of NanoTek LF 1.4 software, a complex of Kryptofix 222/K2CO3/[18F]fluoride was thoroughly dried and allowed to react with 4-N,N,N-trimethylamino-benzaldehyde triflate, 18, in a microreactor (2 m × 100 μ) at 120˚C to obtain 4-[18F]fluorobenz-aldehyde, 19. The labeling efficiency under microfluidic conditions was compared with the previously reported procedures (Table 1). The outlet tube from the reactor was immersed in a reaction vial containing precursor 16 (8 mg) dissolved in a 1:1 mixture of 1 N HCl:MeOH which was then heated at 75˚C for 10 min to obtain [18F]FBAM, 1, in higher overall radiochemical yield (38% ± 4%) when compared to the earlier reports (29% ± 4%). The crude product 1 was subjected to C18 Sep-Pak solid phase extraction before purifying on semi prepara- tive HPLC. [18F]FBAM (29 mCi) was obtained from 100 mCi of [18F] fluoride in 55 min, including HPLC purification, in a radiochemical purity of ≥98%. The identity of the product was confirmed using analytical HPLC by co-elution with 17.
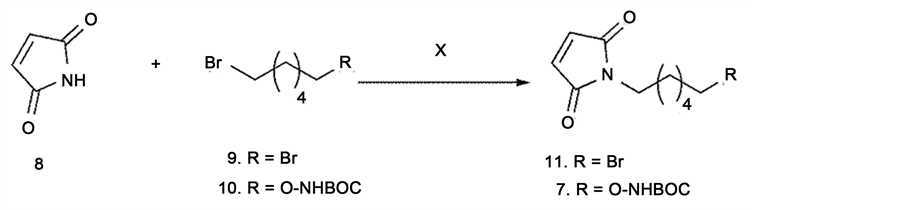
Scheme 2. N-alkylation of maleimde.
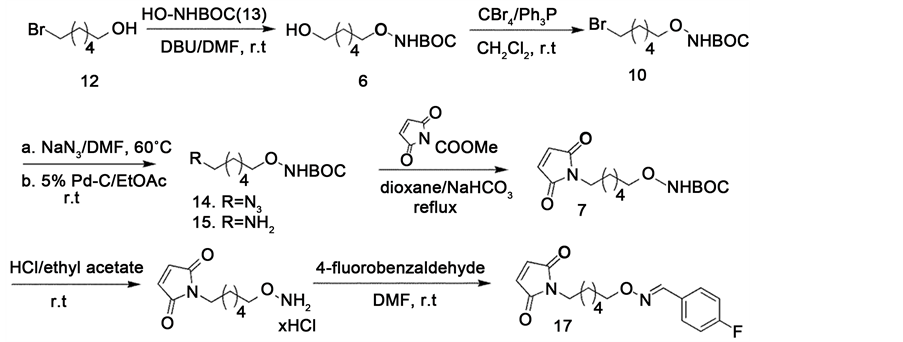
Scheme 3. Synthesis of precursor 16 and the standard 17.
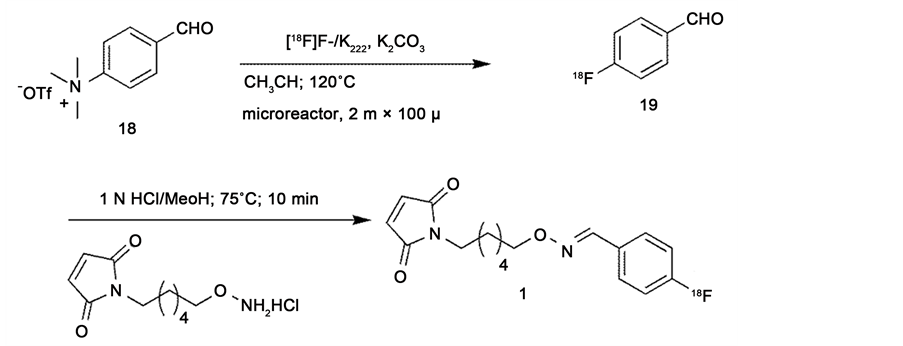
Scheme 4. Synthesis of [18F]FBAM (1).

Table 1. Comparison of amount of precursor 18 used to obtain compound 1.
2.2. Materials and Methods
All reagents and solvents were purchased from Acros or Aldrich and were used as received. Flash column chromatography was performed using silica gel (60 Å, 230 - 400 mesh, Sorbent Technologies, USA) [13] . Analytical thin-layer chromatography (TLC) was performed using 250 μm silica plates (Analtech, Inc., Newark, DE) with a visualization by UV (254 nm) or phosphomolebdic acid spray. 1H and 13C-nuclear magnetic resonance spectra (NMR) were recorded at 300 or 125 MHz, respectively. Chemical shifts for 1H-NMR and 13C-NMR spectra were referenced to the residual protons of the deuterated solvents or to TMS. High resolution mass spectrometry was performed using a JEOL AccuTOF™ DART Mass Spectrometer. tert. Butyl N-[(6-hydroxyhexyl)oxy] carbamate was prepared following the literature procedure [14] . No-carrier-added [18F]F-, produced from recycled [18O] water, was obtained from PetNet (Knoxville, TN). Thin-layer chromatography visualization was performed with radiation detectors using a BioScan AR-2500 radio-TLC reader and Win Scan 1.3 software. Radio-TLC plates were developed using EtOAc/hexane (50/50). Analytical radio-HPLC analyses were performed on an Agilent 1200 series instrument employing a 254 nm UV detector and a Phenomenex Luna C18 column, 5 µ, 4.6 × 250 mm, using 80% acetonitrile/20% 0.1 N ammonium formate at a flow rate of 1mL/min. F-18 labelling was performed in 100 µm × 2 m reactor using Advion NanoTek Microfluidic Synthesis System controlled by NanoTek LF 1.4 Software.
tert-Butyl N-[(6-Hydroxyhexyloxy]carbamate (6)
To a magnetically stirred solution of N-(tert-butyloxycabonyl)hydroxylamine, 13, (5.00 g, 37.6 mmol) and 6-bromohexane-1-ol, 12, (2.71 g, 1.50 mnol) was added DBU (11.4 g, 7.50 mmol) over a period of 5 min. The mixture was allowed to stir for 24 h and then dissolved in CH2Cl2 (200 mL), and the resulting solution was washed sequentially with 1 N HCl (4 × 25 mL) and brine (25 mL). The aqueous portion was discorded and the CH2Cl2 was dried (MgSO4), filtered, and concentrated. The resulting yellow oil was purified by silica gel flash chromatography (3:7, EtOAc:hexane) to provide tert-butyl N-[(6-hydroxyhexyloxy]-carbamate, 6, (8.48 g, 75%). 1H NMR CDCl3 (δ) 1.39 (m, 4H), 1.47 (s, 9H), 1.57 - 1.66 (bm, 4H), 3.62 (t, 2H) and 3.84 (t, 2H); 13C NMR CDCl3 (δ) 25.4, 25.6, 27.9, 28.4, 32.5, 62.7, 76.7, 81.5, 157.2; HRMS (ES) calculated for (M+Na) C11H23NaNO4: 256.1525. Found: 256.1530.
tert-Butyl N-[(6-Bromohexyloxy]carbamate (10)
Triphenylphosphine (9.00 g, 34.0 mmol) and tert-butyl N-[(6-hydroxyhexyloxy] carbamate, 6, (4.40 g, 17.2 mmol) were dissolved in anhydrous dichloromethane (30 mL) under an argon atmosphere. Pyridine (2.8 mL, 34.0 mmol) and carbon tetrabromide (1.90 mL, 17.0 mmol) were added, and the reaction mixture stirred at room temperature for 24 h. The mixture was poured into water, extracted with ether (2 × 50 mL) and the combined organic extracts were dried (anhydrous MgSO4), filtered, and concentrated. The product was purified by silica gel flash chromatography (EtOAc/hexane, 2:7) to provide tert-butyl N-[(6-bromohexyloxy]carbamate, 10, as a colorless oil (4.13 g, 82%). 1H NMR CDCl3 (δ) 1.39 (m, 4H, CH2), 1.47 (s, 9H, CH3), 1.57 - 1.66 (bm, 4H, CH2), 3.26 (t, 2H, CH2Br) and 3.84 (t, 2H, OCH2); 13C NMR CDCl3 (δ) 25.7, 25.9, 28.2, 28.4, 32.5, 32.9, 76.7, 81.5, 157.2; HRMS (ES) calculated for (M + Na) C11H22NaNO3Br: 318.0681. Found: 318.0685.
tert-Butyl N-[(6-Aminohexyloxy]carbamate (15)
A mixture of tert-butyl N-[(6-bromohexyloxy]carbamate, 15 (1.27 g, 4.30 mmol), sodium azide (0.52 g, 8.0 mmol) and 18-crown-6 (0.53 g, 0.10 mmol) in anhydrous benzene (10 mL) was stirred overnight at 60˚C. Insoluble materials were filtered off and benzene was removed under vacuo to obtain the crude azide 14 (0.98 g) which was subjected to hydrogenation without purification. Azide 14 (0.98 g) was dissolved in dry EtOAc (20 mL) and hydrogenated at atmospheric pressure with 5%-Pd/C using a hydrogen balloon. The catalyst was removed by filtration, the solvent evaporated and the product purified using silica gel flash chromatography (EtOAc/hexane, 2:7) to give tert-butyl N-[(6-aminohexyloxy-carbamate)], 15 (0.53 g; 54%). 1H NMR CDCl3 (δ) 1.40-1.78 (m, 8H, CH2), 1.46 (s, 9H, CH3), 2.62 (t, J = 7.3 Hz, 2H, CH2NH2 ) and 3.78 (t, J = 7.3 Hz, 2H, CH2O); 13C NMR CDCl3 (δ) 25.3, 25.8, 27.9, 28.4, 32.5, 41.9, 76.7, 81.5, 157.2; HRMS (ES) calculated for (M + Na) C11H24NaN2O3: 255.1735. Found: 255.1738.
tert-Butyl N-{[6-(1-Maleimidyl)hexyl]oxy}carbamate (7).
To a solution of tert-butyl N-[(6-aminohexyloxy]carbamate, 15, (1.1 g, 5.0 mmol) in THF (100 mL) was added N-methoxycarbonylmaleimide (2.43 g, 25.0 mmol). The mixture was stirred overnight at reflux. A precipitate was formed. After filtration, the filtrate was concentrated to dryness, and the residue purified by flash column chromatography using silica gel and EtOAc/hexane (2:7) to give pure tert-butyl N-{[6-(1-ma- leimidyl)hexyl]oxy}carbamate, 7, (1.21 g, 78%). 1H NMR CDCl3(δ) 1.47 (s, 9H, CH3), 1.21 - 1.61 (bm, 8H, CH2), 3.49 (t, J = 7.4 Hz, 2H, NCH2,), 3.84 (t, J = 6.9 Hz, 2H, OCH2), 6.64 (s, 2H, maleimide) and 7.10 (br s, 1H, NH). 13C NMR CDCl3 (δ) 25.4, 25.6, 27.9, 28.4, 32.5, 62.7, 76.7, 81.5, 157.2; HRMS (ES) calculated for (M+Na) C15H24NaN2O5: 335.3513. Found: 335.3518
N-(6-Aminoxyhexyl)maleimide.HCl (16).
A solution of tert-butyl N-{[6-(1-maleimidyl)hexyl]oxy}carbamate, 7, (1.20 g, 3.84 mmol) in 3 N HCl/EtOAc (1:1, 40 mL) was stirred for 30 min at room temperature. The solvent was removed under reduced pressure, and the residue dissolved in methanol (10 mL). Diethyl ether (100 mL) was added and the white suspension was filtered to obtain N-(6-aminoxyhexyl)maleimide HCl, 16, (0.90 g, 95%), mp =132˚C - 133˚C (lit. 135˚C - 137˚C). 1H NMR DMSO (δ) 1.21 - 1.52 (bm, 4H, CH2), 1.61 - 1.74 (bm, 4H, CH2), 3.48 (t, J = 7.4 Hz, 2H, NCH2), 3.88 (t, J = 6.9 Hz, 2H, OCH2), 6.84 (s, 2H, maleimide) and 7.10 (br s, 1H, NH). 13C NMR CDCl3(δ), 24.34, 26.78, 27.59, 30.72, 48.60, 49.58, 134.30 and 170.8
N-[6-(4-Fluorobenzylidine)aminooxyhexyl]maleimide (17)
A mixture of 4-fluorobenzaldehyde (0.037 g, 0.40 mmol) and N-(6-aminoxyhexyl) maleimide.HCl, 8, (0.05 g, 0.20 mmol) in dimethyl formamide (5 mL) was stirred for 30 min at room temperature. The reaction mixture was diluted with water (30 mL) and extracted with diethyl ether (2 × 25 mL). The ether extracts were washed with brine (10 mL) and dried (anhydrous MgSO4). The solvent was evaporated under reduced pressure and the crude product was purified using silica gel flash chromatography (EtAcO/hexanes, 7:3) to give N-[6-(4-fluorobenzylidine)aminooxyhexyl]maleimide, 17, (0.06 g, 94%) as a white solid. 1H NMR CDCl3 (δ) 1.21 - 1.70 (bm, 8H, CH2), 3.48 (t, J = 7.4 Hz, 2H, NCH2, ), 4.10 (t, J = 6.9 Hz, 2H, OCH2), 6.68 (s, 2H, maleimide), 7.01 (m, 2H, Ar), 7.51 (dd, J = 5.4, 8.2, 2H, Ar) and 8.06 (s, 1H, CHN). 13C NMR CDCl3 (δ) 26.41, 27.05, 30.72, 49.56, 59.38, 115.73, 130.06, 134.31, 134.75, 163.35 and 170.82; HRMS (ES) calculated for (M + Na) C15H24NaN2O3: 322.7864. Found: 322.7868
N-{6-(4-[18F]Fluorobenzylidine)aminooxyhexyl}maleimide (1)
The details of operation of the NanoTek Microfluidic System have been described previously (Pike 2009, 2010). Cyclotron-produced, no-carrier-added [18F]fluoride ion (100 mCi) in [18O]water (225 - 350 μL) was first adsorbed onto an anion exchange resin ORTG cartridge within the concentrator module of a NanoTek apparatus (Advion Biosciences), and then released with a solution of K2CO3 (1.8 mg) plus K2.2.2 (12.0 mg) in MeCN/H2O (9.5:0.5 v/v; 400 μL) into a 5 mL V-vial. The solution was dried by three cycles of azeotropic evaporation with MeCN (0.45 mL) at 100˚C. The dry 18F−-K2.2.2-K+ complex (70 mCi) was dissolved in MeCN (0.5 mL). The isotope solution was then loaded into the loop of the reactor module (431 µL), and 4-N, N, N-trimethyammo- niumbenzaldehyde, 18, (2.5 mg in 0.5 mL) solution was loaded into the other loop on the reagent module (431 µL). These solutions were concurrently infused into a 2 m long micro reactor coil (100 µm) at a combined flow rate of 200 µL/min. The radiofluorinated product exiting the micro reactor was collected in a vial, in the concentrator module, containing maleimide precursor 16 (8 mg) dissolved in a mixture of methanol and 1.0 N HCl (1 mL, 50/50) and the resulting mixture was heated at 75˚C for 10 min. After cooling, the mixture was diluted with water (15 mL) and passed through C18 Sep- Pak cartridge to eliminate water soluble precursor and any unreacted isotope. The product was eluted with acetonitrile (3 mL). The pure product was isolated by semi- preparative HPLC column (Phenomenex Luna reverse phase column, 250 × 10 mm, 10 µ), using gradient elution (A: CH3CN, B: 0.1 M ammonium formate, 0.5 min 40% A and 60% B; 5 - 15 min 40%A - 70% A and 15 - 30 min 70%A, flow rate 4 mL/ min). A fraction (16 - 18 min) was collected, diluted with water (10 mL), and passed through a C18 Sep-Pak cartridge to trap the desired product. The cartridge was then washed with diethyl ether (2 mL) and the solvent evaporated under a stream of dry N2 to afford 29 mCi (38% decay corrected) of [18F]FBAM. The identity of the product was confirmed using analytical HPLC by co-injection with a reference standard, 17, Rt = 6.4 min.
3. Conclusion
[18F]FBAM was successfully synthesized by continuous flow chemistry using an Advion NanoTek Microfludic Synthesis System in high radiochemical yield (38% ± 4%, n = 4; previously reported 29% ± 4%) and radiochemcial purity of ≥98%. The requisite key precursor N-(6-amino-oxyhexyl)maleimide.HCl, 16, was prepared by a different method then that previously reported. Smaller quantities of expensive precursors were used for the synthesis under microfluidic conditions. The overall time for the synthesis was 55 min and the specific activity was determined to be 430 mCi/µmol.
Cite this paper
Akula, M.R., Marepally, S.R., Collier, L.T. and Kabalka, W.G. (2016) Semi-Automated Synthesis of [F-18]FBAM, a Thiol Reactive Prosthetic Group, Using Continuous Flow Chemistry. International Journal of Organic Chemistry, 6, 177-185. http://dx.doi.org/10.4236/ijoc.2016.63018
References
- 1. Wilbur, D.S. (1992) Radiohalogenation of Proteins—An Overview of Radionuclides, Labeling Methods, and Reagents for Conjugate Labeling. Bioconjugate Chemistry, 3, 433-470. http://dx.doi.org/10.1021/bc00018a001
- 2. Sosabowski, J., Melendez-Alafort, L. and Mather, S. (2003) Radiolabelling of Peptides for Diagnosis and Therapy of Non-Oncological Diseases. The Journal of Nuclear Medicine, 47, 223-237.
- 3. Giblin, M.F., Veerrendra, B. and Smith, C.J. (2005) Radiometallation of Receptorspecific Peptides for Diagnosis and Treatment of Human Cancer. In Vi-vo, 19, 9-29.
- 4. Rennen, H.J., Corstens, F.H., Oyen, W.J. and Boerman, O.C. (2001) New Concepts in Infection/Inflammation Imaging. The Journal of Nuclear Medicine, 45, 167-173.
- 5. Brendt, M., Pietzsch, J. and Wuest, F. (2007) Labeling of Low-Density Lipoproteins Using the 18F-Labeled Thiol-Reactive Reagent N-[6-(4-[18F]fluorobenzylidene)aminooxyhexyl] ma-leimide. Nuclear Medicine and Biology, 34, 5-15. http://dx.doi.org/10.1016/j.nucmedbio.2006.09.009
- 6. Kniess, T., Kuchar, M. and Pietzsch, J. (2011) Automated Radiosynthesis of the Thiol- Reactive Labeling Agent N-[6(4-[F-18]fluorobenzylidene)aminooxyhexyl]maleimide ([18-F)]FBAM). Applied Radiation and Isotopes, 69, 1226-1230. http://dx.doi.org/10.1016/j.apradiso.2011.03.043
- 7. Wuest, F., Kohler, L., Berndt, M. and Pietzsch, J. (2009) Systematic Comparison of Two Novel, Thiol Reactive Prosthetic Groups for F-18 Labeling of Peptides and Proteins with the Acylation Agentsuccinimidyl-4-[F-18]fluorobenzoate ([F-18]SFB). Amino Acids, 36, 283-295. http://dx.doi.org/10.1007/s00726-008-0065-2
- 8. Toyokini, T., Walsh, J.C., Domingez, A., Phelps, M.E., Barrio, J.R., Gambhir, S.S. and Satyamurty, N. (2003) Synthesis of a New Heterobi-functional Linker, N-[4-(aminooxy) butyl]maleimide, for Facile Access to a Thiol-Reactive 18F-Labeling Agent. Bioconjugate Chemistry, 14, 1253-1259. http://dx.doi.org/10.1021/bc034107y
- 9. Moore, T., Akula, M. and Kabalka, G.W. (2016) Fluorine-18 Radiochemistry a Novel Thiol Reactive Prosthetic Group [18F]FPyAM. Natural Science, 8, 1-7. http://dx.doi.org/10.4236/ns.2016.81001
- 10. Bruin, B., Kuhnast, Bhinnen, F., Yaouanc, Q., Lamessou, M., Johannes, L., Samson, L., Boisgard, R., Tavitian, B. and Dolle, F. (2005) 1-[3-(2-[18F]Fluoropyridin-3-yloxy)propyl] pyrrole-2,5-dione: Design, Synthesis, and Radiosynthesis of a New [18F]fluoropyridine-Based Maleimide Reagent for the Labeling of Peptides and Proteins. Bioconjugate Chemistry, 16, 406-420.
- 11. Seebach, D., Pichota, A., Beck, A., Pinkerton, A.B., Litz, T., Karjalainen, J. and Gramlich, V. (1999) Preparation of TADDOL Derivatives for New Ap-plications. Organic Letters, 1, 55- 58. http://dx.doi.org/10.1021/ol990060d
- 12. Nakajima, Y., Oda, J. and Inouye, Y. (1978) Stereochemical Studies on Azidation of b-Bromohydrins under Phese Transfer Condition. Tetrahedron Letters, 34, 3107-3110. http://dx.doi.org/10.1016/S0040-4039(01)94955-X
- 13. Still, W.C., Kahn, M. and Mitra, A. (1978) Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution. Journal of Organic Chemistry, 43, 2923-2925. http://dx.doi.org/10.1021/jo00408a041
- 14. Jones, D.S., Ammaker, J.R. and Tedder, M.E. (2001) A Convenient Synthesis of N-(tert- butyloxycarbonyl)aminooxyethers. Tetrahedron Letters, 41, 1531-1533. http://dx.doi.org/10.1016/S0040-4039(99)02331-X